Introduction
Transmissible spongiform encephalopathies (TSEs) or
prion diseases are a family of neurodegenerative disorders caused
by the accumulation of the pathological prion protein, PrPSc
(1,2). PrPSc is a β-sheet rich structure
protein resistant to proteinase K and is derived from the normal
cellular prion protein, PrPc, which is α-helix rich structure
protein and is sensitized to proteinase K (3–5).
The neuronal aggregation of PrPSc or neuronal cells
exposed to the prion protein (PrP) fragment [PrP (106–126)] induces
mitochondrial malfunctions which have been reported as major
hallmarks of neurodegenerative diseases, including Huntington’s
disease and Alzheimer’s disease (6–10).
Previously it has been shown that treatment with gingerol prevents
PrP (106–126)-mediated mitochondrial neurotoxicity through the
regulation of hypoxia-inducible factor-1α (HIF-1α) activation
(11). Xie et al (12) also demonstrated that exposing
neurons to low oxygen activated HIF-1α and inhibited the activation
of the mitochondrial apoptotic pathway induced by nerve growth
factor (NGF) deprivation. These observations suggest that
regulators of mitochondrial homeostasis, including HIF-1α, may be
key factors for the protection against prion-related diseases.
Hypoxic conditions regulate gene transcription in
cellular responses to promote metabolism and angiogenesis through
adaptive processes (13–16). Accordingly, modulation of gene
transcription through HIF-1 plays an important role in hypoxia for
cell survival (17–22). It was previously suggested that
hypoxia protects neuronal cells against PrP (106–126)-mediated
neurotoxicity and that this prevention is associated with
hypoxia-mediated HIF-1α signals (22).
HIF-1 is a heterodimeric transcription factor
comprising the subunits, α and β, of which HIF-1α is widely
expressed in mammalian tissues under hypoxic conditions (19–21,23,24). Recent studies have indicated that
hypoxia-mediated HIF-1α exerts neuroprotective effects (22,23,25). According to a previous study, the
accumulation of HIF-1α attenuates PrP (106-126)-mediated
neurotoxicity by regulating AKT signaling and correlates with the
overexpression of the PRNP gene (22). Cunningham et al (26) also suggested that the upregulation
of HIF-1α protects neural stem/progenitor cells (NSPCs) against
brain injury and stroke through the modulation of Wnt/β-catenin
signals. These data suggest that the upregulation of HIF-1α may
have therapeutic benefits for neuronal damage by promoting survival
signals, such as Wnt/β-catenin pathways.
β-catenin is a protein that constitutes the cadherin
protein complex (27,28). It plays an important role in
various aspects of neurobiology, including neuronal development,
regeneration and neuronal differentiation (29,30). It has been suggested that
β-catenin/Wnt signaling regulates neural stem cell differentiation
by interacting with HIF-1α (31).
These data indicate that HIF-1α may regulate the cellular response
to adaptive processes through Wnt/β-catenin signaling (31).
Furthermore, β-catenin has been shown to play
pivotal roles associated with mitochondrial functions under
pathophysiological conditions (27,32,33). Wei et al (32) showed that exogenous Wnt1 prevents
6-hydroxydopamine (6-OHDA)-mediated mitochondrial damage through
the activation of Wnt/β-catenin signaling pathways in cellular
models of Parkinson’s disease. It has been demonstrated that the
modulation of mitochondrial functions may be a key mechanism behind
the neuroprotective effects of β-catenin signaling against
neurodegenerative diseases.
It has been suggested that HIF-1α prevents neuronal
cells from prion-induced neuronal damage through the activation of
β-catenin signals. Previous studies have shown that prion-mediated
neurotoxicity is blocked by regulating mitochondrial homeostasis
(6,7). However, there is limited knowledge
regarding the neuroprotective mechanisms of HIF-1α activation
associated with mitochondrial homeostasis induced by the activation
of β-catenin. In this study, we examined whether HIF-1α-induced
β-catenin signals protect neuronal cells against PrP
(106-126)-induced neurotoxicity. We also investigated whether the
protective effects are associated with mitochondrial homeostasis
through the regulation of β-catenin signals.
Materials and methods
Cell culture
The SH-SY5Y human neuroblastoma cell line was
obtained from the American Type Culture Collection (ATCC;
Rockville, MD, USA). Cells were cultured in minimum essential
medium (MEM; Gibco-Invitrogen, Carlsbad, CA, USA) that contained
10% fetal bovine serum (FBS) (Sigma-Aldrich, St. Louis, MO, USA)
and gentamycin (0.1 mg/ml) in a humidified incubator which was
maintained at 37°C and 5% CO2. The cells were treated
with melatonin (Sigma-Aldrich) for 12 h and then exposed to 50 μM
of PrP (106-126) with or without 10 μM of the β-catenin inhibitor,
ICG-001 (Axon Medchem BV, Groningen, The Netherlands) for 24 h.
Melatonin was dissolved in ethanol with the final ethanol
concentration in the culture medium not exceeding 0.5%. In
addition, ICG-001 dissolved in dimethylsulfoxide (DMSO) was added
to the culture medium to a final concentration of 0.1% DMSO.
PrP (106-126) treatment
Synthetic PrP (106-126) peptides with the sequence,
Lys-Thr-Asn-Met-Lys-His-Met-Ala-Gly-Ala-Ala-Ala-Ala-Gly-Ala-Val-Val-Gly-Gly-Leu-Gly,
were synthesized by Peptron (Seoul, Korea). The peptides were
dissolved in sterile DMSO at a stock concentration of 10 mM and
stored at −80°C.
Annexin V assay
Apoptosis was assessed in the detached cells using
the Annexin V assay kit (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA) according to the manufacturer’s instructions.
Annexin V measurement was performed by measuring the fluorescence
at excitation (488 nm) and at emission (525 nm) using a Guava
EasyCyte™ HT Flow Cytometer (Millipore, Billerica, MA,
USA).
Terminal deoxynucleotidyl transferase
dUTP nick end-labeling (TUNEL) assay
TUNEL analysis was performed to measure the degree
of cellular apoptosis using an in situ ApoBrdU DNA
fragmentation assay kit (Sigma-Aldrich) following the
manufacturer’s instructions. Cells were washed with
phosphate-buffered saline (PBS) and fixed with paraformaldehyde for
15 min. The cells were pre-incubated with 50 μl DNA-labeling
solution (10 μl TdT reaction buffer, 0.75 μl TdT enzyme, 8 μl
BrdUTP) for 1 h at 37°C. The cells were then incubated with 5 μl
Alexa Fluor 488-labeled-anti-5-bromodeoxyuridine (BrdUrd)-antibody
for 0.5 h at room temperature (20°C). Finally, the cells were
mounted with DakoCytomation fluorescent medium (Dako, Carpintena,
CA, USA) and were visualized under a fluorescence microscope. The
cells were then counterstained with propidium iodide to show all
cell nuclei.
Immunocytochemistry (ICC)
The SH-SY5Y neuroblastoma cells were cultured on
glass coverslips. The cells were washed with PBS and fixed with
cold acetone for 90 sec at room temperature. The cells were washed
again with PBS, blocked with 5% FBS in Tris-buffered saline and
Tween-20 (TBST) and incubated with anti-β-catenin (2 μg/ml)
monoclonal antibody for 48 h at room temperature. The unbound
antibody was removed by an additional PBS wash and the cells were
then incubated with labeled anti-mouse FITC (for anti-β-catenin
antibody) IgG antibodies (4 μg/ml) for 2 h at room temperature.
Finally, the cells were mounted with DakoCytomation fluorescent
medium (Dako) and visualized under a fluorescence microscope.
Western blot analysis
The SH-SY5Y cells were lysed in buffer comprising 25
mM HEPES, 100 mM NaCl, 1 mM EDTA, 5 mM MgCl2, 0.1 mM
dithiothreitol and protease inhibitor mixture, at pH 7.4. Equal
amounts of protein lysates were dissolved in 10–15% sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
electrophoretically transferred onto a nitrocellulose membrane.
Immunoreactivity was detected through sequential incubation with
horseradish peroxidase-conjugated secondary antibodies and enhanced
chemiluminescence (ECL) reagents. The antibodies used for
immunoblotting were β-catenin (Cell Signaling Technology, Inc.,
Beverly, MA, USA), survivin (Santa Cruz Biotechnology, Inc.),
caspase-3 (Cell Signaling Technology, Inc.), Bax (Santa Cruz
Biotechnology, Inc.), cytochrome c (BD Bioscience, Franklin
Lakes, NJ, USA), HIF-1α (Santa Cruz Biotechnology, Inc.) and
β-actin (Sigma-Aldrich). The images were examined using a
Fusion-FX7 imaging system (Vilber Lourmat, Marne-la-Vallée,
France).
Cellular fractionation
The SH-SY5Y cells were re-suspended in mitochondrial
buffer (210 mM sucrose, 70 mM mannitol, 1 mM EDTA and 10 mM HEPES),
broken by a 26-guage needle and subjected to centrifugation at 700
× g for 10 min. The post-nuclear supernatant was centrifuged at
10,000 × g for 30 min. The pellet was then used as a mitochondrial
fraction and the supernatant was used as a cytosolic fraction.
Total proteins were obtained and subjected to western blot
analysis.
Mitochondrial transmembrane potential
(MTP) assay
Alterations in MTP were evaluated using the cationic
fluorescent indicator, JC-1 (Molecular Probes, Eugene, OR, USA).
J-aggregates in intact mitochondria are evident as red fluorescence
with emission at 583 nm, indicating high or normal MTP and as green
fluorescence with emission at 525 nm, indicating low MTP when JC-1
remains in the monomeric form in the cytoplasm. The SH-SY5Y cells
were incubated in MEM containing 10 μM JC-1 at 37°C for 15 min,
washed with PBS and then transferred to a clear 96-well plate. The
Guava EasyCyte HT System (Millipore) was used to measure the JC-1
aggregate fluorescence emission at 583 nm with an excitation
wavelength of 488 nm, while the JC-1 monomer fluorescence intensity
was measured with an excitation and emission wavelength of 488 and
525 nm, respectively. The SH-SY5Y cells were cultured on coverslips
in a 24-well plate, incubated in MEM containing 10 μM JC-1 at 37°C
for 15 min, and then washed with PBS. Finally, the cells were
mounted with DakoCytomation fluorescent medium (Dako) and
visualized under a fluorescence microscope.
Statistical analyses
Data are expressed as the means ± standard deviation
(SD) and were compared using the Student’s t-test, ANOVA and
Duncan’s test using SAS statistical software. P-values <0.05
<0.01 were considered to indicate statistically significant
differences.
Results
Neuroprotective effects of HIF-1α against
prion-induced neuronal apoptosis are dependent on β-catenin
activation
Studies have suggested that exposure to low oxygen
conditions activates the Wnt/β-catenin signaling pathways (26,31). A previous study also showed that
HIF-1α plays a pivotal role as a neuroprotective factor in
prion-mediated neuronal cell death (22). Thus, in this study, we examined
whether HIF-1α induces β-catenin activation and protects SH-SY5Y
neuronal cells against prion peptide-mediated neurotoxicity. The
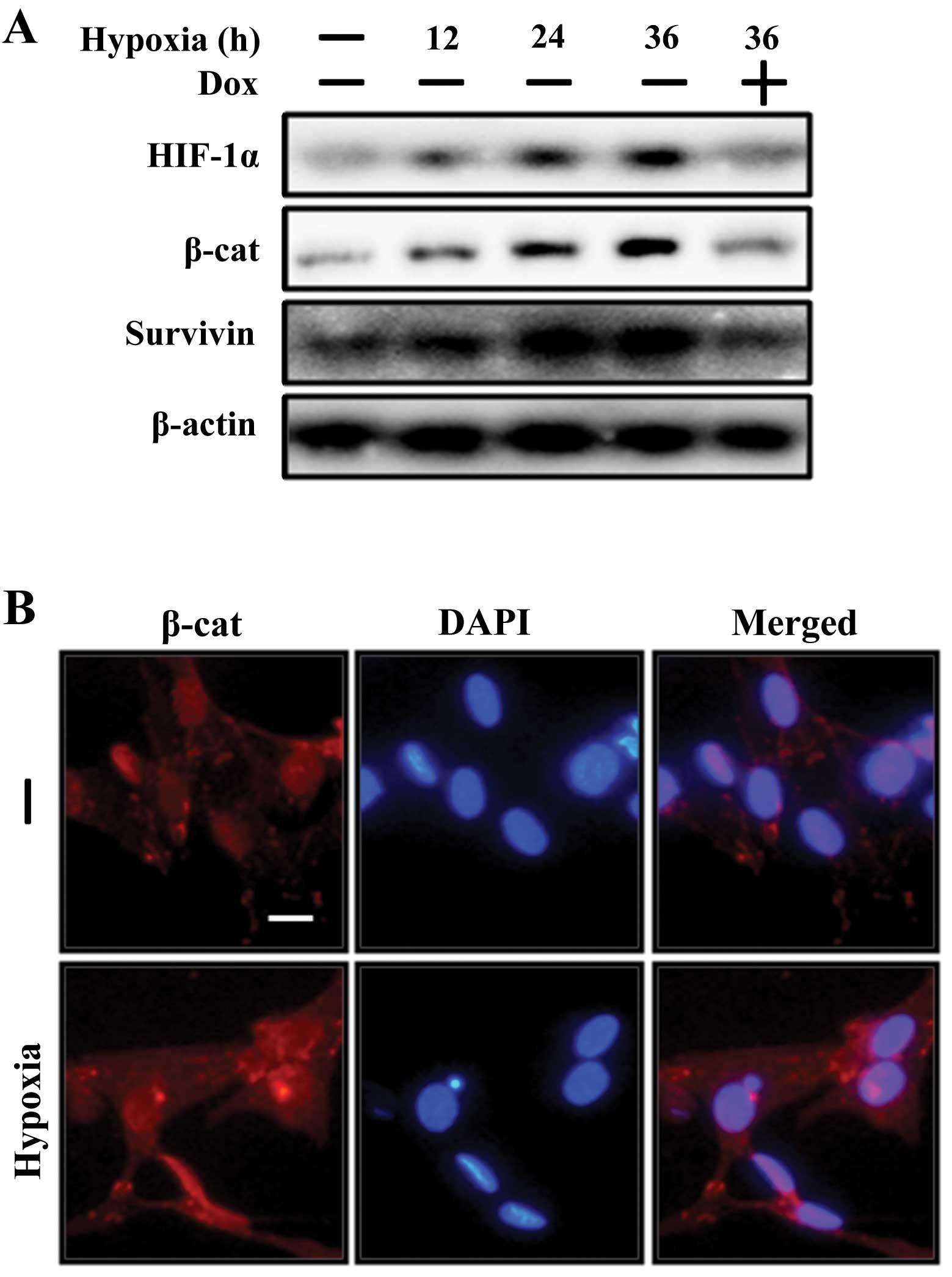
changes in β-catenin and β-catenin target gene survivin expression
levels in the SH-SY5Y cells following exposure to hypoxia with or
without the HIF-1α inhibitor, doxorubicin, were examined. It was
found that the SH-SY5Y cells exposed to hypoxic conditions for 12,
24 and 36 h had increased protein expression levels of β-catenin
and the β-catenin target gene, survivin (Fig. 1A). Consistent with this, the ICC
images revealed that the expression level of β-catenin was
increased under hypoxic conditions (Fig. 1B). However, the HIF-1α inhibitor,
doxorubicin, inhibited HIF-1α, β-catenin and β-catenin target gene
survivin protein levels (Fig.
1A). These data indicate that HIF-1α activates the β-catenin
signaling pathway in neuronal cells.
To determine whether the HIF-1α-mediated
neuroprotective effects on PrP (106-126)-induced neuronal cell
damage are associated with the activation of β-catenin signals, the
cells were exposed to hypoxiac conditions for 12 h and then treated
with 50 μM of PrP (106-126) for 24 h with or without the β-catenin
inhibitor, ICG-001 (10 μM). The results revealed that the apoptotic
cell population (Annexin V-positive) increased in the PrP
(106-126)-treated cells compared with the control groups, whereas
exposure to hypoxic conditions decreased the PrP (106-126)-mediated
apoptotic cell population (Fig. 2A
and B). However, the neuroprotective effects of hypoxia against
PrP (106-126)-mediated neuronal cell death were inhibited by the
β-catenin inhibitor, ICG-001 (Fig. 2A
and B). These results were confirmed by measuring Alexa Fluor
488-labeled anti-BrdUrd with the use of microscopic methods in
TUNEL assay microscopic images (Fig.
2C). Collectively, these results suggest that HIF-1α prevents
neurotoxicity caused by PrP (106-126) through the inhibition of
β-catenin signals.
HIF-1α-induced activation of β-catenin
pathway prevents prion-mediated mitochondrial neurotoxicity under
hypoxic conditions
As hypoxia increases β-catenin activation and
β-catenin regulates mitochondrial functions (27,34), in this study, we investigated
whether hypoxia-induced β-catenin signals exert neuroprotective
effects against PrP (106-126)-induced mitochondrial dysfunction
under hypoxic conditions. The cells were exposed to hypoxic
conditions for 12 h and then treated with 50 μM PrP (106-126) with
or without ICG-001 (10 μM). Exposure to PrP increased the cell
population in which JC-1 existed in its monomeric form, a marker of
lower MTP values, while exposure of the cells to hypoxia blocked
the PrP (106-126)-induced JC-1 monomeric appearance (Fig. 3A). However, the protective effects
of hypoxia on the neuronal cells against PrP (106-126)-mediated
mitochondrial dysfunction were inhibited by the β-catenin
inhibitor, ICG-001 (Fig. 3A).
This result was confirmed by measuring the MTP values using
fluoroscopic methods (Fig. 3B).
Similarly, mitochondrial damage induced by exposure to PrP
(106-126) resulted in the translocation of Bax and in the blockage
of cytochrome c release by hypoxic exposure, whereas ICG-001
blocked the protective effects of hypoxia against the PrP
(106-126)-induced mitochondrial apoptotic signals (Fig. 3C). Collectively, these results
confirm the hypothesis that hypoxia prevents PrP (106-126)-mediated
mitochondrial neurotoxicity by upregulating β-catenin signals.
It has been shown that hypoxia regulates β-catenin
activation through the modulation of HIF-1α stabilization (26,31) and that HIF-1α protects neuronal
cells against PrP (106-126)-induced mitochondrial damage (11). Therefore, in this study, we
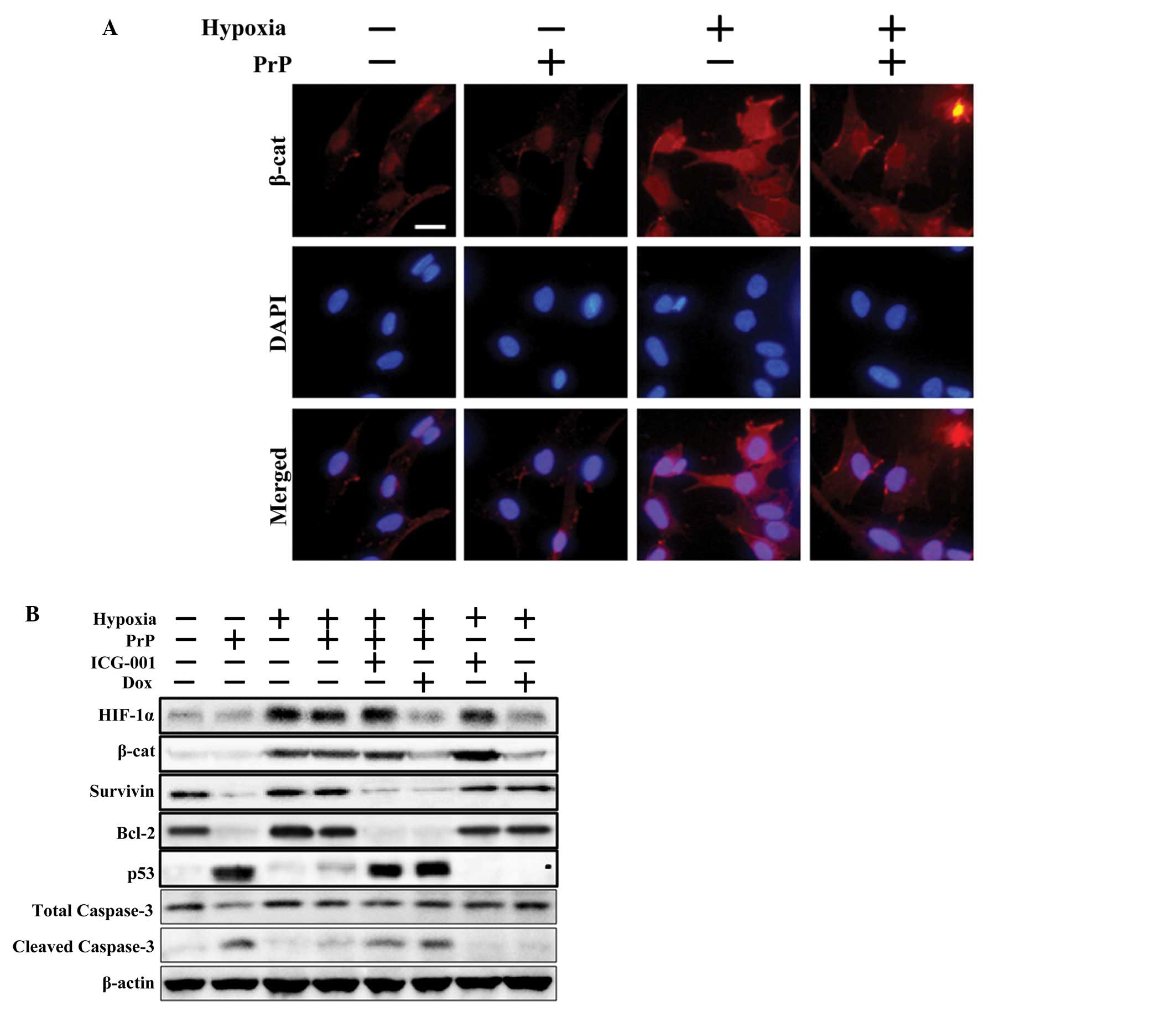
assessed whether HIF-1α has an effect on PrP (106-126)-mediated
mitochondrial toxicity by activating β-catenin signals. Initially,
β-catenin expression levels were examined in the SH-SY5Y cells
following treatment with PrP (106-126) with or without the
β-catenin inhibitor, ICG-001, under hypoxic conditions. ICC images
obtained using fluoroscopic methods revealed that hypoxia-mediated
β-catenin protein upregulation was decreased following treatment
with PrP (106-126) (Fig. 4A).
Consistent with these results, immunoblot assay revealed that the
downregulation of HIF-1α, β-catenin and survivin proteins, induced
by PrP (106-126) treatment, was reversed following exposure to
hypoxic conditions. Furthermore, ICG-001 blocked survivin
expression levels (a marker of β-catenin activation), although the
β-catenin and HIF-1α expression levels were not altered (Fig. 4B). In addition, exposure to
hypoxic conditions increased the levels of the anti-apoptotic
protein, Bcl-2, decreased those of the pro-apoptotic protein, p53,
and cleaved caspase-3 levels in the PrP (106-126)-treated cells.
However, the inhibitory effects of hypoxia against the PrP
(106-126)-induced apoptotic signals were obstructed following
treatment with the β-catenin inhibitor, ICG-001, or the HIF-1α
inhibitor, doxorubicin. Collectively, these data suggest that
HIF-1α prevents PrP (106-126)-induced apoptotic signals by
upregulating β-catenin signals.
Discussion
A previous study showed that hypoxia protects
neuronal cells against prion peptide-induced neurotoxocity by
upregulating HIF-1α (24). It has
also been demonstrated that the protective effects of HIF-1α are
associated with the regulation of mitochondrial functions (11,35). In this study, we demonstrate that
HIF-1α prevents prion-mediated mitochondrial neurotoxicity by
regulating β-catenin signaling pathways.
It has previously been demonstrated that gingerol
protects neuronal cells against PrP (106-126)-induced
neurotoxicity; the protective effects were associated with HIF-1α
activation and correlated with the regulation of mitochondrial
homeostasis (11). The activation
of β-catenin signals has been suggested to protect neuronal cells
from 6-OHDA-induced mitochondrial damage (32). In addition, a recent study
suggested that HIF-1α stabilization, induced by exposure to hypoxic
conditions, activates the Wnt/β-catenin signaling pathways
(31). However, the effects of
HIF-1α on the activation of β-catenin signals has not been reported
as a neuroprotective effect against prion disease. Therefore, in
the current study, we focused on the correlation between
hypoxia-induced HIF-1α and β-catenin signals in prion-mediated
neurotoxicity.
Studies have shown that the activation of β-catenin
prevents neurodegenerative diseases, while several lines of
evidence support a protective role for β-catenin in
neurodegenerative diseases, including Alzheimer’s and Parkinson’s
disease (36,37). L’Episcopo et al (36) suggested that
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) inhibits the
activation of Wnt/β-catenin signaling pathways, whereas
manipulation of β-catenin signaling with the inactivation of
glycogen synthase kinase-3 (GSK-3)β decreases MPTP-induced
neurotoxicity in a mouse model of Parkinson’s disease. Exogenous
Wnt1 has been reported to protect neuronal cells from 6-OHDA (a
dopaminergic neurotoxin), commonly used to generate an experimental
model of Parkinson’s disease, through the upregulation of the
Wnt/β-catenin signaling pathways (32). Consistent with this, the present
study demonstrated that the treatment of cells with PrP (106-126)
induced neuronal apoptosis (Fig.
2) and decreased β-catenin protein levels (Fig. 4B). However, exposure of the cells
to hypoxic conditions inhibited the effects of PrP (106-126)
(Figs. 2 and 4). Hypoxia also increased the Bcl-2
protein expression levels and decreased cleaved caspase-3 protein
expression levels (Fig. 4B).
Consequently, these anti-apoptotic effects of hypoxia were blocked
by the β-catenin inhibitor, ICG-001, or the HIF-1α inhibitor,
doxorubicin (Figs. 2 and 4). These results suggest that the
regulation of β-catenin pathways may be a key mechanism behind the
neuroprotective effects of HIF-1α against prion-mediated neuronal
cell death.
On the other hand, the protective role of HIF-1α in
the cellular response to oxidative damage, including hypoxia has
been supported by several lines of evidence (31,38). Mazumdar et al (31) showed that hypoxia-HIF-1α
stimulates Wnt/β-catenin signaling through β-catenin activation and
the expression of the downstream effectors, lymphoid
enhancer-binding factor-1 (LEF-1) and T cell factor-1 (TCF-1). In
addition, a recent study suggested that HIF-1α upregulates neuronal
stem cell differentiation through the regulation of the Notch and
Wnt/β-catenin signaling pathways (26), while other studies have found that
β-catenin exerts protective effects against mitochondrial
malfunctions in metabolic diseases (27,33). Lehwald et al (27) reported that Wnt/β-catenin
signaling regulates liver metabolism by maintaining mitochondrial
homeostasis. However, the correlation between the protective
effects of hypoxia-induced HIF-1α and β-catenin on mitochondrial
dysfunction in prion-mediated neurotoxicity remains to be
elucidated. Thus, the effects of hypoxia-induced β-catenin on PrP
(106-126)-mediated mitochondrial damage were also investigated in
this study. The results revealed that hypoxia-induced β-catenin
protected the SH-SY5Y neuronal cells from PrP (106-126)-mediated
mitochondrial damage (Fig. 3).
Exposure to hypoxic conditions blocked the PrP (106-126)-induced
reduction in MTP values (Fig. 3A and
B) and inhibited Bax translocation and cytochrome c
release (Fig. 3C). However, these
effects of HIF-1α were blocked by ICG-001 or doxorubicin (Fig. 3). For this reason, this study
supports the hypothesis that the upregulation of β-catenin, caused
by HIF-1α, prevents PrP (106-126)-mediated neurotoxicity through
the modulation of mitochondrial functions.
Collectively, the results from the present study
suggest that HIF-1α prevents the PrP (106-126)-induced activation
of mitochondrial apoptotic pathways in neuronal cells by activating
β-catenin signaling. Our results also suggest that regulators of
Wnt/β-catenin signaling, including HIF-1α or exposure to hypoxic
conditions, may be a potential neurotherapeutic target for the
prevention of prion diseases.
Acknowledgements
This study was supported by the Cooperative Research
Program for Agriculture Science and Technology Development
(PJ907116) of the Rural Development Administration (RDA) and by the
National Research Foundation of the Korea Grant funded by the
Korean Government (2012R1A1B3000463).
References
|
1
|
Prusiner SB: Prions. Proc Natl Acad Sci
USA. 95:13363–13383. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wickner RB: Prion diseases: Infectivity
versus toxicity. Nature. 470:470–471. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Eghiaian F, Grosclaude J, Lesceu S, et al:
Insight into the PrPC→PrPSc conversion from the structures of
antibody-bound ovine prion scrapie-susceptibility variants. Proc
Natl Acad Sci USA. 101:10254–10259. 2004.
|
|
4
|
Turnbaugh JA, Unterberger U, Saá P, et al:
The N-terminal, polybasic region of PrP(C) dictates the efficiency
of prion propagation by binding to PrP(Sc). J Neurosci.
32:8817–8830. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sim VL and Caughey B: Ultrastructures and
strain comparison of under-glycosylated scrapie prion fibrils.
Neurobiol Aging. 30:2031–2042. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jeong JK, Moon MH, Bae BC, et al:
Autophagy induced by resveratrol prevents human prion
protein-mediated neurotoxicity. Neurosci Res. 73:99–105. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jeong JK, Moon MH, Lee YJ, Seol JW and
Park SY: Autophagy induced by the class III histone deacetylase
Sirt1 prevents prion peptide neurotoxicity. Neurobiol Aging.
34:146–156. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Aiken JM, Williamson JL and Marsh RF:
Evidence of mitochondrial involvement in scrapie infection. J
Virol. 63:1686–1694. 1989.PubMed/NCBI
|
|
9
|
Quintanilla RA, Dolan PJ, Jin YN and
Johnson GV: Truncated tau and Aβ cooperatively impair mitochondria
in primary neurons. Neurobiol Aging. 33:619.e25–35. 2012.
|
|
10
|
Freixes M, Rodriguez A, Dalfo E and Ferrer
I: Oxidation, glycoxidation, lipoxidation, nitration, and responses
to oxidative stress in the cerebral cortex in Creutzfeldt-Jakob
disease. Neurobiol Aging. 27:1807–1815. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jeong JK, Moon MH, Park YG, et al:
Gingerol-induced hypoxia-inducible factor 1 alpha inhibits human
prion peptide-mediated neurotoxicity. Phytother Res. 27:1185–1192.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
12
|
Xie L, Johnson RS and Freeman RS:
Inhibition of NGF deprivation-induced death by low oxygen involves
suppression of BIMEL and activation of HIF-1. J Cell Biol.
168:911–920. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Strey CW, Gestrich J, Beckhaus T, et al:
Hypoxia and reoxygenation of primary human hepatocytes induce
proteome changes of glucose metabolism, oxidative protection and
peroxisomal function. Int J Mol Med. 26:577–584. 2010. View Article : Google Scholar
|
|
14
|
Selvendiran K, Bratasz A, Kuppusamy ML,
Tazi MF, Rivera BK and Kuppusamy P: Hypoxia induces chemoresistance
in ovarian cancer cells by activation of signal transducer and
activator of transcription 3. Int J Cancer. 125:2198–2204. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gardner LB and Corn PG: Hypoxic regulation
of mRNA expression. Cell Cycle. 7:1916–1924. 2008. View Article : Google Scholar
|
|
16
|
Malik A, Korol A, Weber M, Hankeln T,
Avivi A and Band M: Transcriptome analysis of the spalax hypoxia
survival response includes suppression of apoptosis and tight
control of angiogenesis. BMC Genomics. 13:6152012. View Article : Google Scholar
|
|
17
|
Ryan HE, Lo J and Johnson RS: HIF-1 alpha
is required for solid tumor formation and embryonic
vascularization. EMBO J. 17:3005–3015. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee SH, Koo KH, Park JW, et al: HIF-1 is
induced via EGFR activation and mediates resistance to anoikis-like
cell death under lipid rafts/caveolae-disrupting stress.
Carcinogenesis. 30:1997–2004. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Adams JM, Difazio LT, Rolandelli RH, et
al: HIF-1: a key mediator in hypoxia. Acta Physiol Hung. 96:19–28.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Koh MY, Spivak-Kroizman TR and Powis G:
HIF-1alpha and cancer therapy. Recent Results Cancer Res.
180:15–34. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ke Q and Costa M: Hypoxia-inducible
factor-1 (HIF-1). Mol Pharmacol. 70:1469–1480. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Jeong JK, Seo JS, Moon MH, Lee YJ, Seol JW
and Park SY: Hypoxia-inducible factor-1α regulates prion protein
expression to protect against neuron cell damage. Neurobiol Aging.
33:1006.e1–10. 2012.
|
|
23
|
Singh N, Sharma G and Mishra V: Hypoxia
inducible factor-1: its potential role in cerebral ischemia. Cell
Mol Neurobiol. 32:491–507. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Seo JS, Seol JW, Moon MH, Jeong JK, Lee YJ
and Park SY: Hypoxia protects neuronal cells from human prion
protein fragment-induced apoptosis. J Neurochem. 112:715–722. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang Q, Qian Z, Pan L, Li H and Zhu H:
Hypoxia-inducible factor 1 mediates the anti-apoptosis of berberine
in neurons during hypoxia/ischemia. Acta Physiol Hung. 99:311–323.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cunningham LA, Candelario K and Li L:
Roles for HIF-1α in neural stem cell function and the regenerative
response to stroke. Behav Brain Res. 227:410–417. 2012.
|
|
27
|
Lehwald N, Tao GZ, Jang KY, et al:
β-Catenin regulates hepatic mitochondrial function and energy
balance in mice. Gastroenterology. 143:754–764. 2012.
|
|
28
|
Flaherty MP, Kamerzell TJ and Dawn B: Wnt
signaling and cardiac differentiation. Prog Mol Biol Transl Sci.
111:153–174. 2012. View Article : Google Scholar
|
|
29
|
Wiedau-Pazos M, Wong E, Solomon E, Alarcon
M and Geschwind DH: Wnt-pathway activation during the early stage
of neurodegeneration in FTDP-17 mice. Neurobiol Aging. 30:14–21.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Meyers J, Hu L, Moses A, Kaboli K,
Papandrea A and Raymond P: β-catenin/Wnt signaling controls
progenitor fate in the developing and regenerating zebrafish
retina. Neural Dev. 7:302012.
|
|
31
|
Mazumdar J, O’Brien WT, Johnson RS, et al:
O2 regulates stem cells through Wnt/β-catenin
signalling. Nat Cell Biol. 12:1007–1013. 2010.
|
|
32
|
Wei L, Sun C, Lei M, et al: Activation of
Wnt/β-catenin pathway by exogenous Wnt1 protects SH-SY5Y cells
against 6-hydroxydopamine toxicity. J Mol Neurosci. 49:105–115.
2013.
|
|
33
|
Liu S, Yeh TH, Singh VP, et al: β-Catenin
is essential for ethanol metabolism and protection against
alcohol-mediated liver steatosis in mice. Hepatology. 55:931–940.
2012.
|
|
34
|
Mitani T, Harada N, Nakano Y, Inui H and
Yamaji R: Coordinated action of hypoxia-inducible factor-1α and
β-catenin in androgen receptor signaling. J Biol Chem.
287:33594–33606. 2012.
|
|
35
|
Jeong JK, Moon MH, Seo JS, Seol JW, Park
SY and Lee YJ: Hypoxia inducing factor-1alpha regulates tumor
necrosis factor-related apoptosis-inducing ligand sensitivity in
tumor cells exposed to hypoxia. Biochem Biophys Res Commun.
399:379–383. 2010. View Article : Google Scholar
|
|
36
|
L’Episcopo F, Tirolo C, Testa N, et al:
Plasticity of subventricular zone neuroprogenitors in MPTP
(1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) mouse model of
Parkinson’s disease involves cross talk between inflammatory and
Wnt/β-catenin signaling pathways: functional consequences for
neuroprotection and repair. J Neurosci. 32:2062–2085.
2012.PubMed/NCBI
|
|
37
|
Liang J, Liu L and Xing D:
Photobiomodulation by low-power laser irradiation attenuates
Aβ-induced cell apoptosis through the Akt/GSK3β/β-catenin pathway.
Free Radic Biol Med. 53:1459–1467. 2012.
|
|
38
|
Hota KB, Hota SK, Srivastava RB and Singh
SB: Neuroglobin regulates hypoxic response of neuronal cells
through Hif-1α- and Nrf2-mediated mechanism. J Cereb Blood Flow
Metab. 32:1046–1060. 2012.PubMed/NCBI
|