1. Introduction
Hypertension is an established risk factor for
coronary heart disease, stroke, congestive heart failure and renal
dysfunction. Despite significant advances in our understanding of
the pathophysiology of hypertension, it remains to be one of the
world’s greatest public health issues (1). It is estimated that one third of the
world’s adult population will be hypertensive by 2025 (2). In particular, in industrialized
countries, the risk of becoming hypertensive [blood pressure (BP)
>140/90 mmHg] during a lifetime exceeds 90% (3). Essential hypertension (EH), or
hypertension due to undetermined causes, accounts for >90% of
cases of hypertension. It is a heterogeneous disorder, with
different patients having different causal factors that lead to
high BP.
To date, the etiology of EH is not well understood
due to multifactorial causes. It is generally believed that EH is a
multifactorial trait involving interactions among genetic,
environmental and demographic factors (4). Of these, hereditary factors account
for 30 to 50% of BP variability (5). Variations in a variety of genes have
shown an association with hypertension in certain studies; however,
these associations are often not reproducible in studies on other
populations. Improved techniques of genetic analysis, particularly
genome-wide linkage analysis, have enabled the search for genes
that contribute to the development of primary hypertension in the
population (6,7). However, the majority of the reported
genetic variants were identified in studies of the nuclear genome
(8–10); only limited insights have been
gained from the investigation of the mitochondrial genome.
This review provides a detailed introduction of the
mitochondrial genome, summarizes the results of studies on the role
of mitochondrial DNA (mtDNA) mutations associated with EH published
thus far, and highlights some of the general conclusions that have
become apparent.
2. Mitochondrial genome and mitochondrial
genetics
Mitochondria are bacterium-sized organelles found in
all nucleated cells (11).
Uniquely, they contain their own genome (mtDNA) and it is widely
accepted that mitochondria originate from aerobic bacteria engulfed
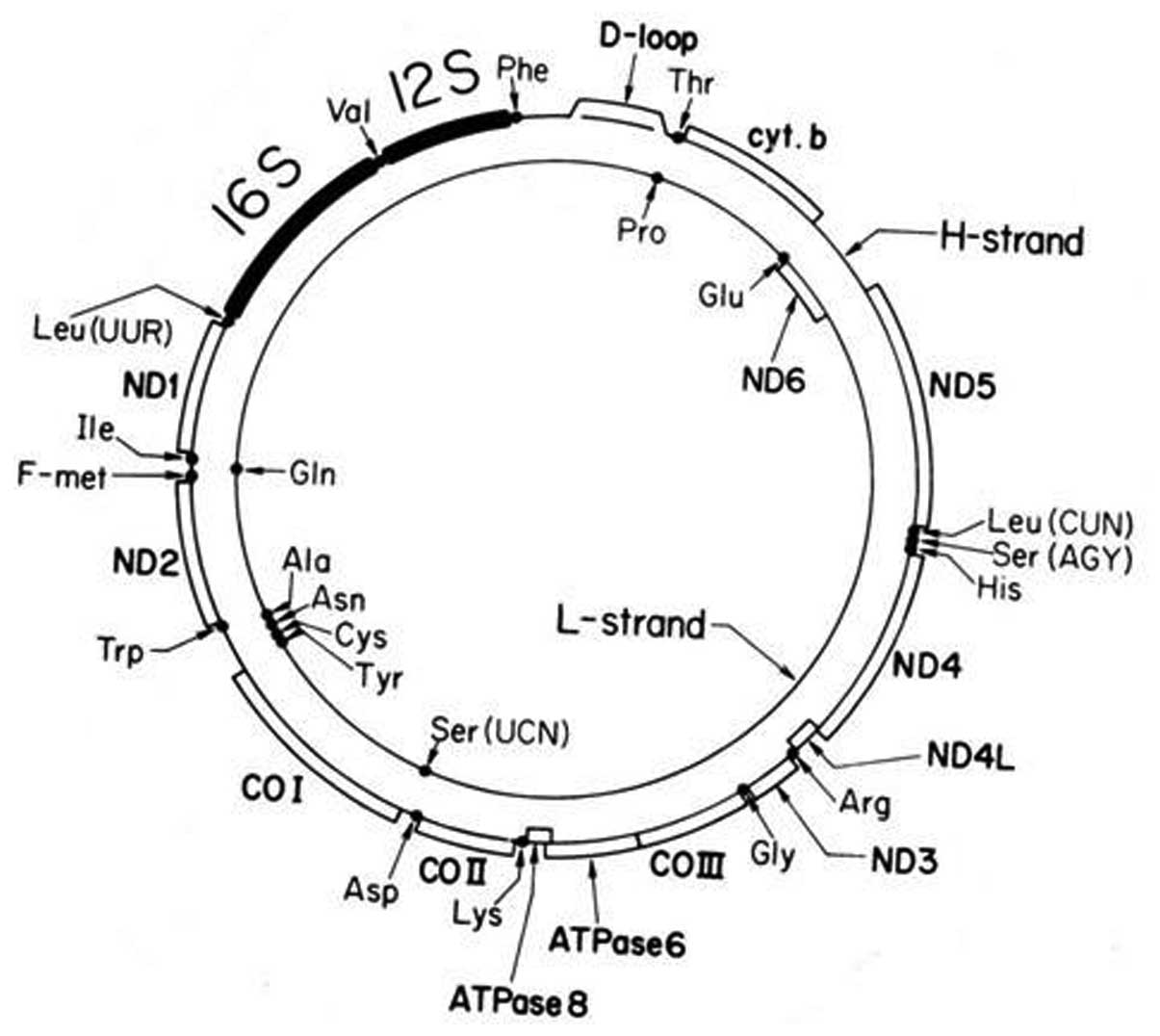
by an anaerobic eukaryotic cell. Mammalian mtDNA encodes 13
proteins that are subunits of the oxidative phosphorylation
(OXPHOS) system and 22 transfer RNAs (tRNAs) and 2 ribosomal RNAs
(rRNAs) (Fig. 1). The
mitochondrial OXPHOS complexes are assembled from genes distributed
between mtDNA and nuclear DNA (nDNA) (12). Unlike nDNA, mtDNAs are maternally
inherited and are present in multiple copies/cell. The numbers vary
according to the bioenergentic needs of each unique tissue and can
range over three orders of magnitude, depending on the cell type
(13).
Each mammalian cell contains hundreds of
mitochondria and thousands of mtDNAs. Since mtDNA is in the
proximity of reactive oxygen species (ROS) generation sites and
mitochondria have less sophisticated DNA protection and repair
systems, mtDNA is hence vulnerable to a high mutation rate
(14). The most prominent is
polyplasmy. Polyplasmy is the basis of heteroplasmy (alternations
of mtDNA may be present in some of the mtDNA molecules) and
homoplasmy (mutations in all of the molecules). Neutral
polymorphisms are usually homoplasmic, whereas pathogenic mutations
are usually heteroplasmic in nature, and EH-associated mtDNA
mutations are commonly homoplasmic or almost homoplasmic.
3. Mitochondrial function and
dysfunction
Mitochondria exert both vital and lethal functions
in physiological and pathological conditions. On the one hand, they
provide the majority of cellular energy in the form of
adenosine-5′-triphosphate (ATP) through OXPHOS (15). Additionally, they are involved in
a variety of processes, including regulation of the cell cycle,
cell signaling, apoptosis and calcium (Ca2+) buffering
(12,16,17).
Oxygen radicals, such as ROS, are also generated
during OXPHOS as a toxic byproduct in the mitochondria, which may
damage the mitochondrial and cellular DNA, protein, lipids and
other molecules, leading to oxidative stress and mitochondrial
dysfunction (18,19). An impairment of normal
mitochondrial function leads to an excessive production of ROS and
a general decrease in ATP levels. Moreover, there is a concomitant
loss of mitochondrial transmembrane potential (20). Excessive ROS and Ca2+
production lead to mitochondrial outer membrane permeabilization
and to the release into the cytosol of cytotoxic proteins normally
confined within the mitochondrial intermembrane space. As a result,
this process induces apoptosis or necrosis (21).
4. Oxidative stress and hypertension
In animal models, oxidative stress has been observed
in spontaneously hypertensive rats (22), renovascular hypertension (23) and salt-sensitive hypertension
(24). Although its pathogenesis
is complex and multifactorial, human hypertension is considered as
a state of increased oxidative stress (25). An excessive endothelial production
of ROS and nitric oxide synthase (NOS) may lead to impaired
endothelium-dependent vasorelaxation in human internal mammary
arteries and the saphenous vein (26), contributing to vascular
pathophysiology by promoting increased vascular tone, cell growth,
as well as the activation of matrix metalloproteinases and the
deposition of extracellular matrix proteins, which are processes
associated with the vascular phenotype of hypertension (27).
5. Role of mtDNA mutations in EH
12S rRNA A1555G mutation
The A1555G mutation in the 12S rRNA gene has been
associated with aminoglycoside-induced and non-syndromic hearing
loss in various ethnic populations (28–31). Chen et al (32) described two Han Chinese families
with hearing loss and hypertension carrying the homoplasmic A1555G
mutation. An A to G transition at this position in the 12S rRNA
gene has been predicted to encode an aminoglycoside binding based
on sequence similarity to Escherichia coli (33) and alters mitochondrial ribosomal
function and translation (34–36), which in turn causes OXPHOS defects
that are thought to contribute to the clinical pathology of
hypertension. Insufficient metabolism caused by mitochondrial
dysfunction may lead to the elevation of systolic BP and may be
involved in the development of hypertension (37).
ND1 T3308C mutation
The homoplasmic ND1 T3308C mutation is a
known disease-associated mutation. This mutation has been suggested
to contribute to the higher penetrance of hearing loss in a large
African family than Japanese and French pedigrees carrying the
tRNASer(UCN) T7511C mutation (38,39). Liu et al (40) reported a Han Chinese family
carrying the ND1 T3308C mutation with EH. The T to C
transition at position 3308 causes translation-initiating
methionine with a threonine in ND1. Thus, the ND1
mRNA is shortened by two amino acids (41). Moreover, the T3308C mutation is
also located in two nucleotides adjacent to the 3′ end of the
tRNALeu(UUR) gene; the T3308C mutation also affects the
processing of the H-strand polycistronic RNA precursors (42).
ND5 T12338C mutation
The well-known T12338C mutation in the ND5
gene, combined with tRNALeu(CUN) A12330G mutation, has
been found in a three generation Han Chinese family with high
penetrance of EH (43). Moreover,
this mutation was also shown to be present in a Chinese family with
hypertrophic cardiomyopathy (44). In fact, the ND5 T12338C
mutation, which is similar to the ND1 T3308C mutation,
causes a replacement of the first amino acid,
translation-initiating methionine with a threonine in the
ND5 polypeptide, and decreases ND5 mRNA levels and
alters the processing of RNA precursors. In addition, this mutation
is located in two nucleotides adjacent to the 3′ end of the
tRNALeu(CUN), and it is anticipated that the T12338C
mutation will lead to a reduction in tRNALeu(CUN)
levels, whereas the A12330G mutation disrupts the highly conserved
base pairing (6T-67A) in the acceptor arm of
tRNALeu(CUN) (45).
Therefore, the combination of T12338C and A12330G mutations may
have contributed to the high penetrance of EH in this Chinese
family (43).
ND6 T14484C mutation
The T14484C mutation in the ND6 gene is one
of the primary mutations associated with Leber’s hereditary optic
neuropathy (LHON) (46). In a
recent study, a large Han Chinese family with maternally
transmitted EH but not presenting any LHON phenotype, was found to
be associated with the T14484C mutation (47). Analysis of the complete mtDNA
sequence of the proband showed the absence of additional pathogenic
mutations, apart from the T14484C mutation. Moreover, analysis of
the mitochondrial function of lymphoblastoid cell lines established
from the family members showed that mitochondrial respiration rate
and membrane potential were significantly reduced when compared
with the control cell lines. In addition, there was an increase in
the levels of ROS and mitochondrial mass in the mutant cell lines.
These data suggest that ND6 T14484C also plays an important
role in the pathogenesis of EH and is also a pathogenic mutation
associated with EH (47).
CyB G15059A mutation
The heteroplasmic G15059A mutation in CyB
gene was first described in a patient with mitochondrial myopathy
(48). This substitution results
in the replacement of a glycine at amino acid position 190 of
CyB with a stop codon leading to a truncated protein that
misses 244 amino acids at the C-terminus of CyB (49). Nikitin et al (50) examined the role of the CyB
G15059A mutation in patients with type 2 diabetes (T2D) with EH. In
addition, the G15059A heteroplasmy level exceeding 39% was
associated with an increased risk of EH, indicating a direct
pathogenic role for this mutation in EH (50).
50-bp deletion
The 50-bp deletion (m.298_347del50) in the mtDNA
control region removes the conserved sequence block II (CSBII) and
the replication primer location (51–53). This deletion, co-occurring with
two novel mutations (ND1 C3519T and ND5 G13204A),
accounted for the complex clinical traits, including hypertension,
T2D and coronary artery disease (CAD) in an Indian family (54). Of note, two short homologous
direct repeats of CCAAACCCC flanked the 50-bp deletion. The CSBII
directs transcription termination and primer formation in mtDNA
replication (55). Thus, it can
be predicted that the 50-bp deletion may reduce the mtDNA copy
number and decrease the levels of cellular energy. However,
conflicting reports have shown that this deletion does not affect
the mtDNA copy number (53); its
functional role requires further elucidation in future studies.
tRNAIle A4295G mutation
The homoplasmic A4295G mutation in the
tRNAIle gene was identified in a three generation Han
Chinese family with maternally inherited EH (56). An A to G transition at nucleotide
position 4295 was shown to be highly evolutionarily conserved, and
was not present in the healthy controls (57). The A4295G mutation is localized at
the 3′ end adjacent to the anticodon (position 37) of
tRNAIle (45);
nucleotide at position 37 is responsible for the stabilization of
functional tRNA (58). Thus, it
has been suggested that this mutation reduces the efficiency with
which tRNAIle can be processed by 3′-tRNase, reducing
the level of functional tRNAIle (59). The functional characterization of
the A4295G mutation shows that this mutation induces a significant
decrease in complex III protein levels, leading to a decrease in
the activity of this complex, which is reflected by the decline in
mitochondrial respiration (60).
tRNAIle A4263G mutation
The A4263G mutation changes the stop codon TAA of
the ND1 mRNA to an equivalent TAG stop codon. This mutation
causes an A to G transition at the 5′ end of the tRNAIle
gene (61,62). Cybrid cells derived from the
proband carrying A4263G mutation show a reduction in
tRNAIle steady-state levels, as well as in the rate of
mitochondrial protein translation. Increased ROS and the decreased
efficiency of 5′ end processing of tRNAIle precursor
indicated that this mutation caused mitochondrial dysfunction that
was responsible for EH in this Han Chinese family (61).
tRNAIle T4291C mutation
The homoplasmic T4291C mutation in the
tRNAIle gene has been associated with a cluster of
metabolic defects, including hypertension, hypercholesterolemia and
hypomagnesaemia in a large family (63). The T4291C mutation occurs
immediately 5′ to the tRNAIle anticodon (position 33),
and is conserved in every sequenced tRNAIle from
bacteria to human mitochondria (64). Biochemical studies with anticodon
stem-loop analogs of tRNAs have been performed and have indicated
that the substitution of cytidine for uridine at this position
markedly impairs ribosomal binding (65), providing evidence of the
functional importance of this mutation.
tRNAMet A4435G mutation
The presence of the A4435G mutation with chronic
progressive external ophthalmoplegia (CPEO) was initially reported
in the study by Jaksch et al (66). Later on, this mutation was found
in patients with LHON (67), as
well as in a Japanese subject with diabetes (68). In addition, the East Asian
haplogroup G2a1-specific A4435G mutation (69) has also been associated with EH in
two Chinese families (70,71).
The homoplasmic A4435G mutation, which is located at immediately 3′
end to the anticodon of tRNAMet (nucleotide position
37), is extremely conserved from bacteria to human mitochondria
(45). In fact, as shown in
previous studies, the A4435G mutation causes ~40–50% reduction in
the steady-state level of tRNAMet and consequently
results in the failure of mt-tRNA metabolism (67,70). Impaired mt-tRNA metabolism
subsequently worsens the mitochondrial protein synthesis, decreases
ATP production and increases ROS levels. Thus, mitochondrial
dysfunction may contribute to the development of EH in these
families carrying the A4435G point mutation (37,63,72).
A4401G mutation in tRNAMet and
tRNAGln
The homoplasmic A4401G mutation was originally
described in a three generation Chinese family with left
ventricular hypertrophy (LVH) (73). This mutation was also present in a
five generation Chinese pedigree with EH (74). The A4401G mutation is localized at
the junction of tRNAMet at the H-strand and
tRNAGln at the L-strand (75). Thus, it is anticipated that this
mutation may lead to defective tRNAMet 5′ end processing
in the H-strand transcripts and reduce the efficiency of
tRNAGln precursor 5′ end cleavage in the L-strand
transcripts. Functional characterization of cell lines derived from
the proband carrying the A4401G mutation shows virtually ~30%
reduction in the levels of tRNAIle and
tRNAGln (74). In
addition, the mutant cell lines present a significant decrease in
the oxygen consumption rate (73). These findings indicate that the
A4401G mutation is involved in the pathogenesis of EH in Han
Chinese families.
T4353C mutation in
tRNAGln
The T4353C mutation, in conjunction with the tRNATrp
C593T mutation, has been shown to account for the high penetrance
of EH in a Han Chinese family (76). Clinical evaluation of this family
showed a typical maternally transmitted pattern. Analysis of the
complete mtDNA sequence identified the homoplasmic T4353C mutation.
This mutation alters a conservative base-pairing (49A-65U) on the T
arm of tRNAGln, thereby affecting tRNA metabolism
(45). Moreover, the T4353C
mutation reduces the steady-state level of tRNAGln,
mutant tRNAGln and tRNAPhe may be
metabolically less stable and more subject to degradation. The
cybrid cell lines carrying the T4353C mutation present
mitochondrial protein synthesis defects. As a result, this mutation
causes the mitochondrial dysfunction responsible for EH.
6. Molecular mechanisms behind mtDNA
mutations associated with EH
In previous studies, we noticed that several
hypertension-associated mitochondrial pathogenic mutations were
located in tRNA genes. Mt-tRNA mutations have structural and
functional effects, including destabilization of the tRNA tertiary
structure, altered processing of RNA precursors, loss of nucleotide
modification and deficient aminoacylation (Fig. 2 and Table I). In particular, these pathogenic
mutations may lead to deficiencies in tRNA 3′ end metabolism
including 3′ end cleavage, CCA addition, aminoacylation or
impairment of critical subunits of respiratory chain functions.
Failures in mt-tRNA metabolism subsequently lead to the impairment
of mitochondrial protein (77–79). However, mutations in protein
coding genes may have the potential to affect OXPHOS and in turn
cellular death, resulting in a failure in mitochondrial protein
synthesis. The increased bioavailability of ROS results in
oxidative stress, which leads to cardiovascular and renal damage,
thus, contributing to EH.
 | Table ISummary of cardiovascular diseases
associated mt-tRNA mutations. |
Table I
Summary of cardiovascular diseases
associated mt-tRNA mutations.
| Position | tRNA species | Allele |
Homoplasmy/heteroplasmy | Clinical
features | Refs. |
|---|
| 29 |
tRNAVal | C1628T | Homoplasmy | Cardiomyopathy | (80) |
| 14 |
tRNALeu(UUR) | A3243G | Heteroplasmy | Cardiomyopathy | (81) |
| 29 |
tRNALeu(UUR) | A3260G | Heteroplasmy | Myopathy,
cardiomyopathy, MELAS | (82–84) |
| 72 |
tRNALeu(UUR) | C3303T | Homoplasmy | Cardiomyopathy,
myopathy, HCM | (85–87) |
| 1 |
tRNAIle | A4263G | Homoplasmy | Hypertension | (61) |
| 7 |
tRNAIle | A4269G | Homoplasmy | Cardiomyopathy | (88) |
| 15 |
tRNAIle | T4277C | Homoplasmy | Cardiomyopathy | (89) |
| 33 |
tRNAIle | T4291C | Homoplasmy | Hypertension,
hypercholesterolemia, hypomagnesaemia | (63) |
| 37 |
tRNAIle | A4295G | Homoplasmy | HCM,
hypertension | (56,57) |
| 42 |
tRNAIle | A4300G | Homoplasmy | HCM | (90) |
| 37 |
tRNAMet | A4435G | Homoplasmy | LHON, CPEO,
hypertension | (66–68,70,71) |
| 1 | tRNAGln
and tRNAMet | A4401G | Homoplasmy | LVH,
hypertension | (73,74) |
| 49 |
tRNAGln | T4353C | Homoplasmy | Hypertension | (76) |
| 58 |
tRNAAla | A5600T | Heteroplasmy | DCM | (91) |
| 54 |
tRNALys | A8348G | Heteroplasmy | Cardiomyopathy | (92) |
| 7 |
tRNAGly | T9997C | Heteroplasmy | HCM | (93) |
| 59 |
tRNAHis | G12192A | Homoplasmy | Cardiomyopathy | (94) |
| 67 |
tRNALeu(CUN) | A12330G | Homoplasmy | Hypertension | (43) |
| 37 |
tRNAGlu | T14709C | Homoplasmy | Cardiomyopathy | (95) |
| 2 |
tRNAThr | T15889C | Heteroplasmy | DCM | (91) |
| 16 |
tRNAThr | A15902G | Heteroplasmy | DCM | (91) |
| 38 |
tRNAThr | T15924C | Homoplasmy | DCM | (96) |
| 51 |
tRNAThr | A15935G | Heteroplasmy | DCM | (91) |
7. Conclusion
EH is a multifactorial syndrome that is
characterized by abnormal energy metabolism and high BP. Recent
studies have identified the mitochondria as the target and origin
of major pathogenic pathways which lead to the progression of
hypertension (97–99). Although existing therapies have
been beneficial, there is a clear need for new approaches to
treatment. Pharmacological targeting of the cellular stresses
underlying mitochondrial dysfunction is showing promise. In
addition, screening for mtDNA mutations for the clinical expression
of hypertension may provide further insight into the understanding
of the pathophysiology for maternally inherited hypertension.
Acknowledgements
This study was supported by grants from the Ministry
of Public Health of Zhejiang Province (2013KYA158), Nanjing Medical
University (no. 2010NJMU011), Hangzhou Bureau of Science and
Technology (no. 20120533Q03) and the Specialized Research Fund for
the Doctoral Program of Higher Education (no. 20124323120004).
References
|
1
|
El Shamieh S, Herbeth B, Azimi-Nezhad M,
et al: Human formyl peptide receptor 1 C32T SNP interacts with age
and is associated with blood pressure levels. Clin Chim Acta.
413:34–38. 2012.PubMed/NCBI
|
|
2
|
Kearney PM, Whelton M, Reynolds K, et al:
Global burden of hypertension: analysis of worldwide data. Lancet.
365:217–223. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Messerli FH, Williams B and Ritz E:
Essential hypertension. Lancet. 370:591–560. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Kunes J and Zicha J: Developmental windows
and environment as important factors in the expression of genetic
information: a cardiovascular physiologist’s view. Clin Sci.
111:295–305. 2006.PubMed/NCBI
|
|
5
|
Timberlake DS, O’Connor DT and Parmer RJ:
Molecular genetics of essential hypertension: recent results and
emerging strategies. Curr Opin Nephrol Hypertens. 10:71–79. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fan JB, Chen X, Halushka MK, et al:
Parallel genotyping of human SNPs using generic high-density
oligonucleotide tag arrays. Genome Res. 10:853–860. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Oliphant A, Barker DL, Stuelpnagel JR and
Chee MS: BeadArray technology: enabling an accurate, cost-effective
approach to high-throughput genotyping. Biotechniques. 56(Suppl
56–58): 60–61. 2002.PubMed/NCBI
|
|
8
|
Levy D, Ehret GB, Rice K, et al:
Genome-wide association study of blood pressure and hypertension.
Nat Genet. 41:677–687. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tobin MD, Tomaszewski M, Braund PS, et al:
Common variants in genes underlying monogenic hypertension and
hypotension and blood pressure in the general population.
Hypertension. 51:1658–1664. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Padmanabhan S, Melander O, Johnson T, et
al: Genome-wide association study of blood pressure extremes
identifies variant near UMOD associated with hypertension. PLoS
Genet. 6:e10011772010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schon EA: Mitochondria. Encyclopedia of
Human Biology. Dulbecco R: 5. 2nd edition. Academic Press; London:
pp. 713–724. 1997
|
|
12
|
Wallace DC: Mitochondrial DNA mutations in
disease and aging. Environ Mol Mutagen. 51:440–450. 2010.PubMed/NCBI
|
|
13
|
Garcia-Rodriguez LJ: Appendix 1. Basic
properties of mitochondria. Methods Cell Biol. 80:809–812. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
DiMauro S and Schon EA: Mitochondrial DNA
mutations in human disease. Am J Med Genet. 106:18–26. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cadenas E and Davies KJ: Mitochondrial
free radical generation, oxidative stress, and aging. Free Radic
Biol Med. 29:222–230. 2000.PubMed/NCBI
|
|
16
|
Schon EA, DiMauro S and Hirano M: Human
mitochondrial DNA: roles of inherited and somatic mutations. Nat
Rev Genet. 13:878–890. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
McBride HM, Neuspiel M and Wasiak S:
Mitochondria: more than just a powerhouse. Curr Biol. 16:R551–R560.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Choksi KB, Boylston WH, Rabek JP, et al:
Oxidatively damaged proteins of heart mitochondrial electron
transport complexes. Biochim Biophys Acta. 1688:95–101. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Murphy MP: Induction of mitochondrial ROS
production by electrophilic lipids: a new pathway of redox
signaling? Am J Physiol Heart Circ Physiol. 290:H1754–H1755. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Abou-Sleiman PM, Muqit MM and Wood NW:
Expanding insights of mitochondrial dysfunction in Parkinson’s
disease. Nat Rev Neurosci. 7:207–219. 2006.
|
|
21
|
Li P, Nijhawan D and Wang X: Mitochondrial
activation of apoptosis. Cell. 116(Suppl 2): S57–S59. 2004.
View Article : Google Scholar
|
|
22
|
Wu L and Juurlink BH: Increased
methylglyoxal and oxidative stress in hypertensive rat vascular
smooth muscle cells. Hypertension. 39:809–814. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lerman LO, Nath KA, Rodriguez-Porcel M, et
al: Increased oxidative stress in experimental renovascular
hypertension. Hypertension. 37:541–546. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Trolliet MR, Rudd MA and Loscalzo J:
Oxidative stress and renal dysfunction in salt-sensitive
hypertension. Kidney Blood Press Res. 24:116–123. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Romero JC and Reckelhoff JF:
State-of-the-Art lecture. Role of angiotensin and oxidative stress
in essential hypertension. Hypertension. 34:943–949. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hamilton CA, Berg G, McIntyre M, et al:
Effects of nitric oxide and superoxide on relaxation in human
artery and vein. Atherosclerosis. 133:77–86. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Paravicini TM and Touyz RM: Redox
signaling in hypertension. Cardiovasc Res. 71:247–258. 2006.
View Article : Google Scholar
|
|
28
|
Prezant TR, Agapian JV, Bohlman MC, et al:
Mitochondrial ribosomal RNA mutation associated with both
antibiotic-induced and non-syndromic deafness. Nat Genet.
4:289–294. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Estivill X, Govea N, Barceló E, et al:
Familial progressive sensorineural deafness is mainly due to the
mtDNA A1555G mutation and is enhanced by treatment of
aminoglycosides. Am J Hum Genet. 62:27–35. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Guan MX: Molecular pathogenetic mechanism
of maternally inherited deafness. Ann NY Acad Sci. 1011:259–271.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Van Camp G and Smith RJ: Maternally
inherited hearing impairment. Clin Genet. 57:409–414. 2000.
|
|
32
|
Chen H, Zheng J, Xue L, et al: The 12S
rRNA A1555G mutation in the mitochondrial haplogroup D5a is
responsible for maternally inherited hypertension and hearing loss
in two Chinese pedigrees. Eur J Hum Genet. 20:607–612. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Hamasaki K and Rando RR: Specific binding
of aminoglycosides to a human rRNA construct based on a DNA
polymorphism which causes aminoglycoside-induced deafness.
Biochemistry. 36:12323–12328. 1997. View Article : Google Scholar
|
|
34
|
Cotney J, McKay SE and Shadel GS:
Elucidation of separate, but collaborative functions of the rRNA
methyltransferase-related human mitochondrial transcription factors
B1 and B2 in mitochondrial biogenesis reveals new insight into
maternally inherited deafness. Hum Mol Genet. 18:2670–2682. 2009.
View Article : Google Scholar
|
|
35
|
Hobbie SN, Bruell CM, Akshay S, et al:
Mitochondrial deafness alleles confer misreading of the genetic
code. Proc Natl Acad Sci USA. 105:3244–3249. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Guan MX, Fischel-Ghodsian N and Attardi G:
Nuclear background determines biochemical phenotype in the
deafness-associated mitochondrial 12S rRNA mutation. Hum Mol Genet.
10:573–580. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Bernal-Mizrachi C, Gates AC, Weng S, et
al: Vascular respiratory uncoupling increases blood pressure and
atherosclerosis. Nature. 435:502–506. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li X, Fischel-Ghodsian N, Schwartz F, et
al: Biochemical characterization of the mitochondrial
tRNASer(UCN) T7511C mutation associated with
nonsyndromic deafness. Nucleic Acids Res. 32:867–877. 2004.
View Article : Google Scholar
|
|
39
|
Li R, Ishikawa K, Deng JH, et al:
Maternally inherited nonsyndromic hearing loss is associated with
the T7511C mutation in the mitochondrial tRNASerUCN gene
in a Japanese family. Biochem Biophys Res Commun. 328:32–37. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Liu Y, Li Z, Yang L, et al: The
mitochondrial ND1 T3308C mutation in a Chinese family with the
secondary hypertension. Biochem Biophys Res Commun. 368:18–22.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Anderson S, Bankier AT, Barrell BG, et al:
Sequence and organization of the human mitochondrial genome.
Nature. 290:457–465. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Guan MX, Enriquez JA, Fischel-Ghodsian N,
et al: The deafness-associated mitochondrial DNA mutation at
position 7445, which affects tRNASer(UCN) precursor
processing, has long-range effects on NADH dehydrogenase subunit
ND6 gene expression. Mol Cell Biol. 18:5868–5879. 1998.PubMed/NCBI
|
|
43
|
Teng L, Zheng J, Leng J and Ding Y:
Clinical and molecular characterization of a Han Chinese family
with high penetrance of essential hypertension. Mitochondrial DNA.
23:461–465. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Liu Z, Song Y, Gu S, et al: Mitochondrial
ND5 12338T>C variant is associated with maternally inherited
hypertrophic cardiomyopathy in a Chinese pedigree. Gene.
506:339–343. 2012.
|
|
45
|
Florentz C, Sohm B, Tryoen-Tóth P, et al:
Human mitochondrial tRNAs in health and disease. Cell Mol Life Sci.
60:1356–1375. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yu-Wai-Man P, Griffiths PG, Hudson G and
Chinnery PF: Inherited mitochondrial optic neuropathies. J Med
Genet. 46:145–158. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Guo H, Zhuang XY, Zhang AM, et al:
Presence of mutation m.14484T>C in a Chinese family with
maternally inherited essential hypertension but no expression of
LHON. Biochim Biophys Acta. 1822:1535–1543. 2012.PubMed/NCBI
|
|
48
|
Andreu AL, Hanna MG, Reichmann H, et al:
Exercise intolerance due to mutations in the cytochrome b gene of
mitochondrial DNA. N Engl J Med. 341:1037–1044. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Andreu AL, Bruno C, Dunne TC, et al: A
nonsense mutation (G15059A) in the cytochrome b gene in a patient
with exercise intolerance and myoglobinuria. Ann Neurol.
45:127–130. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Nikitin AG, Lavrikova EY and Chistiakov
DA: The heteroplasmic 15059G>A mutation in the mitochondrial
cytochrome b gene and essential hypertension in type 2 diabetes.
Diabetes Metab Syndr. 6:150–156. 2012.PubMed/NCBI
|
|
51
|
Chang DD and Clayton DA: Priming of human
mitochondrial DNA replication occurs at the light-strand promoter.
Proc Natl Acad Sci USA. 82:351–355. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kang D, Miyako K, Kai Y, et al: In vivo
determination of replication origins of human mitochondrial DNA by
ligation-mediated polymerase chain reaction. J Biol Chem.
272:15275–15279. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Bi R, Zhang AM, Zhang W, et al: The
acquisition of an inheritable 50-bp deletion in the human mtDNA
control region does not affect the mtDNA copy number in peripheral
blood cells. Hum Mutat. 31:538–543. 2010.PubMed/NCBI
|
|
54
|
Elango S, Govindaraj P, Vishwanadha VP, et
al: Analysis of mitochondrial genome revealed a rare 50 bp deletion
and substitutions in a family with hypertension. Mitochondrion.
11:878–885. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Pham XH, Farge G, Shi Y, et al: Conserved
sequence box II directs transcription termination and primer
formation in mitochondria. J Biol Chem. 281:24647–24652. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Li Z, Liu Y, Yang L, et al: Maternally
inherited hypertension is associated with the mitochondrial
tRNA(Ile) A4295G mutation in a Chinese family. Biochem Biophys Res
Commun. 367:906–911. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Merante F, Myint T, Tein I, et al: An
additional mitochondrial tRNA(Ile) point mutation (A-to-G at
nucleotide 4295) causing hypertrophic cardiomyopathy. Hum Mutat.
8:216–222. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Suzuki T, Nagao A and Suzuki T: Human
mitochondrial tRNAs: biogenesis, function, structural aspects, and
diseases. Annu Rev Genet. 45:299–329. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Levinger L, Giegé R and Florentz C:
Pathology-related substitutions in human mitochondrial tRNA(Ile)
reduce precursor 3′ end processing efficiency in vitro. Nucleic
Acids Res. 31:1904–1912. 2003.PubMed/NCBI
|
|
60
|
Gutiérrez Cortés N, Pertuiset C, Dumon E,
et al: Novel mitochondrial DNA mutations responsible for maternally
inherited nonsyndromic hearing loss. Hum Mutat. 33:681–689.
2012.PubMed/NCBI
|
|
61
|
Wang S, Li R, Fettermann A, et al:
Maternally inherited essential hypertension is associated with the
novel 4263A>G mutation in the mitochondrial tRNAIle
gene in a large Han Chinese family. Circ Res. 108:862–870. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Zhu HY, Wang SW, Liu L, et al: Genetic
variants in mitochondrial tRNA genes are associated with essential
hypertension in a Chinese Han population. Clin Chim Acta.
410:64–69. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Wilson FH, Hariri A, Farhi A, et al: A
cluster of metabolic defects caused by mutation in a mitochondrial
tRNA. Science. 306:1190–1194. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Sprinzl M, Steegborn C, Hübel F and
Steinberg S: Compilation of tRNA sequences and sequences of tRNA
genes. Nucleic Acids Res. 24:68–72. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Ashraf SS, Sochacka E, Cain R, et al:
Single atom modification (O→S) of tRNA confers ribosome binding.
RNA. 5:188–194. 1999.
|
|
66
|
Jaksch M, Kleinle S, Scharfe C, et al:
Frequency of mitochondrial transfer RNA mutations and deletions in
225 patients presenting with respiratory chain deficiencies. J Med
Genet. 38:665–673. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Qu J, Li R, Zhou X, et al: The novel
A4435G mutation in the mitochondrial tRNAMet may
modulate the phenotypic expression of the LHON-associated ND4
G11778A mutation. Invest Ophthalmol Vis Sci. 47:475–483.
2006.PubMed/NCBI
|
|
68
|
Guo LJ, Oshida Y, Fuku N, et al:
Mitochondrial genome polymorphisms associated with type-2 diabetes
or obesity. Mitochondrion. 5:15–33. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Kong QP, Bandelt HJ, Sun C, et al:
Updating the East Asian mtDNA phylogeny: a prerequisite for the
identification of pathogenic mutations. Hum Mol Genet.
15:2076–2086. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Liu Y, Li R, Li Z, et al: Mitochondrial
transfer RNAMet 4435A>G mutation is associated with maternally
inherited hypertension in a Chinese pedigree. Hypertension.
53:1083–1090. 2009.PubMed/NCBI
|
|
71
|
Lu Z, Chen H, Meng Y, et al: The
tRNAMet 4435A>G mutation in the mitochondrial
haplogroup G2a1 is responsible for maternally inherited
hypertension in a Chinese pedigree. Eur J Hum Genet. 19:1181–1186.
2011.
|
|
72
|
Postnov YV, Orlov SN, Budnikov YY, et al:
Mitochondrial energy conversion disturbance with decrease in ATP
production as a source of systemic arterial hypertension.
Pathophysiology. 14:195–204. 2007.
|
|
73
|
Zhu HY, Wang SW, Liu L, et al: A
mitochondrial mutation A4401G is involved in the pathogenesis of
left ventricular hypertrophy in Chinese hypertensives. Eur J Hum
Genet. 17:172–178. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Li R, Liu Y, Li Z, et al: Failures in
mitochondrial tRNAMet and tRNAGln metabolism
caused by the novel 4401A>G mutation are involved in essential
hypertension in a Han Chinese Family. Hypertension. 54:329–337.
2009.PubMed/NCBI
|
|
75
|
Ojala D, Montoya J and Attardi G: tRNA
punctuation model of RNA processing in human mitochondria. Nature.
290:470–474. 1981. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Qiu Q, Li R, Jiang P, et al: Mitochondrial
tRNA mutations are associated with maternally inherited
hypertension in two Han Chinese pedigrees. Hum Mutat. 33:1285–1293.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Levinger L, Mörl M and Florentz C:
Mitochondrial tRNA 3′ end metabolism and human disease. Nucleic
Acids Res. 32:5430–5441. 2004.
|
|
78
|
Kelley SO, Steinberg SV and Schimmel P:
Functional defects of pathogenic human mitochondrial tRNAs related
to structural fragility. Nat Struct Biol. 7:862–865. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
79
|
Kelley SO, Steinberg SV and Schimmel P:
Fragile T-stem in disease-associated human mitochondrial tRNA
sensitizes structure to local and distant mutations. J Biol Chem.
276:10607–10611. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
80
|
Arredondo JJ, Gallardo ME, García-Pavía P,
et al: Mitochondrial tRNA valine as a recurrent target for
mutations involved in mitochondrial cardiomyopathies.
Mitochondrion. 12:357–362. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Vilarinho L, Santorelli FM, Rosas MJ, et
al: The mitochondrial A3243G mutation presenting as severe
cardiomyopathy. J Med Genet. 34:607–609. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Zeviani M, Gellera C, Antozzi C, et al:
Maternally inherited myopathy and cardiomyopathy: association with
mutation in mitochondrial DNA tRNA(Leu)(UUR). Lancet. 338:143–147.
1991. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Sweeney MG, Brockington M, Weston MJ, et
al: Mitochondrial DNA transfer RNA mutation Leu(UUR)A→G 3260: a
second family with myopathy and cardiomyopathy. Q J Med.
86:435–438. 1993.
|
|
84
|
Nishino I, Komatsu M, Kodama S, et al: The
3260 mutation in mitochondrial DNA can cause mitochondrial
myopathy, encephalopathy, lactic acidosis, and strokelike episodes
(MELAS). Muscle Nerve. 19:1603–1604. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Goldstein JD, Shanske S, Bruno C and
Perszyk AA: Maternally inherited mitochondrial cardiomyopathy
associated with a C-to-T transition at nucleotide 3303 of
mitochondrial DNA in the tRNA(Leu(UUR)) gene. Pediatr Dev Pathol.
2:78–85. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Silvestri G, Santorelli FM, Shanske S, et
al: A new mtDNA mutation in the tRNA(Leu(UUR)) gene associated with
maternally inherited cardiomyopathy. Hum Mutat. 3:37–43. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
87
|
Palecek T, Tesarova M, Kuchynka P, et al:
Hypertrophic cardiomyopathy due to the mitochondrial DNA mutation
m.3303C>T diagnosed in an adult male. Int Heart J. 53:383–387.
2012.PubMed/NCBI
|
|
88
|
Taniike M, Fukushima H, Yanagihara I, et
al: Mitochondrial tRNA(Ile) mutation in fatal cardiomyopathy.
Biochem Biophys Res Commun. 186:47–53. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Giordano C, Perli E, Orlandi M, et al:
Cardiomyopathies due to homoplasmic mitochondrial tRNA mutations:
morphologic and molecular features. Hum Pathol. 44:1262–1270. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Taylor RW, Giordano C, Davidson MM, et al:
A homoplasmic mitochondrial transfer ribonucleic acid mutation as a
cause of maternally inherited hypertrophic cardiomyopathy. J Am
Coll Cardiol. 41:1786–1796. 2003. View Article : Google Scholar
|
|
91
|
Arbustini E, Diegoli M, Fasani R, et al:
Mitochondrial DNA mutations and mitochondrial abnormalities in
dilated cardiomyopathy. Am J Pathol. 153:1501–1510. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Terasaki F, Tanaka M, Kawamura K, et al: A
case of cardiomyopathy showing progression from the hypertrophic to
the dilated form: association of Mt8348A→G mutation in the
mitochondrial tRNA(Lys) gene with severe ultrastructural
alterations of mitochondria in cardiomyocytes. Jpn Circ J.
65:691–694. 2001.PubMed/NCBI
|
|
93
|
Merante F, Tein I, Benson L and Robinson
BH: Maternally inherited hypertrophic cardiomyopathy due to a novel
T-to-C transition at nucleotide 9997 in the mitochondrial
tRNA(glycine) gene. Am J Hum Genet. 55:437–446. 1994.PubMed/NCBI
|
|
94
|
Shin WS, Tanaka M, Suzuki J, et al: A
novel homoplasmic mutation in mtDNA with a single evolutionary
origin as a risk factor for cardiomyopathy. Am J Hum Genet.
67:1617–1620. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
95
|
Van Hove JL, Freehauf C, Miyamoto S, et
al: Infantile cardiomyopathy caused by the T14709C mutation in the
mitochondrial tRNA glutamic acid gene. Eur J Pediatr. 167:771–776.
2008.PubMed/NCBI
|
|
96
|
Ruppert V, Nolte D, Aschenbrenner T, et
al: Novel point mutations in the mitochondrial DNA detected in
patients with dilated cardiomyopathy by screening the whole
mitochondrial genome. Biochem Biophys Res Commun. 318:535–543.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
97
|
Dikalov S: Cross talk between mitochondria
and NADPH oxidases. Free Radic Biol Med. 51:1289–1301. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
98
|
Leone TC and Kelly DP: Transcriptional
control of cardiac fuel metabolism and mitochondrial function. Cold
Spring Harb Symp Quant Biol. 76:175–182. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
99
|
Cottrill KA and Chan SY: Metabolic
dysfunction in pulmonary hypertension: the expanding relevance of
the Warburg effect. Eur J Clin Invest. 43:855–865. 2013. View Article : Google Scholar : PubMed/NCBI
|