Introduction
Diabetic cardiomyopathy is characterized by
phenotypic alterations in cardiac muscle, independent of micro- and
macrovascular disease, coronary artery disease and hypertension
(1–3). Several factors have been reported to
be involved in the pathogenesis of this disease, including
hyperglycemia, insulin resistance and oxidative stress-induced
insults (3,4,5).
Among these factors, hyperglycemia is considered one of the most
important factors in the onset of diabetic cardiomyopathy (4,6).
Thus, studies on the response of cardiac cells to acutely high
levels of glucose may provide information for the prevention and
treatment of the cumulative effects of high glucose (HG).
It is speculated that most of the effects of
excessive glucose are associated with metabolism (7). An increase in glycolysis and hence,
pyruvate, enhances the rate of oxidative phosphorylation, which
results in reactive oxygen species (ROS) production. The increased
ROS production contributes to oxidative stress, leading to
myocardial damage (5,8,9).
For this reason, curcumin, which has antioxidant effects, has been
used to prevent diabetic cardiomyopathy in rats with streptozotocin
(STZ)-induced diabetes (10).
Accumulating evidence suggests that the activation of the
mitogen-activated protein kinase (MAPK) signaling pathway
attributes to the development of diabetic complications, including
cardiac damage (10–13). Mammals express at least 3 distinct
groups of MAPKs, including p38 MAPK, extracellular signal-regulated
protein kinase 1/2 (ERK1/2) and c-Jun N-terminal kinase (JNK). MAPK
signaling pathway is activated by HG stimulation in several types
of cell model. Fang et al (14) demonstrated that the exposure of
rat mesangial cells to HG (25 mM) significantly upregulated
phosphorylated (p) expression levels of p38 MAPK and ERK1/2. The
increased expression of p-p38 MAPK and p-ERK1/2 was inhibited by
pre-treatment with N-acetyl-L-cysteine (NAC; a ROS scavenger),
indicating the involvement of ROS in the HG-induced activation of
p38 MAPK and ERK1/2 in rat mesangial cells (14). In STZ-induced diabetic rats, the
expression levels of p-p38 MAPK and p-ERK1/2 are enhanced; curcumin
has been shown to prevent diabetic cardiomyopathy by inhibiting the
activation of p38 MAPK and ERK1/2 (10). Thus, it is reasonable to assume
that molecules with antioxidant and inhibitory effects on MAPK
activation may protect against HG-induced cardiomyocyte injury. One
of these candidate molecules is hydrogen sulfide
(H2S).
H2S, a well-known toxic gas with a
characteristic smell of rotten eggs, has been qualified as the
third gasotransmitter along with nitric oxide (NO) and carbon
monoxide (CO) (15,16). H2S has been considered
as an important cardioprotective agent. Exogenous H2S
has been shown to reduce myocardial necrosis and rescue contractile
activity in isoproterenol-treated rat hearts (17). Chronic H2S therapy
improves survival and prevents ischemic-induced heart failure
(18). In our previous studies,
we demonstrated that exogenous H2S protects H9c2 cardiac
cells against chemical hyoxia-induced injury by inhibiting
oxidative stress and upregulating heat shock protein 90 (HSP90)
expression (19,20). To date, the role of H2S
in diabetes-induced cardiac damage has attracted considerable
attention due to to its antioxidant (9,19–21) and modulatory effects on the
signaling pathways, including MAPK pathways (22,23). Recently, H2S has been
shown to provide protection against cardiomyopathy and vascular
dysfunction in models of STZ-induced diabetes (24). In the diabetic db/db mouse heart
model, Peake et al (9)
demonstrated the protective effects of H2S against
ischemic-reperfusion injury by activating nuclear factor
(erythroid-derived 2)-like 2 (Nrf2) signaling. However, whether
exogenous H2S protects cardiomyocytes against HG-induced
injury by inhibiting p38 MAPK and ERK1/2 activation remains
unclear. To examine this, in the present study, H9c2 cardiac cells
were treated with 35 mM glucose (high glucose, HG) to establish a
HG-induced cardiomyocyte injury model. We then focused on i) the
effects of HG on the activation of p38 MAPK and ERK1/2; ii) the
roles of the activation of p38 MAPK and ERK1/2 in HG-induced
cardiomyocyte injury; iii) the effects of exogenous H2S
on the HG-induced increase in p38 MAPK and ERK1/2 activation; iv)
the roles of p38 MAPK and ERK1/2 activation in the protective
effects of exogenous H2S against HG-induced injury in
H9c2 cells. In the current study, to our knowledge, we provide the
first evidence that exogenous H2S provides protection
against HG-induced injury [including cytotoxicity, apoptosis and
the loss of mitochondrial membrane potential (MMP)] by inhibiting
the activation of p38 MAPK and ERK1/2 and preventing oxidative
stress in H9c2 cells.
Materials and methods
Materials
Sodium hydrogen sulfide (NaHS),
2′,7′-dichlorofluorescein diacetate (DCFH-DA), Hoechst 33258 and
NAC were purchased from Sigma-Aldrich (St. Louis, MO, USA). The
Cell Counting Kit-8 (CCK-8) and rhodamine 123 (Rh123) were supplied
by Dojindo Laboratories (Kumamoto, Japan). Anti-p-ERK1/2 antibody,
anti-ERK1/2 antibody, anti-p38 antibody, anti-p-p38 antibody, U0126
and SB203580 were purchased from Cell Signaling Technology (Boston,
MA, USA). Anti-β-actin antibody, horseradish peroxidase
(HRP)-conjugated secondary antibody and the BCA Protein Assay kit
were obtained from KangChen Bio-tech (Shanghai, China). Fetal
bovine serum (FBS) and Dulbecco’s modified Eagle’s medium
(DMEM)-F12 medium were obtained from Gibco BRL (Grand Island, NY,
USA). The enhanced chemiluminescence (ECL) solution was purchased
from KeyGen Biotech (Nanjing, China). The H9c2 cardiac cells were
supplied by the Sun Yat-sen University Experimental Animal Center
(Guangzhou, China).
H9c2 cell culture and treatments
The H9c2 cardiac cell line was acquired from the Sun
Yat-sen University Experimental Animal Center. The cells were
cultured in DMEM, supplemented with 10% FBS in a humidified
atmosphere of 95% air and 5% CO2 at 37°C. The culture
medium was replaced with fresh medium every 2–3 days and expanded
to new culturewares when the cells reached approximately 80%
confluency.
To explore the protective effects of H2S
on HG-induced injury, the H9c2 cells were pre-conditioned with 400
μM NaHS for 30 min prior to exposure to HG for 24 h. To
further determine whether the protective effects of NaHS were
associated with the inhibition of p38 MAPK or ERK1/2 activity, the
H9c2 cells were pre-conditioned with SB203580 (a selective
inhibitor of p38 MAPK), or U0126 (a selective inhibitor of ERK1/2)
60 min prior to exposure to 35 mmol/l glucose for 24 h. To confirm
whether the protective effects of NaHS were associated with its
antioxidant action, H9c2 cells were pre-treated with NAC (a ROS
scavenger).
Cell viability assay
The H9c2 cells in the logarithmic growth phase were
cultured in plates at a concentration of 1×104 cells/ml.
The CCK-8 assay was then employed to assess the viability of the
H9c2 cardiac cells. After the indicated treatments, the cells were
washed with PBS and 10 μl CCK-8 solution at a 10% dilution
was added to each well and then the plate was incubated for
approximately 90 min in an incubator. Absorbance at 450 nm was
measured using a microplate reader (Molecular Devices, Sunnyvale,
CA, USA). The means of the optical density (OD) of 3 wells in the
indicated groups were used to calculate the percentage of cell
viability according to the following formula: cell viability (%) =
(ODtreatment group/ODcontrol group) ×100%.
The experiment was repeated 5 times.
Hoechst 33258 nuclear staining for the
assessment of apoptosis
Apoptotic cell death was assessed using the Hoechst
33258 staining method. The H9c2 cells were plated in 35-mm dishes
at a density of 1×106 cells/well. At the end of the
indicated treatments, the cells were harvested and fixed with
paraformaldehyde in 0.1 mol/l PBS for 10 min. After rinsing with
PBS, the nuclear DNA was stained with 5 mg/ml Hoechst 33258 dye for
10 min before being rinsed briefly with PBS and then visualized
under a fluorescence microscope (Bx50-FLA; Olympus, Tokyo, Japan).
Viable cells displayed a uniform blue fluorescence throughout the
nucleus, whereas apoptotic cells showed fragmented and condensed
nuclei. The experiment was repeated 3 times.
Measurement of MMP
MMP was assessed using a fluorescent dye, Rh123, a
cell-permeable carionic dye that preferentially enters the
mitochondria based on the highly negative MMP. The depolarization
of MMP results in the loss of Rh123 from the mitochondria and a
decrease in intracellular green fluorescence. H9c2 cardiac cells
were cultured on a slide with Eagle’s minimal essential medium
(EMEM)-F12. After the indicated treatments, the slides were washed
4 times with PBS. The H9c2 cells were incubated with 1 mg/l Rh123
at 37°C for 30 min in an incubator and washed briefly with PBS 4
times. Then Rh123 fluorescence was measured over the entire field
of vision using a fluorescence microscope connected to an imaging
system (BX50-FLA; Olympus). The mean fluorescence intensity (MFI)
of Rh123 from 5 random fields was analyzed using ImageJ 1.41o
software and was taken as an index of the levels of MMP. The
experiment was carried out 5 times.
Western blot analysis
After being subjected to the indicated treatments,
H9c2 cardiac cells were harvested and lysed with cell lysis
solution. Total proteins in the cell lysates were quantified using
the BCA Protein Assay kit. Loading buffer was added to the
cytosolic extracts and after boiling for approximately 5 min, equal
amounts of supernatant from each sample were fractionated by 10%
sodium dodecyl sulphate-polyacrylamide gel electrophoresis
(SDS-PAGE). Total proteins in the gel were transferred onto
polyvinylidene difluoride (PVDF) membranes. The membranes were
blocked for approximately 90 min at room temperature in fresh
blocking buffer [0.1% Tween-20 in Tris-buffered saline (TBS-T)
containing 5% milk] and then incubated with either anti-p38
(1:1,000 dilution), anti-p-p38 (1:1,000 dilution), anti-p-ERK1/2
(1:1,000 dilution) or anti-ERK1/2 (1:1,000 dilution) antibodies in
freshly prepared TBS-T with 3% fat-free milk overnight with slow
agitation at 4°C temperature. Following 3 washes with TBS-T, the
membranes were incubated with HRP-conjugated goat anti-rabbit
secondary antibody (1:2,500 dilution; KangChen Bio-tech) in TBS-T
with 3% fat-free milk for 90 min at room temperature. The membranes
were washed 3 times with TBS-T solution, developed in ECL solution
and visualized using X-ray film. Each experiment was repeated 3
times. For quantification, the films were scanned and analyzed
using ImageJ 1.47i software.
Measurement of intracellular ROS
levels
The determination of intracellular ROS levels was
performed by measuring a fluorescent product formed by the
oxidation of DCFH-DA. Briefly, the culture medium was removed and
the cells were washed with PBS 3 times. Following the addition of
fresh culture medium, the cells were incubated with DCFH-DA at the
final concentration of 10 μmol/l for 30 min at 37°C. The
cells were then washed again with PBS 3 times and the relative
amount of fluorescent product was assessed using a fluorescence
microscope connected to an imaging system (BX50-FLA; Olympus). The
MFI from 5 random fields was measured using ImageJ 1.41o software
and the MFI was used as an index of the amount of ROS. The
experiment was carried out 5 times.
Statistical analysis
All data are presented as the means ± SEM.
Differences between groups were analyzed one-way analysis of
variance (ANOVA) using SPSS 13.0 software (SPSS, Chicago, IL, USA)
followed by the LSD post hoc comparison test. A P-value <0.05
was considered to indicate a statistically significant
difference.
Results
Exogenous H2S inhibits the
HG-induced upregulation of the expression of p-p38 MAPK in H9c2
cells
As illustrated in Fig.
1, treatment of the H9c2 cells with 35 mM glucose (HG) for 24 h
significantly upregulated the expression of p-p38 MAPK in the H9c2
cells. However, this increase in the expression of p-p38 MAPK was
attenuated by pre-treatment of the cells with 400 μM NaHS (a
donor of H2S) for 30 min prior to exposure to HG. NaHS
at 400 μM alone did not alter the basal expression of p-p38
MAPK in the H9c2 cells (Fig.
1).
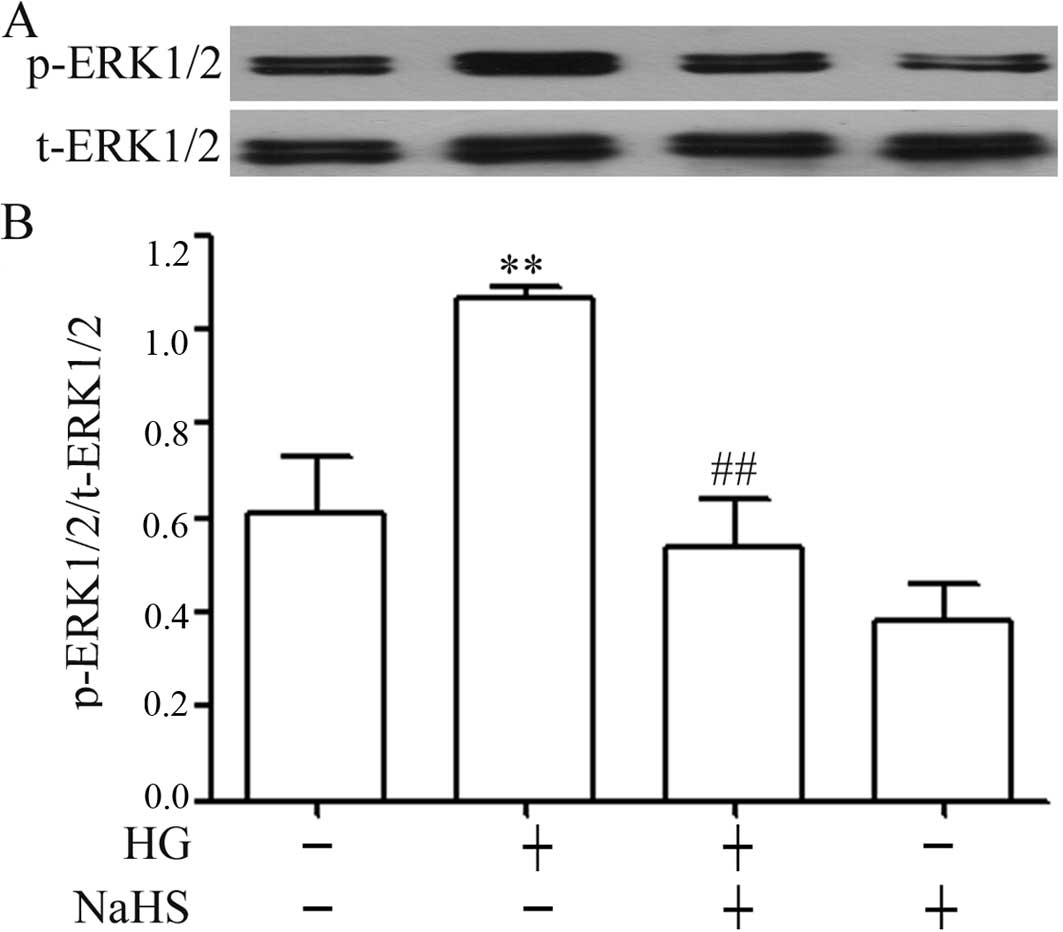
Exogenous H2S attenuates the
HG-induced increase in the expression of p-ERK1/2 in H9c2
cells
The results from western blot analysis revealed that
the exposure of H9c2 cells to 35 mM glucose for 24 h markedly
elevated the expression levels of p-ERK1/2 (Fig. 2), but did not induce significant
changes in the expression of total (t)-ERK1/2. Additionally,
similar to the inhibitory effects of NaHS on p-p38 MAPK expression,
pre-treatment of the H9c2 cells with 400 μM NaHS for 30 min
prior to exposure to 35 mM glucose markedly reduced the increased
expression of p-ERK1/2 induced by HG for 24 h (Fig. 2). Alone, NaHS at 400 μM did
not alter the basal expression of p-ERK1/2 in the H9c2 cells.
NaHS, p38 MAPK inhibitor and ERK1/2
inhibitor ameliorate HG-induced cytotoxicity in H9c2 cells
Firstly, we examined whether exogenous
H2S protects H9c2 cells against HG-induced cytotoxicity.
The H9c2 cells were pre-treated with 400 μM NaHS for 30 min
followed by exposure to 35 mM glucose for 24 h. Our results
revealed that pre-treatment with NaHS for 30 min significantly
decreased HG-induced cytotoxicity, as evidenced by an increase in
cell viability (Fig. 3). Since
the above results (Figs. 1 and
2) showed that the expression
levels of p-p38 MAPK and p-ERK1/2 were enhanced by HG treatment, we
then wished to confirm whether p-p38 MAPK and p-ERK1/2 activation
contributed to HG-induced cytotoxicity. As shown in Fig. 3, pre-treatment of the H9c2 cells
with either 3 μM SB203580 (an inhibitor of p38 MAPK) or
U0126 (a selective inhibitor of ERK1/2) for 60 min prior to
exposure to 35 mM glucose markedly suppressed HG-induced
cytotoxicity, leading to an increase in cell viability. These
findings suggest that the activation of p38 MAPK and ERK1/2 is
involved in HG-induced cytotoxicity.
NaHS, p38 MAPK inhibitor and ERK1/2
inhibitor diminish HG-induced apoptosis of H9c2 cells
We further explored the effects of exogenous
H2S, p38 MAPK inhibitor and ERK1/2 inhibitor on
HG-induced apoptosis. As shown in Fig. 4B, the exposure of the H9c2 cells
to 35 mM glucose for 24 h induced characteristics typical of
apoptosis, as evidenced by tbe condensation of chromatin, the
shrinkage of nuclei and apoptotic bodies. Of note, pre-treatment of
the cells with 400 μM NaHS for 30 min prior to HG exposure
significantly attenuated the HG-induced increase in the number of
cells with nuclear condensation and fragmentation (Fig. 4C). Similarly, pre-treatment of the
cells with either 3 μM SB203580 or 15 μM U0126 for 60
min prior to HG treatment inhibited HG-induced apoptosis (Fig. 4D and E). Alone, NaHS, SB203580 or
U0126 did not significantly affect cell morphology or the
percentage of apoptotic H9c2 cells (Fig. 4F–H). The above results indicate
that exogenous H2S protects H9c2 cells against
HG-induced apoptosis which is associated, at least in part, with
the activation of p38 MAPK and ERK1/2.
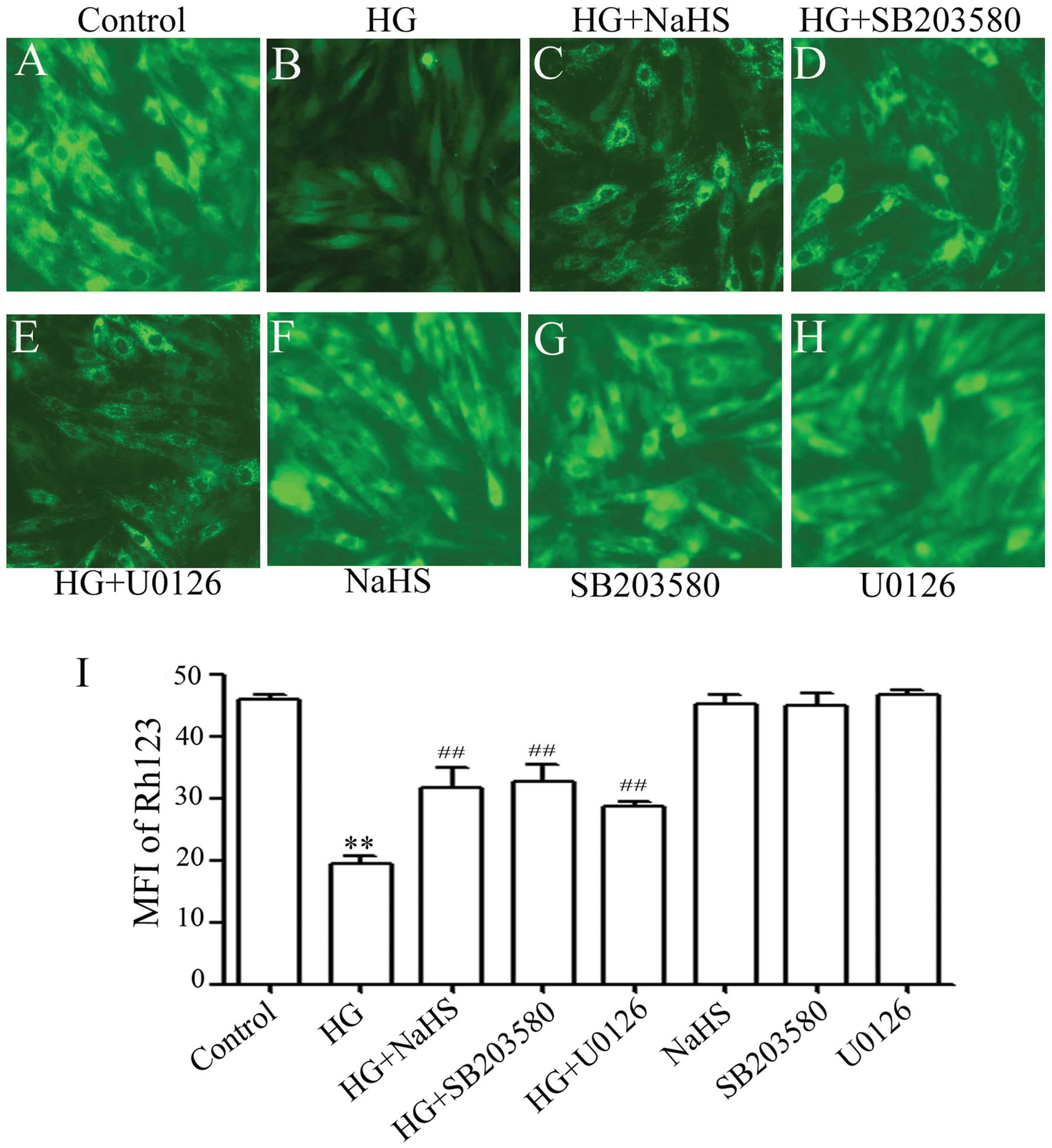
NaHS, p38 MAPK inhibitor and ERK1/2
inhibitor reduce HG-triggered oxidative stress in H9c2 cells
Accumulating evidence has indicated that the
generation of ROS participates in HG-induced cardiac injury
(25). Therefore, in this study,
we observed the effects of exogenous H2S on HG-induced
ROS generation in H9c2 cells. It was shown that treatment of the
cells with 35 mM glucose for 24 h considerably increased the
generation of ROS (Fig. 5B). The
increased ROS generation was diminished by pre-treatment of the
cells with 400 μM NaHS for 30 min prior to exposure to HG
(Fig. 5C), illustrating the
inhibitory effects of exogenous H2S on HG-induced
oxidative stress. To investigate whether the activation of p38 MAPK
and ERK1/2 is involved in HG-induced oxidative stress, the H9c2
cells were pre-treated with 3 μM SB203580 or 15 μM
U0126 for 60 min prior to exposure to HG. As shown in Fig. 5, pre-treatment with SB203580
(Fig. 5D) or U0126 (Fig. 5E) markedly reduced the HG-induced
increase in ROS generation, suggesting that the activation of p38
MAPK and ERK1/2 contributes to the overproduction of ROS induced by
HG.
NaHS, p38 MAPK inhibitor and ERK1/2
inhibitor suppress the HG-induced dissipation of MMP in H9c2
cells
Previous studies have shown that ROS induces
mitochondrial damage, which has been implicated in HG-induced
cardiac insults (25). Thus, we
explored whether exogenous H2S prevents the loss of MMP
in HG-treated H9c2 cells. As shown in Fig. 6, treatment of the cells with 35 mM
glucose for 24 h markedly induced mitochondrial damage, as
evidenced by the dissipation of MMP (Fig. 6B). Importantly, the dissipation of
MMP was ameliorated by pre-conditioning with 400 μM NaHS for
30 min prior to treatment of the cells with 35 mM glucose (Fig. 6C and I), suggesting the protective
effects of exogenous H2S against the HG-induced loss of
MMP. In addition, pre-treatment of the cells with either 3
μM SB203580 (Fig. 6D) or
15 μM U0126 (Fig. 6E) for
60 min prior to exposure to 35 mM glucose antagonized the
HG-induced dissipation of MMP, indicating the involvement of the
activation of p38 MAPK and ERK1/2 in HG-induced mitochondrial
insults in H9c2 cells. Alone, NaHS at 400 μM or 3 μM
SB203580 or 15 μM U0126 did not induce the loss of MMP
(Fig. 6F–I).
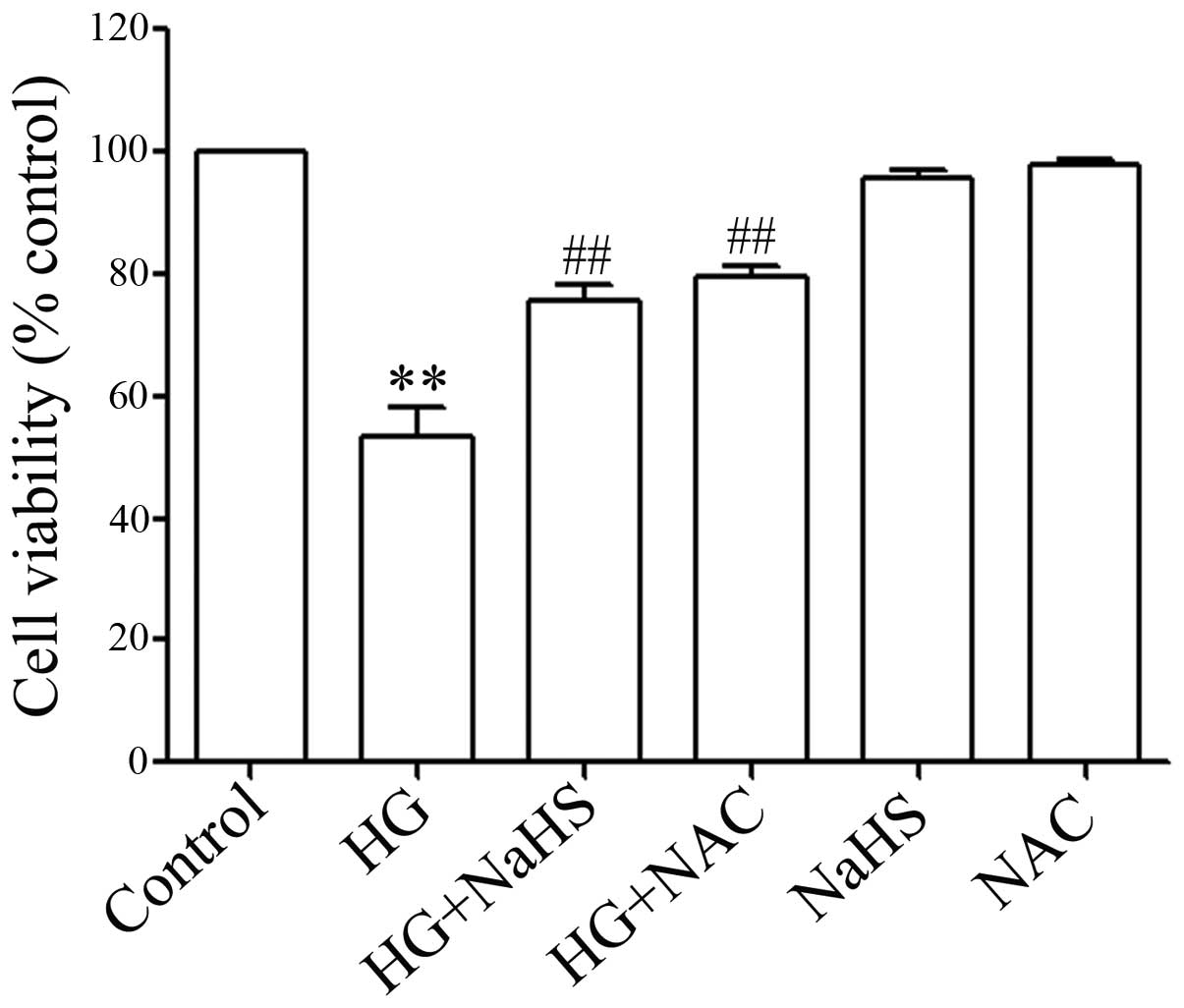
ROS scavenger decreases the HG-induced
cytotoxicity in H9c2 cells
In order to demonstrate whether the role of
exogenous H2S (NaHS) in inhibiting HG-induced
cytotoxicity is associated with its antioxidant effects, the H9c2
cells were pre-treated with 1,000 μM NAC (a ROS scavenger)
for 60 min prior to esposure to 35 mM glucose for 24 h. Similar to
the inhibitory effects of NaHS pre-treatment on HG-induced
cytotoxicity, NAC pre-treatment significantly inhibited HG-induced
cytotoxicity, resulting in an increase in cell viability (Fig. 7), revealing that the antioxidant
effects of H2S contribute, at least in part, to the
inhibitory effects of exogenous H2S on cytotoxicity
induced by HG in the H9c2 cells.
Discussion
Increasing clinical studies have shown that
hyperglycemia is a risk factor for the development of diabetic
cardiomyopathy. However, the mechanisms responsible for HG-induced
cardiac damage are not yet fully understood. Consistent with the
data from previous studies (4,5,8–10),
our results revealed that the exposure of H9c2 cardiac cells to 35
mM (HG) induced significant injury, as evidenced by a decrease in
cell viability, an increase in the number of apoptotic cells and
ROS production, as well as the dissipation of MMP. In order to
clarify the correlation between the increased ROS levels and
cytotoxicity, the H9c2 cells were pre-treated with NAC, a scavenger
of ROS, prior to exposure to HG. The results of this study
demonstrated that NAC pre-treatment markedly inhibited HG-induced
cytotoxicity, leading to an increase in cell viability, suggesting
the critical role of ROS in HG-elicited cardiomyocyte insults,
which is supported by the results of previous studies (5,8–10).
Since the activation of p38 MAPK and ERK1/2 has been
shown to contribute to the development of diabetic complications,
such as diabetic nephropathy (14) and retinopathy (26), in this study, we first examined
the effects of HG on the phosphorylation of p38 MAPK and ERK1/2 in
H9c2 cells. Our results revealed that exposure of the cells to 35
mM glucose markedly enhanced the expression levels of p-p38 MAPK
and p-ERK1/2, indicating the activation of p38 MAPK and ERK1/2 by
HG. Similar to our results, p38 MAPK and ERK1/2 are activated in
HG-induced human retinal pigmented epithelial cells (26). In addition, the chronic exposure
of human mesangial cells to HG activates the p38 MAPK pathway
(27). Our data are comparable
with those of previous studies (26,27). Based on our findings, as well as
those of previous studies (26,27), it can by hypothesized that HG may
be an inducer of MAPK pathway activation.
Secondly, we investigated the roles of p38 MAPK and
ERK1/2 in HG-induced cardiomyocyte injury. p38 MAPK and ERK1/2 are
activated by cellular stress and have been reported to participate
in cardiomycyte apoptosis and cardiac pathologies (28,29). The present study demonstrated that
the pre-treatment of H9c2 cells with SB203580 (a selective
inhibitor of p38 MAPK) markedly ameliorated HG-induced injury
(including cytotoxicity, apoptosis, overproduction of ROS and the
loss of MMP), as evidenced by an increase in cell viability and a
decrease in the number of apoptotic cells and ROS production, as
well as the attenuation of the dissipation of MMP. These results
suggest that the activation of p38 MAPK contributes to HG-induced
cytotoxicity, apoptosis and MMP loss, which may be an important
mechanism underlying HG-triggered cardiac injury. Our findings
support the notion that in the complex signaling events of
apoptosis, the activation of p38 MAPK attenuates the mitochondrial
function induced by changes in the ratio of pro-apoptotic (Bax) and
anti-apoptotic (Bcl-2) members of the mitochondria, causing MMP
loss, the release of cytochrome c and the activation of
caspases, thus leading to apoptosis (30).
In addition, the role of ERK1/2 in HG-induced injury
has gained much attention. The activation of ERK1/2 is stimulated
by various stimuli and targets different downstream molecules, and
therefore performs different functions. For example, the
phosphorylation of ERK1/2/Akt in cardiomyocytes during early
reperfusion has been found to serve as a defense mechanism against
ischemia (31). Blockage with the
ERK1/2-specific inhibitors, PD98059 and U0126, has been shown to
inhibit ischemic pre-conditioning-induced cardioprotection
(32). U0126 also attenuates the
cardioprotective effects induced by fibroblast growth factor-1
(FGF-1) (33). However, the
effects of ERK1/2 on cell survival are controversial, as the
inhibition of ERK1/2 with U0126 and the downregulation of ERK1/2
using RNA interference has been shown to significantly reduce
H2O2-induced apoptosis in neuronal cell lines
(PC12 and SH-SY5Y cells) (34).
In addition, the activation of the ERK1/2 pathway has been shown to
be involved in chemical hypoxia-induced cardiomyocyte insults
(23) and STZ-induced cardiac
dysfunction (10). In accordance
with these studies (10,23,34), in this study, we demonstrate that
ERK1/2 activation contributes to HG-induced injury. This is
supported by the following findings: i) HG upregulated the
expression levels of p-ERK1/2 and ii) U0126 protected H9c2 cells
against HG-induced injury, including cytotoxicity, apoptosis, ROS
overproduction and the loss of MMP. These findings suggest that
stimulating the activation of ERK1/2 may be another key mechanism
responsible for HG-induced cardiomyocyte insults.
Importantly, in this study, we demonstrate the
potential protective effects of H2S in HG-treated H9c2
cardiac cells. H2S has been considered as a novel
gasomolecule with cardioprotective effects. H2S is
produced enzymatically in mammalian species via the action of 3
enzymes in the cysteine biosynthesis pathway: cystathionine-γ-lyase
(CSE), cystathionine-β-synthase (CBS) and 3-mercaptopyruvate
sulfurtransferase (3-MST). Changes in H2S levels in
diabetes have attracted attention. There is clear evidence that
circulating levels of H2S are reduced in animal models
of diabetes (24,35–37). Notably, lower circulating
H2S levels have been measured in plasma samples obtained
from patients with type 2 diabetes mellitus (T2DM) (35,38). These findings have promoted
researchers to explore the protective effects of exogenous
H2S against diabetes-induced cardiac injury. Increasing
evidence shows that exogenous H2S exerts protective
effects against several models of myocardial injury in the setting
of type 1 diabetes by preventing apoptosis and oxidative stress
(24,39). In db/db mice, H2S
therapy in the form of NaHS has been shown to attenuate myocardial
ischemia-reperfusion injury (9).
In agreement with these studies (9,24,39), we found that exogenous
H2S exerted multiple protective effects, including
anticytotoxic, anti-apoptotic and antioxidant effects, as well as
mitochondrial protective effects (alleviating the loss of MMP),
against HG-induced injury in H9c2 cells. One of the mechanisms
underlying these protective effects may be associated the
antioxidant effects of H2S (lowering ROS production), as
NAC, a ROS scavenger, exerted protective effects similar to those
of H2S against HG-induced cytotoxicity.
Since we have previously demonstrated the inhibitory
effects of exogenous H2S on the chemical hypoxia-induced
activation of p38 MAPK and ERK1/2 in PC12 cells (22) and the activation of ERK1/2 in H9c2
cells (23), in this study, we
explored the modulatory effects of exogenous H2S on the
HG-stimulated activation of p38 MAPK and ERK1/2 in H9c2 cells. We
found that pre-treatment of the H9c2 cells with NaHS attenuated not
only the expression level of p-p38 MAPK, but also that of p-ERK1/2
induced by HG in H9c2 cells, revealing that exogenous
H2S inhibits the HG-induced activation of p38 MAPK and
ERK1/2, which may be one of important mechanisms responsible for
the cardioprotective effects of exogenous H2S against
HG-induced injury in H9c2 cells. The involvement of hte inhibition
of the p38 MAPK pathway in the cytoprotective effects of exogenous
H2S has also been reported by other studies. Hu et
al indicated that H2S suppresses LPS-induced
inflammation by inhibiting p38 MAPK in microglia (40) and that H2S protects
SH-SY5Y cells against rotenone-induced apoptosis through the
suppression of p38 MAPK activation (41). Additionally, exogenous
H2S reduces doxorubicin-induced cardiotoxicity by
inhibiting the activation of the p38 MAPK pathway (29). Our results, as well as those from
previous studies (22,29,40,41) suggest that exogenous
H2S may be a potential inhibitor of p38 MAPK.
However, reports on the effects of H2S on
the activation of ERK1/2 are controversial. The ERK1/2 activation
has been shown to be increased (42–46), decreased (22,23,29,47–49) or unaltered (50,51) following exposure to
H2S. In the present study, we provide clear evidence
that the activation of ERK1/2 contributes to HG-induced injury in
H9c2 cells and that exogenous H2S protects H9c2 cells
against HG-induced injury by inhibiting ERK1/2 activation, which is
supported by our previous study [Dong et al (23)]. Although the mechanisms through
which ERK1/2 induces either cell death or survival and the reasons
for the differential effects induced by exogenous H2S on
the activation of ERK1/2 are unclear, it is generally accepted that
the duration and magnitude of ERK1/2 activation may be an important
factor in determining cell survival or apoptosis (52). In addition, exploring the
interaction between ERK1/2 and survival or apoptotic regulators may
provide important information on the mechanisms underlying the
differential effects of exogenous H2S on ERK1/2
activation.
In conclusion, to our knowledge, the findings of
present study demonstrate for the first time that exogenous
H2S protects against HG-induced injury by attenuating
the activation of p38 MAPK and ERK1/2 in H9c2 cardiac cells. Our
results also provide important evidence that the activation of p38
MAPK and ERK1/2 is involved in HG-induced multiple cardiomyocyte
injury, including cytotoxicity, apoptosis, ROS overproduction and
the dissipation of MMP. These findings may provide a rationale for
designing effective therapeutic strategies for the treatment of
heart disease in the setting of diabetes.
Acknowledgements
The present study was supported by grants from the
Science and Technology Planning Project of Guangdong of China
(2010B080701035, 2010B080701105 and 2012B031800358) and the
National Natural Science Foundation of China (H0208).
References
|
1
|
Grundy SM, Benjamin IJ, Burke GL, et al:
Diabetes and cardiovascular disease: a statement for healthcare
professionals from the American Heart Association. Circulation.
100:1134–1146. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Cai L and Kang YJ: Oxidative stress and
diabetic cardiomyopathy: a brief review. Cardiovasc Toxicol.
1:181–193. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Francis GS: Diabetic cardiomyopathy: fact
or fiction? Heart. 85:247–248. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rodrigues B, Cam MC and McNeill JH:
Metabolic disturbances in diabetic cardiomyopathy. Mol Cell
Biochem. 180:53–57. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Privratsky JR, Wold LE, Sowers JR, Quinn
MT and Ren J: AT1 blockade prevents glucose-induced cardiac
dysfunction in ventricular myocytes: role of the AT1 receptor and
NADPH oxidase. Hypertension. 42:206–212. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ren J and Davidoff AJ: Diabetes rapidly
induces contractile dysfunctions in isolated ventricular myocytes.
Am J Physiol. 272:H148–H158. 1997.PubMed/NCBI
|
|
7
|
Rahimi R, Nikfar S, Larijani B and
Abdollahi M: A review on the role of antioxidants in the management
of diabetes and its complications. Biomed Pharmacother. 59:365–373.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cai H and Harrison DG: Endothelial
dysfunction in cardiovascular diseases: the role of oxidant stress.
Circ Res. 87:840–844. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peake BF, Nicholson CK, Lambert JP, et al:
Hydrogen sulfide preconditions the db/db diabetic mouse heart
against ischemia-reperfusion injury by activating Nrf2 signaling in
an Erk-dependent manner. Am J Physiol Heart Circ Physiol.
304:H1215–H1224. 2013. View Article : Google Scholar
|
|
10
|
Soetikno V, Sari FR, Sukumaran V, et al:
Curcumin prevents diabetic cardiomyopathy in streptozotocin-induced
diabetic rats: possible involvement of PKC-MAPK signaling pathway.
Eur J Pharm Sci. 47:604–614. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Evans JL, Goldfine ID, Maddux BA and
Grodsky GM: Oxidative stress and stress-activated signaling
pathways: a unifying hypothesis of type 2 diabetes. Endocr Rev.
23:599–622. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Igarashi M, Wakasaki H, Takahara N, et al:
Glucose or diabetes activates p38 mitogen-activated protein kinase
via different pathways. J Clin Invest. 103:185–195. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yan J, Young ME, Cui L, Lopaschuk GD, Liao
R and Tian R: Increased glucose uptake and oxidation in mouse
hearts prevent high fatty acid oxidation but cause cardiac
dysfunction in diet-induced obesity. Circulation. 119:2818–2828.
2009. View Article : Google Scholar
|
|
14
|
Fang S, Jin Y, Zheng H, et al: High
glucose condition upregulated Txnip expression level in rat
mesangial cells through ROS/MEK/MAPK pathway. Mol Cell Biochem.
347:175–182. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Łowicka E and Bełtowski J: Hydrogen
sulfide (H2S) - the third gas of interest for
pharmacologists. Pharmacol Rep. 59:4–24. 2007.
|
|
16
|
Moore PK, Bhatia M and Moochhala S:
Hydrogen sulfide: from the smell of the past to the mediator of the
future? Trends Pharmacol Sci. 24:609–611. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Geng B, Chang L, Pan C, et al: Endogenous
hydrogen sulfide regulation of myocardial injury induced by
isoproterenol. Biochem Biophys Res Commun. 318:756–763. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Calvert JW, Jha S, Gundewar S, et al:
Hydrogen sulfide mediates cardioprotection through Nrf2 signaling.
Circ Res. 105:365–374. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chen SL, Yang CT, Yang ZL, et al: Hydrogen
sulphide protects H9c2 cells against chemical hypoxia-induced
injury. Clin Exp Pharmacol Physiol. 37:316–321. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang Z, Yang C, Xiao L, et al: Novel
insights into the role of HSP90 in cytoprotection of H2S
against chemical hypoxia-induced injury in H9c2 cardiac myocytes.
Int J Mol Med. 28:397–403. 2011.PubMed/NCBI
|
|
21
|
Nicholson CK and Calvert JW: Hydrogen
sulfide and ischemia-reperfusion injury. Pharmacol Res. 62:289–297.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lan A, Liao X, Mo L, et al: Hydrogen
sulfide protects against chemical hypoxia-induced injury by
inhibiting ROS-activated ERK1/2 and p38MAPK signaling pathways in
PC12 cells. PLoS One. 6:e259212011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dong XB, Yang CT, Zheng DD, et al:
Inhibition of ROS-activated ERK1/2 pathway contributes to the
protection of H2S against chemical hypoxia-induced
injury in H9c2 cells. Mol Cell Biochem. 362:149–157. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Suzuki K, Olah G, Modis K, et al: Hydrogen
sulfide replacement therapy protects the vascular endothelium in
hyperglycemia by preserving mitochondrial function. Proc Natl Acad
Sci USA. 108:13829–13834. 2011. View Article : Google Scholar
|
|
25
|
Malhotra A, Vashistha H, Yadav VS, et al:
Inhibition of p66ShcA redox activity in cardiac muscle cells
attenuates hyperglycemia-induced oxidative stress and apoptosis. Am
J Physiol Heart Circ Physiol. 296:H380–H388. 2009. View Article : Google Scholar
|
|
26
|
Yuan Z, Feng W, Hong J, Zheng Q, Shuai J
and Ge Y: p38MAPK and ERK promote nitric oxide production in
cultured human retinal pigmented epithelial cells induced by high
concentration glucose. Nitric Oxide. 20:9–15. 2009. View Article : Google Scholar
|
|
27
|
Wilmer WA, Dixon CL and Hebert C: Chronic
exposure of human mesangial cells to high glucose environments
activates the p38 MAPK pathway. Kidney Int. 60:858–871. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Sugden PH and Clerk A: ‘Stress-responsive’
mitogen-activated protein kinases (c-Jun N-terminal kinases and p38
mitogen-activated protein kinases) in the myocardium. Circ Res.
83:345–352. 1998.
|
|
29
|
Guo R, Lin J, Xu W, et al: Hydrogen
sulfide attenuates doxorubicin-induced cardiotoxicity by inhibition
of the p38 MAPK pathway in H9c2 cells. Int J Mol Med. 31:644–650.
2013.PubMed/NCBI
|
|
30
|
Yang J, Liu X, Bhalla K, et al: Prevention
of apoptosis by Bcl-2: release of cytochrome c from mitochondria
blocked. Science. 275:1129–1132. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yue TL, Wang C, Gu JL, et al: Inhibition
of extracellular signal-regulated kinase enhances
Ischemia/Reoxygenation-induced apoptosis in cultured cardiac
myocytes and exaggerates reperfusion injury in isolated perfused
heart. Circ Res. 86:692–699. 2000. View Article : Google Scholar
|
|
32
|
Strohm C, Barancik T, Brühl ML, Kilian SA
and Schaper W: Inhibition of the ER-kinase cascade by PD98059 and
UO126 counteracts ischemic preconditioning in pig myocardium. J
Cardiovasc Pharmacol. 36:218–229. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Buehler A, Martire A, Strohm C, et al:
Angiogenesis-independent cardioprotection in FGF-1 transgenic mice.
Cardiovasc Res. 55:768–777. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen L, Liu L, Yin J, Luo Y and Huang S:
Hydrogen peroxide-induced neuronal apoptosis is associated with
inhibition of protein phosphatase 2A and 5, leading to activation
of MAPK pathway. Int J Biochem Cell Biol. 41:1284–1295. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jain SK, Bull R, Rains JL, et al: Low
levels of hydrogen sulfide in the blood of diabetes patients and
streptozotocin-treated rats causes vascular inflammation? Antioxid
Redox Signal. 12:1333–1337. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Yusuf M, Kwong Huat BT, Hsu A, Whiteman M,
Bhatia M and Moore PK: Streptozotocin-induced diabetes in the rat
is associated with enhanced tissue hydrogen sulfide biosynthesis.
Biochem Biophys Res Commun. 333:1146–1152. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Ahmad FU, Sattar MA, Rathore HA, et al:
Exogenous hydrogen sulfide (H2S) reduces blood pressure
and prevents the progression of diabetic nephropathy in
spontaneously hypertensive rats. Ren Fail. 34:203–210.
2012.PubMed/NCBI
|
|
38
|
Whiteman M, Gooding KM, Whatmore JL, et
al: Adiposity is a major determinant of plasma levels of the novel
vasodilator hydrogen sulphide. Diabetologia. 53:1722–1726. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Gao Y, Yao X, Zhang Y, et al: The
protective role of hydrogen sulfide in myocardial
ischemia-reperfusion-induced injury in diabetic rats. Int J
Cardiol. 152:177–183. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Hu LF, Wong PT, Moore PK and Bian JS:
Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation
by inhibition of p38 mitogen-activated protein kinase in microglia.
J Neurochem. 100:1121–1128. 2007. View Article : Google Scholar
|
|
41
|
Hu LF, Lu M, Wu ZY, Wong PT and Bian JS:
Hydrogen sulfide inhibits rotenone-induced apoptosis via
preservation of mitochondrial function. Mol Pharmacol. 75:27–34.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Adhikari S and Bhatia M:
H2S-induced pancreatic acinar cell apoptosis is mediated
via JNK and p38 MAP kinase. J Cell Mol Med. 12:1374–1383. 2008.
|
|
43
|
Zhi L, Ang AD, Zhang H, Moore PK and
Bhatia M: Hydrogen sulfide induces the synthesis of proinflammatory
cytokines in human monocyte cell line U937 via the ERK-NF-kappaB
pathway. J Leukoc Biol. 81:1322–1332. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Mukherjee S, Lekli I, Goswami S and Das
DK: Freshly crushed garlic is a superior cardioprotective agent
than processed garlic. J Agric Food Chem. 57:7137–7144. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hu Y, Chen X, Pan TT, et al:
Cardioprotection induced by hydrogen sulfide preconditioning
involves activation of ERK and PI3K/Akt pathways. Pflugers Arch.
455:607–616. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yang G, Wu L, Bryan S, Khaper N, Mani S
and Wang R: Cystathionine gamma-lyase deficiency and
overproliferation of smooth muscle cells. Cardiovasc Res.
86:487–495. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Yang G, Yang W, Wu L and Wang R:
H2S, endoplasmic reticulum stress, and apoptosis of
insulin-secreting beta cells. J Biol Chem. 282:16567–16576.
2007.PubMed/NCBI
|
|
48
|
Lu M, Hu LF, Hu G and Bian JS: Hydrogen
sulfide protects astrocytes against H(2)O(2)-induced neural injury
via enhancing glutamate uptake. Free Radic Biol Med. 45:1705–1713.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Kloesch B, Liszt M and Broell J:
H2S transiently blocks IL-6 expression in rheumatoid
arthritic fibroblast-like synoviocytes and deactivates p44/42
mitogen-activated protein kinase. Cell Biol Int. 34:477–484.
2010.
|
|
50
|
Cai WJ, Wang MJ, Moore PK, Jin HM, Yao T
and Zhu YC: The novel proangiogenic effect of hydrogen sulfide is
dependent on Akt phosphorylation. Cardiovasc Res. 76:29–40. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Bliksøen M, Kaljusto ML, Vaage J and
Stensløkken KO: Effects of hydrogen sulphide on
ischaemia-reperfusion injury and ischaemic preconditioning in the
isolated, perfused rat heart. Eur J Cardiothorac Surg. 34:344–349.
2008.PubMed/NCBI
|
|
52
|
Marshall CJ: Specificity of receptor
tyrosine kinase signaling: transient versus sustained extracellular
signal-regulated kinase activation. Cell. 80:179–185. 1995.
View Article : Google Scholar
|