Introduction
The receptor for advanced glycation end products
(RAGE) belongs to the immunoglobulin superfamily of cell surface
receptors. It comprises of three domains, an extracellular domain
(with one V-type and two C-type domains), a transmembrane-spanning
domain and a cytoplasmic domain (1). RAGE is activated by binding to a
diverse repertoire of ligands (2). These ligands include advanced
glycation end products (AGEs) (3), S100 family proteins (4–6),
high-mobility group box 1 (HMGB1) (5,7)
and amyloid β (Aβ) peptides (8).
Activated RAGE triggers multiple intracellular pathways, such as
the production of reactive oxygen species, the activation of
p21ras, Erk1/2 (p44/p42) mitogen-activated protein kinases, p38 and
stress-activated protein kinase (SAPK)/c-Jun N-terminal kinase
(JNK) mitogen-activated protein kinases, rhoGTPases,
phosphatidylinositol-3 kinase and Janus kinase (JAK)/signal
transducers and activators of transcription (STAT), eventually
leading to the activation of nuclear factor-κB (NF-κB), activator
protein-1 (AP-1) and STAT-3 (9–12).
RAGE is physiologically expressed in multiple
tissues during embryonic development, but is mostly downregulated
as cells reach homeostasis in adult life. Therefore, with the
exception of the skin and lungs, RAGE expression is kept at low
levels in the adult body (13,14). The binding of ligands and the
increased expression of RAGE potentially lead to
inflammation-associated pathological conditions, such as those
found in neurodegenerative diseases (8,15,16), cardiovascular and renal diseases
(17,18), pulmonary diseases (19), diabetes and metabolic disorders
(20,21), as well as cancer (22). Under such conditions, NF-κB
activated by RAGE signaling enhances the expression of RAGE, thus
composing a self-sustaining positive feedback loop. Therefore, RAGE
is one of the most promising targets for the development of
therapeutic methods.
Antagonistic RAGE peptides (23,24), RAGE-blocking antibodies (25) and ligand-binding drugs (26) have been examined in an effort to
inhibit or abrogate RAGE signaling. These studies focused on the
extracellular domain of RAGE and its various ligands. On the other
hand, we have previously found that the short cytoplasmic domain of
RAGE is phosphorylated at serine 391 (Ser391) by protein kinase C,
zeta (PKCζ) upon ligand binding. The phosphorylated domain then
recruits Toll-interleukin 1 receptor domain-containing adaptor
protein (TIRAP), an adaptor protein for TLR2/4 (27), and transduces a signal to
downstream molecules. Since the RAGE-TIRAP interaction is commonly
induced by diverse ligands, such as AGEs, S100 proteins and HMGB1,
an inhibitory tool for the interaction between RAGE and TIRAP may
efficiently abrogate RAGE-mediated signaling regardless of ligand
species. In the present study, we developed inhibitor decoy
peptides for RAGE signaling (RAGE-I) by mimicking the
phosphorylated cytosolic domain of RAGE. RAGE-I was cationized by
polyethylenimine (PEI) for its efficient delivery into target
cells, as previously described (28). We showed that RAGE-I, delivered
intracellularly, bound to TIRAP and mitigated RAGE-mediated
signaling. Neuronal cell death induced by an excess amount of S100B
and the migration and invasion of glioma cells were suppressed by
the application of RAGE-I in vitro. Our results indicate
that RAGE-I provides a powerful therapeutic tool for the blocking
of RAGE-mediated multiple signaling.
Materials and methods
Preparation of PEI-avidin and
biotinylated RAGE-I
To efficiently deliver inhibitor peptides into
cells, PEI-avidin was used as the vehicle as previously described
(28). Avidin was coupled with
PEI600 (both from Wako Chemicals, Osaka, Japan) by
1-ethyl-3-(3-dimethylaminopropyl) carbodiimide hydrochloride (EDC;
Pierce, Rockford, IL, USA) [avidin (2.5 mg/ml) in PEI600-solution
(100 mg/ml, pH 5.0) and 0.1 mg/ml EDC] for 16 h at room
temperature. After the reaction, the solution was exhaustively
dialyzed against PBS. Inhibitor peptides were designed to function
as a decoy by binding to TIRAP, mimicking the cytoplasmic sequence
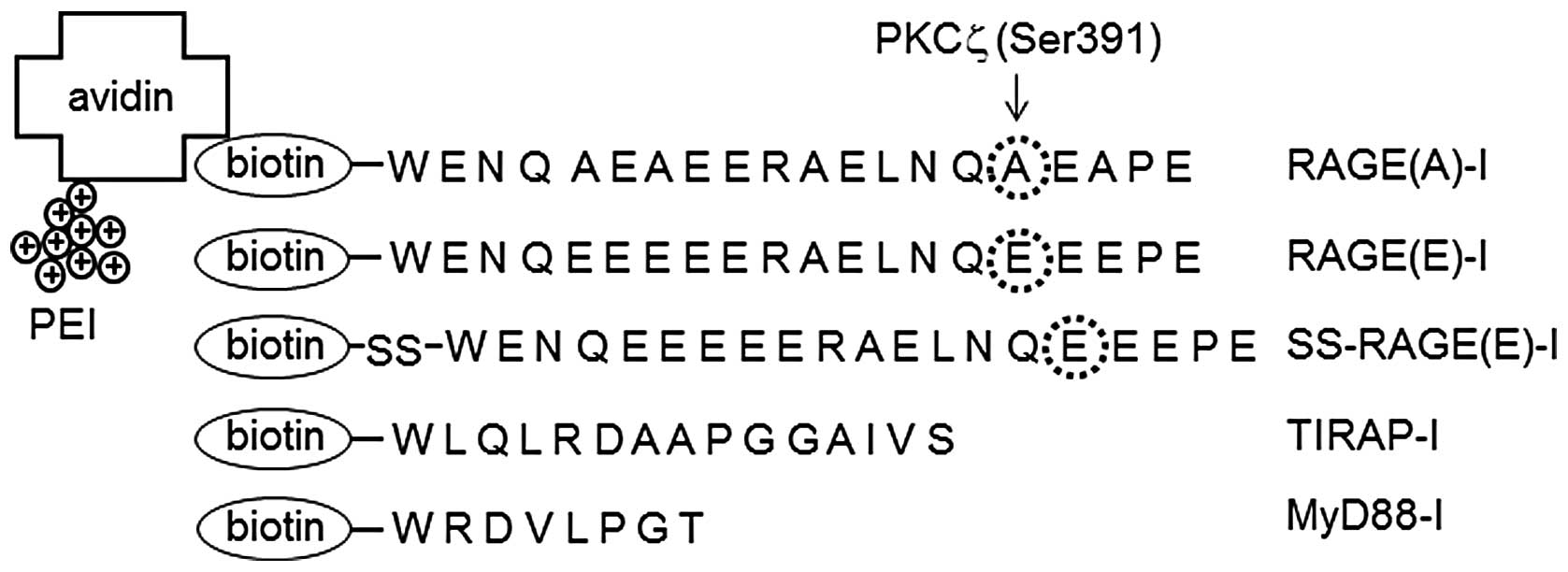
of RAGE (387–395 amino acids). Ser391 was replaced with alanine
[RAGE(A)-I, non-phosphorylatable] or with glutamic acid [RAGE(E)-I,
phosphorylation mimic] (Fig. 1).
The peptides were biotinylated under conventional conditions. In
SS-RAGE(E)-I, a disulfide bond was introduced between the peptide
and biotin to facilitate the release of RAGE(E)-I from the
PEI-avidin vehicle under intracellular reducing conditions as
previously described (29,30).
Cells, chemicals and antibodies
SH-SY5Y human neuroblastoma, U-87MG human
glioblastoma, B16-BL6 mouse melanoma and HEK293 human embryonic
kidney cells were cultured in D/F medium supplemented with 10%
fetal bovine serum (both from Life Technologies, Carlsbad, CA,
USA).
The antibodies used were as follows: goat polyclonal
antibody against glutathione S-transferase (GST; GE Healthcare,
Waukesha, WI, USA); mouse monoclonal antibody against Myc and
HRP-labeled anti-mouse secondary antibody (Cell Signaling
Technologies, Beverly, MA, USA). The Rac/Cdc42 Activation Assay kit
was purchased from Millipore (Billerica, MA, USA). Endo-Porter was
purchased from Gene Tools, LLC (Philomath, OR, USA).
Purification of recombinant proteins
GST, GST-TIRAP, GST-myeloid differentiation protein
88 (MyD88), GST-thyroid receptor activator molecule (TRAM) and
GST-S100B proteins were purified from the bacterial lysates of BL21
competent cells transformed with pGEX6P1 vectors containing each
cDNA using glutathione sepharose beads (GE Healthcare) according to
the standard procedures outlined by the manufacturer. For the
purification of S100B, GST-S100B was incubated with PreScission
protease (1 U/100 μg of GST-S100B; GE Healthcare) for the
cleavage of GST. GST was removed using glutathione sepharose beads.
The purity of the proteins was determined by SDS-PAGE.
Pull-down assay and western blot
analysis
Biotinylated peptides were incubated with
GST-proteins or cell lysates for 2 h. The pull-down of biotinylated
peptide-protein complexes was carried out using
streptavidin-coupled Dynabeads (Life Technologies).
Western blot analysis was performed under
conventional conditions after lysing the cells with M-PER Mammalian
Protein Extraction Reagent (Thermo Scientific, Lafayette, CO, USA)
with PhosphoSTOP (Roche Applied Science, Indianapolis, IN,
USA).
Apoptosis assay, migration assay and
invasion assay
For apoptosis assay, the SH-SY5Y cells were cultured
with D/F-0.5% FBS for 24 h prior to incubation with GST or S100B at
0–10 μM for 24 h. Apoptotic cells were identified after
staining with Hoechst 33342 (Life Technologies) for 30 min. For
migration assay, 10,000 U-87MG cells incubated with inhibitor
peptides in advance were inoculated into the top wells of Boyden
chambers (pore size, 8 μm; BD Falcon, Franklin Lakes, NJ,
USA). The filters were stained after incubation for 8 h and the
migrated cells were counted. For invasion assay, 25,000 U-87MG
cells incubated with inhibitor peptides in advance were inoculated
into the top wells of Boyden chambers pre-coated with growth
factor-reduced Matrigel (Life Technologies). The filters were
stained after incubation for 8 h and invaded cells were
counted.
Cell counting and cell growth assay
Cells were counted using a haemocytometer after
staining with 0.5% trypan blue. Each experiment was repeated at
least three times. For cell growth assay, the CellTiter-Glo
Luminescent Cell Viability Assay system (Promega Biosciences, San
Luis Obispo, CA, USA) was used. The experiment was carried out
according to the manufacturer’s instructions.
Rac/Cdc42 activation assay
The experiment was performed using the Rac1/Cdc42
Activation kit (Millipore) according to the manufacturer’s
instructions. U-87MG cells were treated with
PEI-avidin/biotin-inhibitor peptides for 24 h. The activation of
Cdc42 (GTP bound form) was determined by pull-down assay using
GST-PAK-1 (containing 67–150 amino acids) and glutathione sepharose
beads followed by western blot analysis.
Statistical analysis
Prior to statistical analysis, each experiment was
repeated at least three times. The results are expressed as the
means ± SD. For comparison, analysis of variance (ANOVA) was used.
If ANOVA showed significant differences, the Bonferroni procedure
was used as a post hoc test. P-values <0.05 were considered to
indicate statistically significant differences.
Results
Preparation and intracellular delivery of
RAGE-I
In our previous study, we showed that the
phosphorylation of Ser391 of the intracellular domain of RAGE by
PKCζ was critical for recruiting the downstream signal transducer,
TIRAP (27). In this study, we
therefore designed decoy peptides covering the serine residue,
i.e., 387–395 of RAGE. As shown in Fig. 1, we prepared three peptides,
RAGE(A)-I, RAGE(E)-I and SS-RAGE(E)-I (see Materials and methods).
Two inhibitor peptides for TIRAP (TIRAP-I) and MyD88 (MyD88-I),
which were already reported to function as a decoy for TLR2/4 by
binding to the TIR domain of TIRAP and MyD88 (31,32), were included for comparison. The
peptides were biotinylated for intracellular delivery using avidin
conjugated with PEI-avidin. PEI cationization is a powerful tool
used to deliver a protein or peptide into cells (28). To confirm the efficient
transduction of biotinylated molecules by PEI-avidin, we examined
the intracellular delivery of biotin-GFP using B16-BL6 mouse
melanoma, SH-SY5Y human neuroblastoma and U-87MG human glioblastoma
cells. As shown in Fig. 2, the
PEI-avidin/biotin-GFP complex, but not the biotin-GFP alone was
delivered efficiently into the cells. Cationized molecules are
known to incorporate into cells through the macropinocytosis
pathway (33) and Endo-Porter is
known to enhance the release of incorporated molecules from the
endosome to the cytoplasm (30).
The fluorescence from intracellular GFP was intensified and showed
a more diffused pattern in the cytoplasm by application of the
cells with Endo-Porter (Fig. 2).
Therefore, we used the combination of PEI-avidin and Endo-Porter
for the intracellular delivery in the following experiments.
Direct binding of RAGE(E)-I to TIRAP
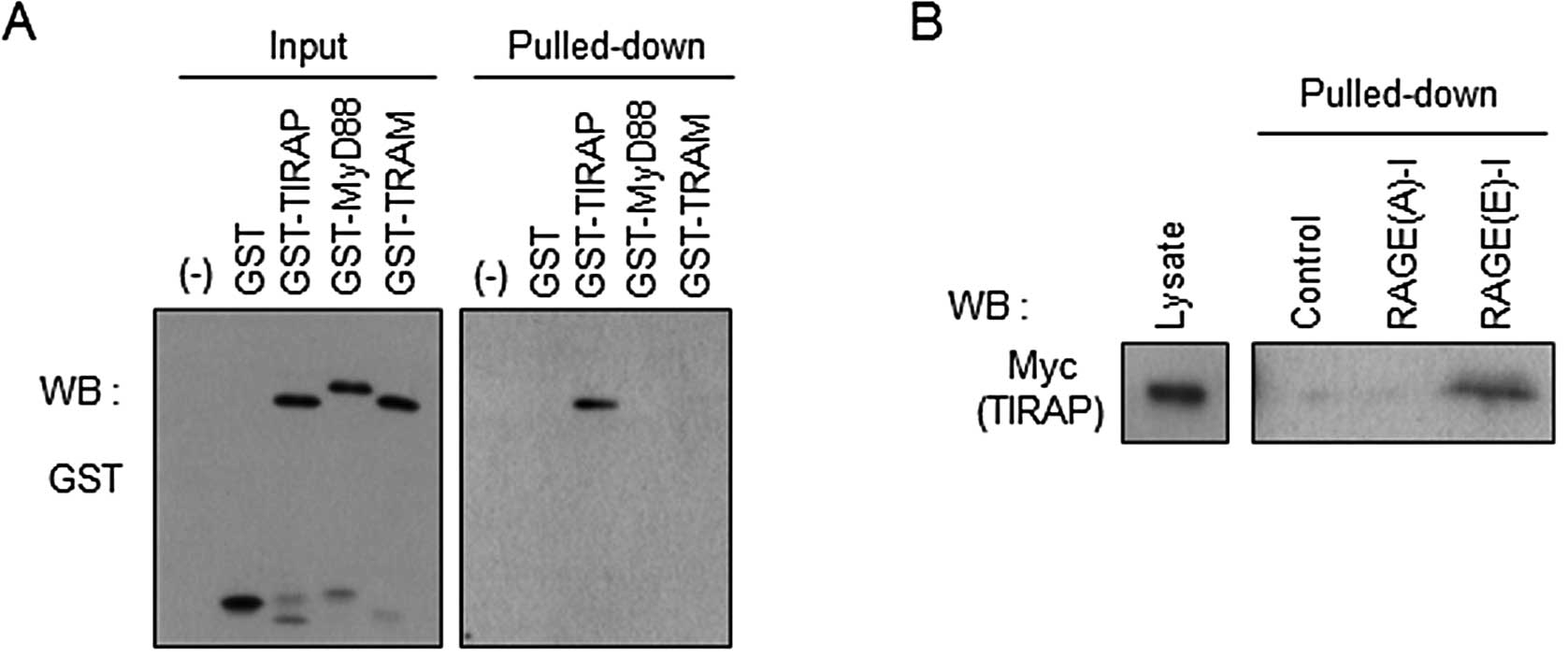
We prepared GST-fused TIRAP and other adaptor
proteins of Toll-like receptors, MyD88 and TRAM, and incubated them
with RAGE(E)-I in vitro. We detected only TIRAP in the
pulled-down fraction of RAGE(E)-I, indicating the direct binding of
the inhibitor peptide to TIRAP, but not to MyD88 and TRAM (Fig. 3A). We then examined the
significance of the phosphorylation site at Ser391 of RAGE-I for
interaction with TIRAP. Biotinylated RAGE-I was incubated with cell
lysates expressing Myc-TIRAP, pulled-down with streptavidin beads,
and analyzed by western blot analysis. As shown in Fig. 3B, phosphorylation-mimic RAGE(E)-I
strongly interacted with TIRAP, whereas non-phosphorylatable
RAGE(A)-I did not show any appreciable binding with TIRAP. These
results indicate that RAGE(E)-I is a promising tool for interfering
with RAGE signaling through a decoy function for TIRAP.
RAGE-I protects cell death induced by
S100B
Huttunen et al (5) reported that relatively high
concentrations (5 μM) of S100B induced apoptosis in neuronal
cells. In this study, after confirming the cytotoxic effects of 10
μM S100B on SH-SY5Y cells (Fig. 4B), we examined whether the
prophylactic application of RAGE-I reduces S100B-induced
cytotoxicity in SH-SY5Y cells. We highly purified recombinant human
S100B protein (Fig. 4A, lane 7)
and treated the cells with the protein. By monitoring nuclear
shrinkage with Hoechst staining, an assessment of apoptotic cell
death, we found that the excess amount of S100B (10 μM)
induced an increase in apoptotic cell death (Fig. 4B). By contrast, the prophylactic
addition of RAGE(E)-I, SS-RAGE(E)-I, TIRAP-I and MyD88-I, but not
RAGE(A)-I attenuated the apoptotic ratio (Fig. 4C). SS-RAGE(E)-I showed almost the
same effect in comparison with TIRAP-I and MyD88-I. These results
indicate that the signaling from RAGE-TIRAP interaction plays a
critical role in the induction of S100B-mediated cell death and
that this may be prevented by pre-treatment of the cells with RAGE
inhibitor peptides.
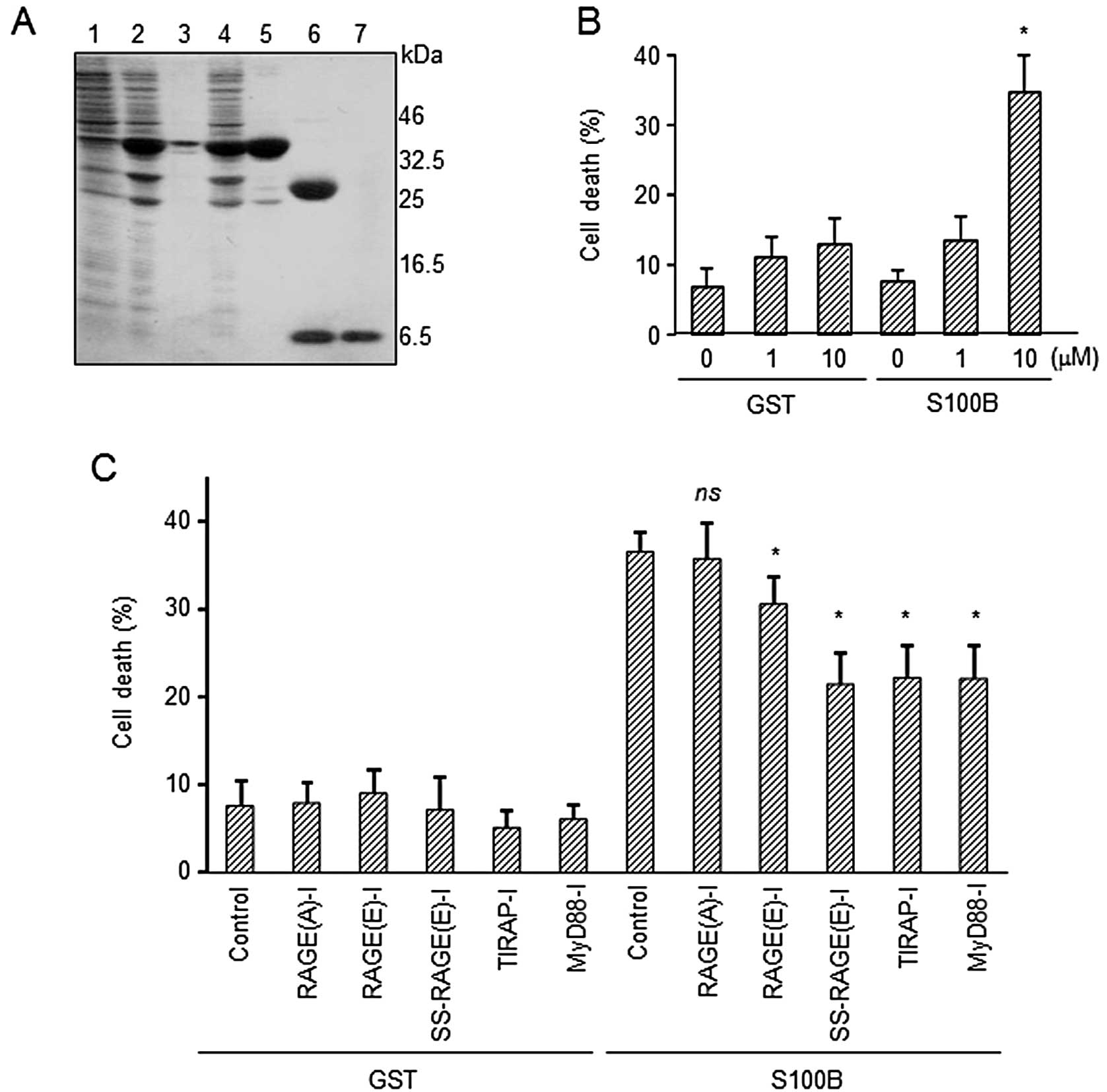 | Figure 4Mitigation of S100B-related cell
death by inhibitor peptides for receptor for advanced glycation end
products (RAGE) signaling (RAGE-I). (A) Expression and purification
of S100B from E. coli. The purity of S100B was confirmed by
SDS-PAGE. Lane 1, E. coli lysates without isopropyl
β-D-1-thiogalactopyranoside (IPTG) induction; lane 2, induction of
GST-S100B with IPTG; lane 3, precipitates after lysis; lane 4,
supernatants after lysis; lane 5, GST-S100B after purification with
Sephadex 4B-column; lane 6, cleavage of GST from S100B by
PreScission protease; lane 7, purified S100B. (B) Induction of
apoptosis by S100B. SH-SY5Y cells were incubated with control GST
or S100B for 24 h. Apoptotic cells were identified after staining
with Hoechst 33342. *P<0.05, significantly different
from the control group. (C) Polyethylenimine
(PEI)-avidin/biotin-inhibitor peptides (1 μM) were added to
the SH-SY5Y cells for 12 h prior to treatment with GST or S100B (10
μM, 24 h). Apoptotic cells were identified after staining
with Hoechst 33342. Statistically significant differences were
determined by comparing with the cells without S100B.
*P<0.05, significantly different from the control
group with S100B; ns, not significant. |
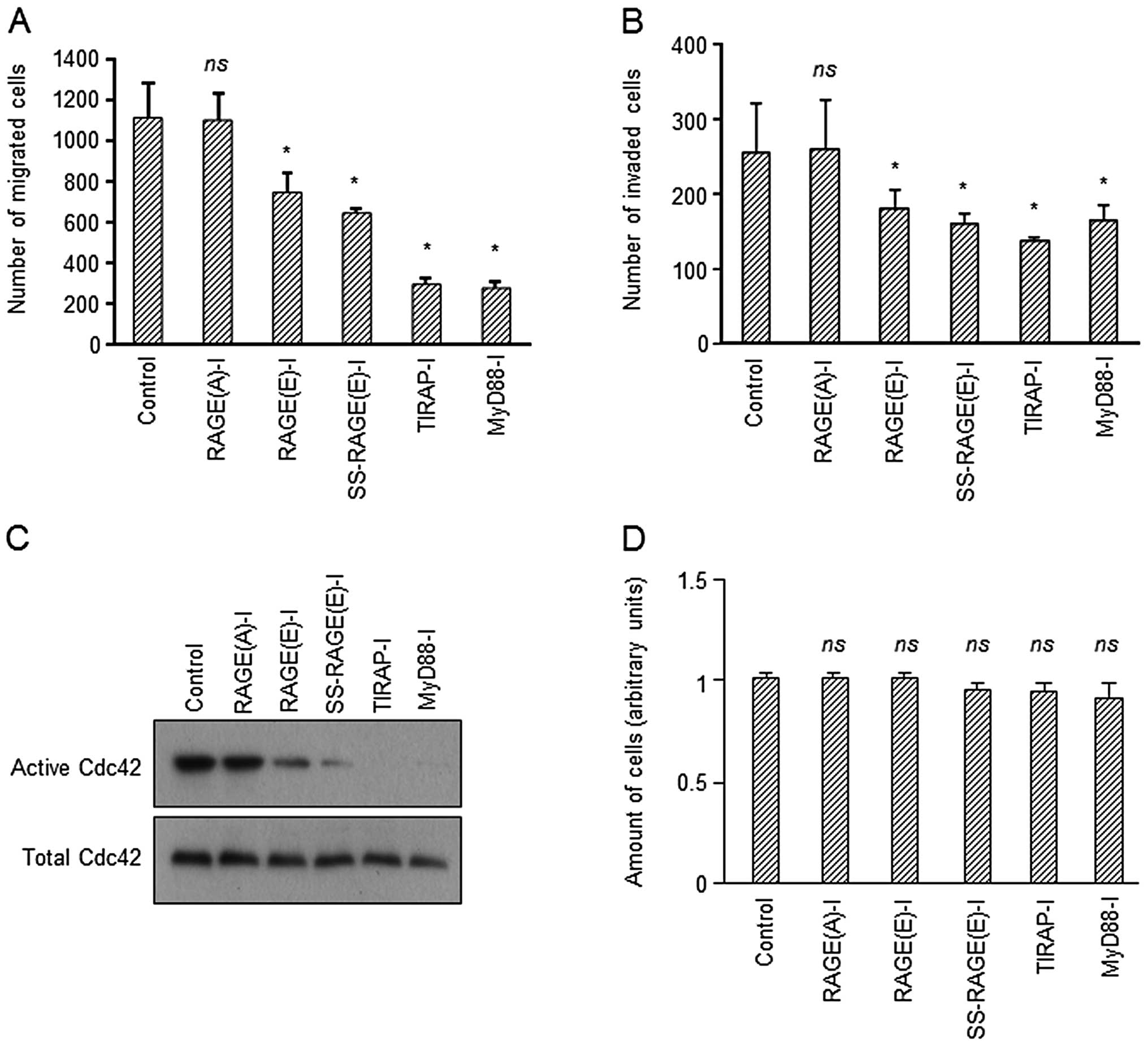
RAGE-I inhibits cell migration and
invasion, but not cell growth
It is known that the activation of RAGE results in
enhanced migration and invasion of various types of cells (34,35). Therefore, we also investigated
whether RAGE-I inhibits the migration and invasion of U-87MG
glioblastoma cells in vitro. When the cells were treated
with RAGE-I, the migration and invasion of the cells were
significantly suppressed (Fig. 5A and
B). In addition, these results correlated with the inactivation
of Cdc42 (Fig. 5C), which is
closely associated with RAGE-mediated cell migration (36,37). A higher suppression was observed
in the SS-RAGE(E)-I-treated group compared with the
RAGE(E)-I-treated group (Fig.
5A–C). This possibly occurred as RAGE(E)-I was released from
biotin-PEI-avidin and more efficiently captured endogenous TIRAP.
Under similar conditions, the growth of U-87MG cells was not
significantly affected as assayed by determining the intracellular
adenosine triphosphate (ATP) content (Fig. 5D), or by counting the number of
cells (Table I). These results
indicate that RAGE-I effectively abrogates the downstream signaling
of ligand-activated RAGE in glioblastoma cells in terms of
migration and invasion.
 | Table ITreatment with inhibitor peptides did
not affect the growth of U-87MG cells. |
Table I
Treatment with inhibitor peptides did
not affect the growth of U-87MG cells.
| Groups | No. of cells
(×105) |
|---|
| Control | 10.9±0.8 |
| RAGE(A)-I | 10.7±0.5 |
| RAGE(E)-I | 10.5±0.5 |
| SS-RAGE(E)-I | 10.8±0.7 |
| TIRAP-I | 10.9±0.8 |
| MyD88-I | 10.7±0.7 |
Discussion
RAGE plays pivotal roles in a variety of
physiological contexts, including early development. On the other
hand, the aberrant hyperfunctional state of RAGE due to the
overexpression of RAGE and/or the overstimulation of RAGE by
ligands is involved in a variety of pathological conditions,
including diabetic syndrome, cancer and neurodegenerative diseases.
Thus, interfering with RAGE function is considered a potent
therapeutic measure. Abe et al (25) reported that RAGE was overexpressed
in human melanoma cells and that AGE stimulated the growth and
migration of the cells. They showed that the application of an
antibody against the extracellular domain of RAGE suppressed the
growth of melanoma cells in vitro and in vivo.
Huttunen et al (23)
demonstrated that a C-terminal motif of amphoterin (150–183 amino
acids) bound to RAGE and inhibited the migration and invasion of
human fibrosarcoma cells. Arumugam et al (24) prepared S100P-derived small
peptides. The peptide inhibited the interaction of S100P, S100A4
and HMGB-1 with RAGE and abrogated the growth of pancreatic tumors
and glioma. In Alzheimer’s disease (AD), the Aβ peptide accumulates
in plaque in the brain. RAGE mediates Aβ-induced perturbations in
cerebral vessels, neurons and microglia in AD. Deane et al
(38) identified a high-affinity
RAGE-specific inhibitor (FPS-ZM1) that blocked Aβ binding to the V
domain of RAGE and inhibited Aβ40- and Aβ42-induced cellular stress
in RAGE-expressing cells in vitro and in the mouse brain
in vivo.
These studies focused on the extracellular domain of
RAGE and its various ligands. On the other hand, we focused on the
intracellular domain of RAGE based on our previous findings of
RAGE-TIRAP interaction (27).
Inhibitor peptides were designed by mimicking the phosphorylated
cytosolic domain of RAGE. Since the RAGE-TIRAP interaction is a
common converged event for diverse ligands such as AGE, S100
proteins and HMGB1, we hypothesized that an inhibitory tool for the
interaction between RAGE and TIRAP may efficiently abrogate
RAGE-mediated signaling regardless of ligand species.
RAGE has been demonstrated to play a significant
role in the development of several degenerative and
inflammation-related diseases (8,15,17–22). In the present study, we monitored
cellular behavior related with two pathological conditions,
neuronal cell death and the migration and invasion of cancer cells.
Griffin et al (39) and
Mrak and Griffinbc (40)
demonstrated that S100B contributes to the progression of AD. In
the present study, we demonstrated that the addition of S100B
induced the apoptosis of SH-SY5Y cells and that the pre-treatment
of the cells with RAGE inhibitor peptides resulted in the
mitigation of apoptosis. The decrease in apoptosis suggests that
RAGE-TIRAP interaction is one of the targets for the prevention of
neurodegeneration. In cancer cells, cell migration and invasion are
considered, at least in part, to occur due to the hyperfunction of
RAGE (25). Since the high
expression of RAGE and its ligands have previously been found in
many cancer cases (22), in this
study, we tried to inhibit cell migration and invasion by RAGE-I
using U-87MG human glioblastoma cells. As expected, the treatment
of U-87MG cells with RAGE-I resulted in the suppression of
migration and invasion. The reduced migration and invasion appeared
not be due to cytotoxicity as the cell growth was not affected
(Table I) (Fig. 5D).
Protein transduction (PTD) sequences such as TAT,
polyarginine and antennapedia are widely used for the intracellular
delivery of proteins and peptides (33). Although the inhibitor peptides for
TIRAP and MyD88 conjugated with the PTD sequence (Imgenex) are
available, the recommended concentration for application is ~100
μM. The necessity for high concentrations of the agents
hampers the practical application of these peptides for future
clinical, as well as experimental use. We demonstrated that
PEI-avidin delivers biotinylated peptides at 1 μM into cells
and that the delivered peptides can function sufficiently. The
addition of disulfide bonds to facilitate the release of the
peptide from PEI-avidin under intracellular reducing conditions
enhanced the efficiency of the inhibitor peptides. These data
indicate that our approach provides a promising tool, not only for
the analysis of the etiology of RAGE-related disorders, but also
for the development of therapeutic measures against such
diseases.
Acknowledgements
The present study was supported in part by grants
(AS242Z01065Q) from the Japan Science and Technology Agency (to
H.M.), from the Ministry of Health, Labor and Welfare (Research for
Intractable Diseases; to N.H.), from the Ministry of Education,
Culture, Sports, Science and Technology of Japan (Grant-in-Aid for
Scientific Research on Innovation Areas; to M.S.), from The Naito
Foundation (to M.S.), from The Research Foundation for
Pharmaceutical Sciences (to M.S.) and from The Ichiro Kanehara
Foundation (to M.S.).
Abbreviations:
|
RAGE
|
receptor for advanced glycation end
products
|
|
RAGE-I
|
inhibitor peptides for RAGE
signaling
|
|
TIRAP
|
Toll-interleukin 1 receptor
domain-containing adaptor protein
|
|
MyD88
|
myeloid differentiation protein 88
|
|
PEI
|
polyethylenimine
|
References
|
1
|
Neeper M, Schmidt AM, Brett J, et al:
Cloning and expression of a cell surface receptor for advanced
glycosylation end products of proteins. J Biol Chem.
267:14998–15004. 1992.PubMed/NCBI
|
|
2
|
Schmidt AM, Yan SD, Yan SF and Stern DM:
The biology of the receptor for advanced glycation end products and
its ligands. Biochim Biophys Acta. 1498:99–111. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Schmidt AM, Mora R, Cao R, et al: The
endothelial cell binding site for advanced glycation end products
consists of a complex: an integral membrane protein and a
lactoferrin-like polypeptide. J Biol Chem. 269:9882–9888.
1994.PubMed/NCBI
|
|
4
|
Hofmann MA, Drury S, Fu C, et al: RAGE
mediates a novel proinflammatory axis: a central cell surface
receptor for S100/calgranulin polypeptides. Cell. 97:889–901. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huttunen HJ, Kuja-Panula J, Sorci G,
Agneletti AL, Donato R and Rauvala H: Coregulation of neurite
outgrowth and cell survival by amphoterin and S100 proteins through
receptor for advanced glycation end products (RAGE) activation. J
Biol Chem. 275:40096–40105. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Leclerc E, Fritz G, Weibel M, Heizmann CW
and Galichet A: S100B and S100A6 differentially modulate cell
survival by interacting with distinct RAGE (receptor for advanced
glycation end products) immunoglobulin domains. J Biol Chem.
282:31317–31331. 2007. View Article : Google Scholar
|
|
7
|
Li J, Qu X and Schmidt AM: Sp1-binding
elements in the promoter of RAGE are essential for
amphoterin-mediated gene expression in cultured neuroblastoma
cells. J Biol Chem. 273:30870–30878. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yan SD, Chen X, Fu J, et al: RAGE and
amyloid-beta peptide neurotoxicity in Alzheimer’s disease. Nature.
382:685–691. 1996.
|
|
9
|
Huang JS, Guh JY, Chen HC, Hung WC, Lai YH
and Chuang LY: Role of receptor for advanced glycation end-product
(RAGE) and the JAK/STAT-signaling pathway in AGE-induced collagen
production in NRK-49F cells. J Cell Biochem. 81:102–113. 2001.
View Article : Google Scholar
|
|
10
|
Yeh CH, Sturgis L, Haidacher J, et al:
Requirement for p38 and p44/p42 mitogen-activated protein kinases
in RAGE-mediated nuclear factor-kappaB transcriptional activation
and cytokine secretion. Diabetes. 50:1495–1504. 2001. View Article : Google Scholar
|
|
11
|
Lander HM, Tauras JM, Ogiste JS, Hori O,
Moss RA and Schmidt AM: Activation of the receptor for advanced
glycation end products triggers a p21(ras)-dependent
mitogen-activated protein kinase pathway regulated by oxidant
stress. J Biol Chem. 272:17810–17814. 1997. View Article : Google Scholar
|
|
12
|
Huttunen HJ, Kuja-Panula J and Rauvala H:
Receptor for advanced glycation end products (RAGE) signaling
induces CREB-dependent chromogranin expression during neuronal
differentiation. J Biol Chem. 277:38635–38646. 2002. View Article : Google Scholar
|
|
13
|
Brett J, Schmidt AM, Yan SD, et al: Survey
of the distribution of a newly characterized receptor for advanced
glycation end products in tissues. Am J Pathol. 143:1699–1712.
1993.PubMed/NCBI
|
|
14
|
Beccafico S, Riuzzi F, Puglielli C, et al:
Human muscle satellite cells show age-related differential
expression of S100B protein and RAGE. Age. 33:523–541. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Srikanth V, Maczurek A, Phan T, et al:
Advanced glycation endproducts and their receptor RAGE in
Alzheimer’s disease. Neurobiol Aging. 32:763–777. 2011.
|
|
16
|
Maczurek A, Shanmugam K and Münch G:
Inflammation and the redox-sensitive AGE-RAGE pathway as a
therapeutic target in Alzheimer’s disease. Ann NY Acad Sci.
1126:147–151. 2008.PubMed/NCBI
|
|
17
|
Koyama H and Nishizawa Y: AGEs/RAGE in
CKD: irreversible metabolic memory road toward CVD? Eur J Clin
Invest. 40:623–635. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Reiniger N, Lau K, McCalla D, et al:
Deletion of the receptor for advanced glycation end products
reduces glomerulosclerosis and preserves renal function in the
diabetic OVE26 mouse. Diabetes. 59:2043–2054. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu L, Ma L, Nicholson LF and Black PN:
Advanced glycation end products and its receptor (RAGE) are
increased in patients with COPD. Respir Med. 105:329–336. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ramasamy R, Yan SF and Schmidt AM:
Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis
of diabetes and its complications. Ann NY Acad Sci. 1243:88–102.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Su XD, Li SS, Tian YQ, Zhang ZY, Zhang GZ
and Wang LX: Elevated serum levels of advanced glycation end
products and their monocyte receptors in patients with type 2
diabetes. Arch Med Res. 42:596–601. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Leclerc E, Heizmann CW and Vetter SW: RAGE
and S100 protein transcription levels are highly variable in human
melanoma tumors and cells. Gen Physiol Biophys. 28:F65–F75.
2009.PubMed/NCBI
|
|
23
|
Huttunen HJ, Fages C, Kuja-Panula J,
Ridley AJ and Rauvala H: Receptor for advanced glycation end
products-binding COOH-terminal motif of amphoterin inhibits
invasive migration and metastasis. Cancer Res. 62:4805–4811.
2002.PubMed/NCBI
|
|
24
|
Arumugam T, Ramachandran V, Gomez SB,
Schmidt AM and Logsdon CD: S100P-derived RAGE antagonistic peptide
reduces tumor growth and metastasis. Clin Cancer Res. 18:4356–4364.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Abe R, Shimizu T, Sugawara H, et al:
Regulation of human melanoma growth and metastasis by AGE-AGE
receptor interactions. J Invest Dermatol. 122:461–467. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arumugam T, Ramachandran V and Logsdon CD:
Effect of cromolyn on S100P interactions with RAGE and pancreatic
cancer growth and invasion in mouse models. J Natl Cancer Inst.
98:1806–1818. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sakaguchi M, Murata H, Yamamoto K, et al:
TIRAP, an adaptor protein for TLR2/4, transduces a signal from RAGE
phosphorylated upon ligand binding. PLoS One. 6:e231322011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kitazoe M, Murata H, Futami J, et al:
Protein transduction assisted by polyethylenimine-cationized
carrier proteins. J Biochem. 137:693–701. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Murata H, Sakaguchi M, Futami J, et al:
Denatured and reversibly cationized p53 readily enters cells and
simultaneously folds to the functional protein in the cells.
Biochemistry. 45:6124–6132. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Futami M, Watanabe Y, Asama T, et al:
Uniformly cationized protein efficiently reaches the cytosol of
mammalian cells. Bioconjug Chem. 23:2025–2031. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schilling D, Thomas K, Nixdorff K, Vogel
SN and Fenton MJ: Toll-like receptor 4 and Toll-IL-1 receptor
domain-containing adapter protein (TIRAP)/myeloid differentiation
protein 88 adapter-like (Mal) contribute to maximal IL-6 expression
in macrophages. J Immunol. 169:5874–5880. 2002. View Article : Google Scholar
|
|
32
|
Loiarro M, Sette C, Gallo G, et al:
Peptide-mediated interference of TIR domain dimerization in MyD88
inhibits interleukin-1-dependent activation of NF-{kappa}B. J Biol
Chem. 280:15809–15814. 2005.PubMed/NCBI
|
|
33
|
Kaplan IM, Wadia JS and Dowdy SF: Cationic
TAT peptide transduction domain enters cells by macropinocytosis. J
Control Release. 102:247–253. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bassi R, Giussani P, Anelli V, et al:
HMGB1 as an autocrine stimulus in human T98G glioblastoma cells:
role in cell growth and migration. J Neurooncol. 87:23–33. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kataoka K, Ono T, Murata H, et al: S100A7
promotes the migration and invasion of osteosarcoma cells via the
receptor for advanced glycation end products. Oncol Lett.
3:1149–1153. 2012.PubMed/NCBI
|
|
36
|
Hudson BI, Kalea AZ, Del Mar Arriero M, et
al: Interaction of the RAGE cytoplasmic domain with diaphanous-1 is
required for ligand-stimulated cellular migration through
activation of Rac1 and Cdc42. J Biol Chem. 283:34457–34468. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yamamoto K, Murata H, Putranto EW, et al:
DOCK7 is a critical regulator of the RAGE-Cdc42 signaling axis that
induces formation of dendritic pseudopodia in human cancer cells.
Oncol Rep. 29:1073–1079. 2013.PubMed/NCBI
|
|
38
|
Deane R, Singh I, Sagare AP, et al: A
multimodal RAGE-specific inhibitor reduces amyloid β-mediated brain
disorder in a mouse model of Alzheimer disease. J Clin Invest.
122:1377–1392. 2012.PubMed/NCBI
|
|
39
|
Griffin WS, Sheng JG, McKenzie JE, et al:
Life-long overexpression of S100beta in Down’s syndrome:
implications for Alzheimer pathogenesis. Neurobiol Aging.
19:401–405. 1998.PubMed/NCBI
|
|
40
|
Mrak RE and Griffinbc WS: The role of
activated astrocytes and of the neurotrophic cytokine S100B in the
pathogenesis of Alzheimer’s disease. Neurobiol Aging. 22:915–922.
2001.PubMed/NCBI
|