Introduction
Non-alcoholic fatty liver disease (NAFLD) includes
non-alcoholic fatty liver (NAFL) and non-alcoholic steatohepatitis
(NASH), and NASH may lead to hepatic cirrhosis or hepatocellular
carcinoma (HCC) (1,2). NAFLD/NASH can be viewed as a
metabolic syndrome phenotype involving the liver, and the incidence
of NAFLD has increased with corresponding increases in risk factors
for metabolic syndrome, such as obesity, diabetes and hypertension.
Such metabolic risk factors may promote the pathological
progression of NAFLD/NASH, and NAFLD advances more rapidly in
patients with a greater number of metabolic risk factors (3,4).
The incidence of hypertension is significantly
higher in patients with NAFLD compared with the general population
(5). Furthermore, a higher
incidence of NASH has been observed in patients with NAFLD who also
have hypertension and these patients are likely to progress to
advanced hepatic fibrosis (6),
which suggests that hypertension may promote the onset or
progression of hepatitis and hepatic fibrosis. However, the
possible association between hypertension and the pathological
progression of NAFLD has not been fully established.
Rats fed a choline-deficient L-amino acid-defined
(CDAA) diet initially develop fatty liver and hepatitis, and may
subsequently progress to hepatic fibrosis and HCC (7). These findings indicate that the rat
fed a CDAA diet is an appropriate animal model of the pathology of
NASH. The spontaneously hypertensive rat (SHR) is produced by the
mating of rats with hypertension. These rats spontaneously develop
hypertension at a rate of 100% between the ages of 7 to 15 weeks,
even without a high-salt diet, although a high salt load further
increases the blood pressure (8).
Carbon tetrachloride (CCl4)-induced hepatic fibrosis
in the SHR model is more severe than that in normotensive
Wistar-Kyoto (WKY) rats used as controls (9), and SHRs receiving a
choline-deficient (CD) diet show severe hepatic steatosis compared
with WKY rats receiving the same diet (10). SHRs and WKY rats both originate
from Wistar rats, but they diverged sufficiently long ago to now be
essentially separate background strains (11). This is a limitation of the
comparison of effects in these 2 types of rats. In addition,
several proteins differ in the livers of SHRs and WKY rats,
including proteins that induce oxidative stress (11). For this reason, we investigated
the effects of hypertension induced by a high-salt diet on the
severity of hepatic steatosis, liver injury and hepatic fibrosis
induced by a CDAA diet in the SHR model.
Materials and methods
Animals and diets
Six-week-old male SHRs were obtained from Charles
River Laboratories (Kanagawa, Japan). The rats were allowed to
acclimatize to the laboratory conditions for at least 7 days at a
constant temperature of 24ºC with a 12-h light-dark cycle, and fed
standard chow (control diet) containing 0.27% NaCl (CE-2; Kyudo,
Kumamoto, Japan) and water ad libitum. All animal
experiments were approved by the Institutional Animal Care and Use
Committee of Kagoshima University, Kagoshima, Japan. After the
acclimatization period, the rats were placed in groups that were
fed 3 different diets ad libitum: standard chow with high
salt (8.0% NaCl) or normal salt (0.27% NaCl) for 7 weeks, followed
by a CDAA diet containing a high (8.0% NaCl) or normal salt (0.25%
NaCl) level for an additional 8 or 24 weeks (high- and normal-salt
groups, respectively); or standard chow with normal salt (0.27%
NaCl) for 15 or 31 weeks (control groups). Diets were obtained from
Dyets Inc. (Bethlehem, PA, USA) and 30 g per day was administered
to equalize the total food intake.
The resected livers were weighed and used for RNA
extraction, or thin slices were immersed in 10% formalin and
embedded in paraffin to make 4-μm-thick sections for staining with
hematoxylin and eosin (H&E), Oil red O, Azan or Sirius Red.
Blood was collected by vena cava puncture following a 12-h fast and
then centrifuged. The resulting serum was stored at −80ºC.
Measurement of systolic blood pressure
and serum markers
Systolic blood pressure (SBP) was monitored from 7
to 31 weeks using the tail-cuff method (model MK-1030; Muromachi
Co., Ltd., Tokyo, Japan). At each time point, SBP was measured for
each rat and the mean was calculated. Serum alanine
aminotransferase (ALT) and alkaline phosphatase (ALP) levels were
determined using Spotchem II-liver function 2 (Arkray Inc., Kyoto,
Japan). Serum triglyceride and free fatty acid levels were
determined by ELISA at SRL, Inc. (Tokyo, Japan). Fasting blood
glucose (FBG) and serum immunoreactive insulin (IRI) levels were
determined by ELISA (Morinaga Institute of Biological Science,
Kanagawa, Japan).
Assessment of hepatic steatosis and
hepatic fibrosis
Hepatic steatosis was assessed using hepatic
triglyceride levels and Oil red O staining. Hepatic lipids were
extracted with chloroform-methanol and measured enzymatically using
a commercial kit (L-type Wako TGH; Wako Pure Chemical Industries,
Osaka, Japan). Oil red O staining was performed to evaluate the
accumulation of fat droplets in hepatocytes in the frozen liver
sections. Sirius Red or Azan-stained sections were analyzed to
evaluate fibrosis in the liver. Five fields were selected randomly
from each right lobe per sample, and samples from 6 to 11 rats in
each group were examined. Thus, a total of 30 to 55 fields were
analyzed for each group. The ratios of the Oil red O-, Azan- or
Sirius Red-stained area to the total area were quantified by image
analysis. A spot RT color camera (Visitron Systems Inc., Puchheim,
Germany) was used to capture and analyze the fields at ×100
magnification. Image analysis was performed using Quick Grain
Standard ver. 5.0.3. software (Inotech, Hiroshima, Japan).
Assessment of hepatic mRNA levels for
genes associated with liver inflammation and fibrosis
The relative levels of specific mRNAs in the
resected livers were assessed by real-time polymerase chain
reaction (qPCR) using a One Step SYBR PrimeScript RT-PCR kit II
(Takara Bio Inc., Shiga, Japan). Total RNA was extracted from the
livers using isogen (Nippon Gene, Tokyo, Japan). The expression
levels of target genes were calculated relative to the expression
of glyceraldehyde-3-phosphate dehydrogenase (GAPDH), which was used
as an endogenous control gene to normalize the target gene
expression levels. All procedures were performed according to
manufacturer’s instructions. mRNA levels were determined for tumor
necrosis factor (TNF)-α, interleukin (IL)-6, IL-10, heme oxygenase
(HO)-1, α-smooth muscle actin (SMA), transforming growth factor
(TGF)-β1 and tissue inhibitor of metalloproteinase (TIMP)-1. PCR
primers were obtained from Takara Bio Inc. The primer sequences
used are listed in Table I.
 | Table IOligonucleotide sequences used for
qPCR. |
Table I
Oligonucleotide sequences used for
qPCR.
| Gene | GenBank number | Primer
sequences |
|---|
| TNF-α | NM_012675 | F:
5-TCAGTTCCATGGCCCAGAC-3
R: 5-GTTGTCTTTGAGATCCATGCCATT-3 |
| IL-6 | NM_012589 |
F:5-CCACTTCACAAGTCGGAGGCTTA-3
R: 5-GTGCATCATCGCTGTTCATACAATC-3 |
| IL-10 | NM_012854 | F:
5-CAGACCCACATGCTCCGAGA-3
R: 5-CAAGGCTTGGCAACCCAAGTA-3 |
| HO-1 | NM_012580 | F:
5-AGGTGCACATCCGTGCAGAG-3
R: 5-TCCAGGGCCGTATAGATATGGTACA-3 |
| α-SMA | NM_031004.2 | F:
5-AGCCAGTCGCCATCAGGAAC-3
R: 5-GGGAGCATCATCACCAGCAA-3 |
| TGF-β1 | NM_021578 | F:
5-TGCGCCTGCAGAGATTCAAG-3
R: 5-ACGTAACGCCAGGAATTGTTGCTA-3 |
| TIMP-1 | NM_053819.1 | F:
5-CGAGACCACCTTATACCAGCGTTA-3
R: 5-TGATGTGCAAATTTCGTTCC-3 |
| GAPDH | NM_017008.3 | F:
5-GGCACAGTCAAGGCTGAGAATG-3
R: 5-ATGGTGAAGACGCCAGTA-3 |
Statistical analysis
A statistical comparison among groups was performed
using the Tukey-Kramer test. A Student’s t-test was used for a
comparison between 2 groups. A value of P<0.05 was considered to
indicate a statistically significant difference. Data are presented
as the means ± standard deviation (SD).
Results
Systolic blood pressure is affected by
salt levels, but not by the CDAA diet
SBP gradually increased until 15 weeks in the high-
and normal-salt groups. The increase in SBP was significantly
greater after 7 weeks on the standard diet in the high-salt group
compared with the normal-salt group and this difference continued
after the CDAA diet commenced. By contrast, SBP did not differ
between the normal-salt groups fed a CDAA or standard diet
(Fig. 1).
Metabolic parameters after 8 weeks on the
CDAA diet
There were no differences in dietary intake between
the high- and normal-salt groups due to dietary restrictions during
the study period; however, body weight was significantly lower and
the liver/body weight ratio was significantly higher in the
high-salt group compared with the normal-salt and control groups
after 8 weeks on the CDAA diet (both P-values <0.05). FBG levels
were significantly higher in the high-salt group compared with the
other 2 groups. Serum insulin levels tended to be higher in the
high-salt group compared with the normal-salt group after 8 weeks;
however, these levels in the CDAA groups were not significantly
higher than those in the control group. Serum triglyceride levels
were significantly higher in the CDAA groups compared with the
control group; however, there was no difference between the high-
and normal-salt groups. Serum free fatty acids were significantly
higher in the normal-salt group compared with the high-salt group,
and lowest in the control group (Table II).
| Table IIMetabolic parameters after 8 weeks on
the CDAA diet. |
Table II
Metabolic parameters after 8 weeks on
the CDAA diet.
| | CDAA diet |
|---|
| |
|
|---|
| Parameters | Control (n=6) | Normal-salt
(n=11) | High-salt
(n=11) |
|---|
| Body weight
(g) | 349.1 (17.5) | 374.6 (17.8) | 313.0
(36.4)b |
| Liver weight
(g) | 9.79 (0.34) | 10.69 (1.35) | 10.08 (1.39) |
| Liver weight/body
weight (%) | 2.81 (0.06) | 2.85 (0.32) | 3.22 (0.10)b |
| Fasting blood
glucose (mg/dl) | 96.1 (8.9) | 89.5 (17.2) | 129.8
(27.4)b |
| Serum insulin
(ng/ml) | 1.20 (0.60)
(n=4) | 0.69 (0.36) | 1.24 (1.13)
(n=9) |
| Triglyceride
(mg/dl) | 15.8 (2.4) | 29.0 (4.8)a | 29.2 (8.6)a |
| Free fatty acids
(mEq/l) | 520.8 (46.3) | 772.0
(95.2)a | 551.0
(108.0)c |
Severity of hepatic steatosis after 8
weeks on the CDAA diet
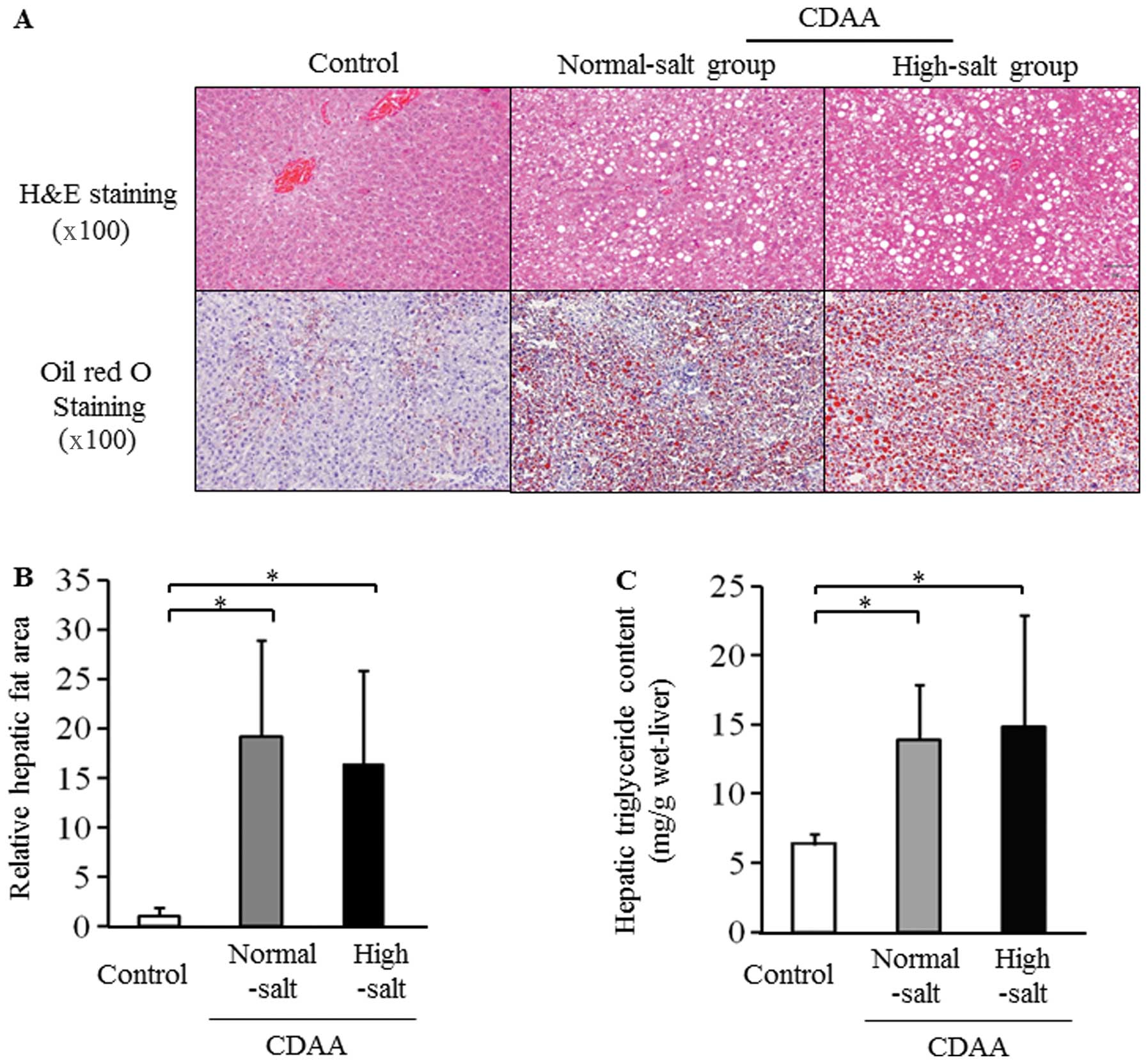
Micro- and macrovesicular steatosis was clearly
visible in the H&E-stained liver sections from the rats fed a
CDAA diet with normal- and high-salt for 8 weeks (Fig. 2A). The hepatic fat area based on
Oil red O staining and the hepatic triglyceride content was
significantly higher in the CDAA groups compared with the control
group (Fig. 2B and C). However,
hepatic steatosis did not differ between the 2 salt groups fed a
CDAA diet (Fig. 2B and C).
Liver injury after 8 weeks on the CDAA
diet
Serum ALT levels were significantly higher in the
high-salt group compared with the normal-salt group (P<0.05)
(Fig. 3). Serum ALP levels were
significantly higher in the high-salt group compared with the
control group and showed a tendency to be higher in the high-salt
group compared with the normal-salt group (Fig. 3).
Hepatic mRNA levels after 8 weeks on the
CDAA diet with high or low salt
The mRNA expression levels for TNF-α and IL-6 did
not differ between the 2 salt groups after 8 weeks on the CDAA
diet; however, IL-6 levels were relatively higher in the high-salt
group compared with the normal-salt and control groups (Fig. 4). By contrast, hepatic mRNA levels
for IL-10 and HO-1 were significantly lower in the high-salt group
compared with the normal-salt group (Fig. 4). However, the hepatic mRNA level
for IL-10 was higher and that for HO-1 was lower in the normal-salt
group compared with the control group.
Metabolic parameters after 24 weeks on
the CDAA diet
Body weight was significantly lower and the
liver/body weight ratio was significantly higher in the high-salt
group compared with the normal-salt group after 24 weeks on the
CDAA diet. FBG levels were relatively higher in the high-salt group
compared with the normal-salt and control groups. Serum insulin
levels tended to be higher in the high-salt group compared with the
normal-salt group; however, these levels were lower than those in
the control group. Serum triglyceride levels were significantly
higher in the CDAA groups compared with the control group, but did
not differ between the high- and normal-salt groups. The levels of
serum free fatty acids were significantly higher in the normal-salt
group compared with those in the control group (Table III).
| Table IIIMetabolic parameters after 24 weeks
on the CDAA diet. |
Table III
Metabolic parameters after 24 weeks
on the CDAA diet.
| | CDAA diet |
|---|
| |
|
|---|
| Parameters | Control (n=6) | Normal-salt
(n=11) | High-salt
(n=11) |
|---|
| Body weight
(g) | 375.7 (17.9) | 399.6 (17.5) | 303.9
(16.5)b |
| Liver weight
(g) | 12.48 (0.91) | 10.97
(0.46)a | 9.47 (0.95)b |
| Liver/body weight
ratio (%) | 3.33 (0.29) | 2.75 (0.09)a | 3.11 (0.21)c |
| Fasting blood
glucose (mg/dl) | 86.7 (16.4) | 103.7 (16.6) | 119.2 (41.2) |
| Serum insulin
(ng/ml) | 2.29 (0.63) | 0.20 (0.16) | 0.56 (0.27) |
| Triglyceride
(mg/dl) | 20.5 (3.6) | 45.0 (1.3)a | 38.2 (14.3)a |
| Free fatty acids
(mEq/l) | 537.7 (167.5) | 804.2
(90.3)a | 668.0 (210.2) |
Severity of hepatic fibrosis after 24
weeks on the CDAA diet with high or low salt
The CDAA diet was administered from week 7 to week
31 (a total of 24 weeks) with or without high salt for the
induction of liver fibrosis (Fig.
1). Liver fibrosis assessed by Sirius Red and Azan staining was
significantly more severe in the high-salt group compared with the
normal-salt and control groups (Fig.
5). The hepatic mRNA levels for α-SMA were significantly higher
in the high-salt group compared with the normal-salt and control
groups (Fig. 6), and hepatic
TGF-β1 and TIMP-1 mRNA levels tended to be higher in the high-salt
group compared with the normal-salt group. By contrast, hepatic
mRNA levels for IL-10 and HO-1 were significantly lower in the
high-salt group compared with those in the normal-salt group,
whereas the hepatic mRNA level of IL-10 was significantly higher
and that of HO-1 tended to be lower in the normal-salt group
compared with the control group (Fig.
6).
Discussion
The prevalence of NAFLD including NASH has increased
with an increase in the incidence of metabolic syndrome, which
includes obesity, diabetes, dyslipidemia and hypertension (12–16). Hypertension, high levels of ALT
and insulin resistance have all been associated with NASH and liver
fibrosis (6); however, the effect
of hypertension on NAFLD or NASH at the molecular level is
uncertain. In this study, we investigated the mechanisms through
which hypertension affects the degree of hepatic steatosis, liver
injury and hepatic fibrosis induced by a CDAA diet in a
hypertensive rat model. The results revealed that hypertension
induced by a high-salt diet had a negligible effect on the
progression of hepatic steatosis in rats, but may be a potential
risk factor for liver injury and hepatic fibrosis due to glucose
intolerance and decreased IL-10-mediated and HO-1-induced
anti-inflammatory effects.
Ikuta et al concluded that hypertension may
have an effect on the progression of NASH (10) and Hsu also suggested that
hypertension may induce severe hepatic fibrosis (9). However, these reports are limited
due to their dependence on comparing SHRs and normotensive WKY
rats, which are clearly different despite having a similar origin
(11). By contrast, we adjusted
blood pressure using 2 salt levels in the SHR model. In our study,
SBP gradually increased over the study period and the increase in
SBP was significantly more severe in the high-salt group compared
with the normal-salt group. By contrast, SBP did not differ between
the normal-salt group fed a CDAA diet and the controls fed a
standard diet with a normal-salt level. SBP was also suppressed by
an anti-hypertensive agent (data not shown). Thus, these results
demonstrate that the increase in SBP in the SHR model was
independent of diet, apart from the salt level, and therefore our
results reveal a direct mechanistic effect of hypertension on
NAFLD/NASH.
Hypertension has been shown to reduce the plasma
levels of IL-10 (17) and we also
found that hypertension inhibited the expression of IL-10 in the
livers of CDAA-fed rats. Treatment with IL-10 in vivo has
been shown to significantly reduce SPB in hypertensive mice
(18). IL-10 is a potent
anti-inflammatory cytokine that plays a pivotal role in the
regulation of immune and inflammatory responses. The inhibition of
IL-10 has been shown to promote the expression of inflammatory
cytokines, reduce insulin signaling and activate gluconeogenic and
lipidogenic pathways in an animal model of diet-induced fatty liver
disease (19). In addition, the
upregulation of HO-1 has been shown to induce a reduction in the
levels of cytokines, adhesion molecules, chemokines and neutrophil
accumulation, and to ameliorate organ injury in a state of shock
(20–22). As previously demonstrated, the
induction of HO-1 reduces hepatic ischemia reperfusion injury
(23), hepatic injury after
trauma hemorrhage (24) and renal
oxidative stress in diabetic SHRs (25). The inhibition of HO-1 may also be
associated with acetaminophen-induced liver injury (26). Thus, HO-1 is thought to play a
protective role in several organs under various deleterious
conditions. IL-10 also inhibits hepatitis (27,28) and this inhibition may occur
through a mechanism involving increased HO-1 expression induced by
an increase in IL-10 levels (29). Furthermore, in our study, in the
CDAA-fed rats, the normal-salt group showed higher levels of IL-10
and lower levels of HO-1 compared with the control group. This
higher level of IL-10 may be due to a feedback mechanism against
hepatitis induced by CDAA, although the CDAA diet may reduce HO-1
expression. By contrast, hypertension inhibited IL-10 and HO-1
expression, with a resultant increase in ALT to a level higher than
that caused by the CDAA diet alone. In addition, indirect evidence
indicates that IL-10 exerts a protective effect on endothelial
function in diabetes and hypertension (17,30). IL-10 has also been shown to
inhibit the activation of hepatic stellate cells and decrease
hepatic fibrosis and apoptosis in a CCl4-induced rat liver fibrosis
model (31,28). These findings suggest that
CDAA-induced hepatic fibrosis progresses more in SHRs fed a
high-salt diet compared with those fed a normal-salt diet due to
the inhibition of IL-10 and reduced HO-1 expression. Thus, we
speculate that the normal-salt group had increased IL-10 expression
to compensate for liver injury and hepatic fibrogenesis induced by
the CDAA diet, but the high-salt group could not compensate in the
same manner due to the inhibition of IL-10 and HO-1 by
hypertension.
Hypertension is associated with NAFLD/NASH,
increased diastolic blood pressure has been linked to hepatic fat
content (32), and the incidence
of NAFLD in patients with systolic hypertension is twice that in
the general population (33).
Donati et al also found that the prevalence of fatty liver
in patients with hypertension without diabetes and obesity was
3-fold higher than that in patients with normal blood pressure
(34). These patients have higher
homeostasis model assessment-insulin resistance (HOMA-IR) scores,
and hypertension may trigger insulin resistance, which may induce
fatty liver. However, our study on rats showed that hypertension
caused by a higher salt load did not influence the degree of fatty
deposition in the liver. The CD diet model does not present with
insulin resistance (10) and
hyperinsulinemia was not observed in CDAA loading in the SHR model
with or without high salt, compared with the control group fed a
standard diet in the current study. By contrast, FBG and serum
insulin levels were higher in the high-salt group compared with the
normal-salt group. HOMA-IR calculated from FBG and serum insulin
levels in the high-salt group was higher than that in the
normal-salt group (data not shown). Therefore, hypertension may
induce glucose intolerance or insulin resistance in the SHR model.
Hepatic steatosis induced by a CDAA diet may generally be severe,
but insulin resistance should not be critical in hepatic steatosis
induced by a CDAA diet, which may mask the effect of hypertension
on hepatic steatosis. Therefore, the effect of hypertension on the
severity of hepatic steatosis requires further investigation using
other fatty liver models.
There are well known associations between
hypertension and insulin resistance (35–37). Higher insulin resistance may lead
to hypertension due to a decrease in nitric oxide (NO), the
facilitation of a sympathetic nerve response and an increase in
vascular smooth muscle contraction, while hypertension induced by a
high-salt diet can increase insulin resistance (38,39). In our study, there was a possible
association between hypertension induced by a higher salt load and
glucose intolerance or insulin resistance in the CDAA-fed SHR
model. Insulin resistance is linked to hepatic fibrosis (40), and peroxisome
proliferator-activated receptor (PPAR)-γ agonists that improve
insulin resistance also improve the pathology of NAFLD/NASH
(41). The finding that
hypertension induced by a higher salt load contributed to the
progression of hepatic fibrosis suggests that glucose intolerance
or insulin resistance induced by hypertension may be factors in the
acceleration of hepatic fibrosis.
The pathological progression of NAFLD/NASH is not
yet fully understood (42), but
the ‘two hit theory’ is widely supported as the pathogenic
mechanism of NASH (43). In this
theory, it is proposed that the accumulation of triglycerides in
hepatic cells (first hit) and then oxidative stress, lipid
peroxidation and endotoxins increase cytokine levels and insulin
resistance, which then leads to liver injury and progression to
hepatic fibrosis (second hit). Our results suggest that the reduced
expression of IL-10 and/or HO-1 with a consequent reduction in the
inhibition of anti-inflammatory effects, and increased glucose
intolerance or insulin resistance, may be involved in the
hypertension-mediated exacerbation of liver injury and the
acceleration of hepatic fibrosis. Thus, hypertension may be a
contributing factor to the second hit rather than the first hit in
NASH. Furthermore, our results suggest that aggressive
anti-hypertensive therapy is required in patients with NAFLD/NASH
accompanied by hypertension for the prevention of cardiovascular
diseases and the inhibition of the progression of hepatic
diseases.
Acknowledgements
We thank Yuko Morinaga, Etsuko Horiguchi and Ayaka
Hamabe for their technical assistance. This study was supported in
part by grants from the Ministry of Education, Culture, Sports,
Science and Technology of Japan (no. 23590981), and the Ministry of
Health, Labour and Welfare of Japan (H20-Hepatitis-general-008) and
the Takeda Science Foundation.
References
|
1
|
Ascha MS, Hanouneh IA, Lopez R, Tamimi TA,
Feldstein AF and Zein NN: The incidence and risk factors of
hepatocellular carcinoma in patients with nonalcoholic
steatohepatitis. Hepatology. 51:1972–1978. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yasui K, Hashimoto E, Tokushige K, et al:
Clinical and pathological progression of non-alcoholic
steatohepatitis to hepatocellular carcinoma. Hepatol Res.
42:767–773. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Adams LA, Lymp JF, St Sauver J, et al: The
natural history of nonalcoholic fatty liver disease: a
population-based cohort study. Gastroenterology. 129:113–121. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hamaguchi M, Kojima T, Takeda N, et al:
The metabolic syndrome as a predictor of nonalcoholic fatty liver
disease. Ann Intern Med. 143:722–728. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
López-Suárez A, Guerrero JM,
Elvira-González J, Beltrán-Robles M, Cañas-Hormigo F and
Bascuñana-Quirell A: Nonalcoholic fatty liver disease is associated
with blood pressure in hypertensive and nonhypertensive individuals
from the general population with normal levels of alanine
aminotransferase. Eur J Gastroenterol Hepatol. 23:1011–1017.
2011.
|
|
6
|
Dixon JB, Bhathal PS and O’Brien PE:
Nonalcoholic fatty liver disease: predictors of nonalcoholic
steatohepatitis and liver fibrosis in the severely obese.
Gastroenterology. 121:91–100. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Nakae D, Yoshiji H, Mizumoto Y, et al:
High incidence of hepatocellular carcinomas induced by a choline
deficient L-amino acid defined diet in rats. Cancer Res.
52:5042–5045. 1992.PubMed/NCBI
|
|
8
|
Matsui H, Ando K, Kawarazaki H, et al:
Salt excess causes left ventricular diastolic dysfunction in rats
with metabolic disorder. Hypertension. 52:287–294. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hsu CT: Ultrastructural changes in liver
damage induced by carbon tetrachloride in spontaneously
hypertensive rats and Wistar-Kyoto rats. J Auton Nerv Syst.
70:79–83. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ikuta T, Kanno K, Arihiro K, et al:
Spontaneously hypertensive rats develop pronounced hepatic
steatosis induced by choline-deficient diet: Evidence for
hypertension as a potential enhancer in non-alcoholic
steatohepatitis. Hepatol Res. 42:310–320. 2012. View Article : Google Scholar
|
|
11
|
Svoboda DS and Kawaja MD: Changes in
hepatic protein expression in spontaneously hypertensive rats
suggest early stages of non-alcoholic fatty liver disease. J
Proteomics. 75:1752–1763. 2012. View Article : Google Scholar
|
|
12
|
Amarapurkar DN, Hashimoto E, Laurentius
LA, et al: Howcommon is non-alcoholic fatty liver disease in the
Asia-Pacific region and are there local differences? J
Gastroenterol Hepatol. 22:788–793. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Lewis JR and Mohanty SR: Nonalcoholic
fatty liver disease: a review and update. Dig Dis Sci. 55:560–578.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kojima S, Watanabe N, Numata M, Ogawa T
and Matsuzaki S: Increase in the prevalence of fatty liver in Japan
over the past 12 years: analysis of clinical background. J
Gastroenterol. 38:954–961. 2003.PubMed/NCBI
|
|
15
|
Radu C, Grigorescu M, Crisan D, Lupsor M,
Constantin D and Dina L: Prevalence and associated risk factors of
non-alcoholic fatty liver disease in hospitalized patients. J
Gastrointestin Liver Dis. 17:255–260. 2008.PubMed/NCBI
|
|
16
|
Jimba S, Nakagami T, Takahashi M, et al:
Prevalence of non-alcoholic fatty liver disease and its association
with impaired glucose metabolism in Japanese adults. Diabet Med.
22:1141–1145. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matrougui K, Abd Elmageed Z, Kassan M, et
al: Natural regulatory T cells control coronary arteriolar
endothelial dysfunction in hypertensive mice. Am J Pathol.
178:434–441. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kassan M, Galan M, Partyka M, Trebak M and
Matrougui K: Interleukin-10 released by CD4(+)CD25(+) natural
regulatory T cells improves microvascular endothelial function
through inhibition of NADPH oxidase activity in hypertensive mice.
Arterioscler Thromb Vasc Biol. 31:2534–2542. 2011.
|
|
19
|
Cintra DE, Pauli JR, Araújo EP, et al:
Interleukin-10 is a protective factor against diet-induced insulin
resistance in liver. J Hepatol. 48:628–637. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shen XD, Ke B, Zhai Y, et al: Toll-like
receptor and heme oxygenase-1 signaling in hepatic
ischemia/reperfusion injury. Am J Transplant. 5:1793–1800. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yu HP, Choudhry MA, Shimizu T, et al:
Mechanism of the salutary effects of flutamide on intestinal
myeloperoxidase activity following trauma-hemorrhage: up-regulation
of estrogen receptor-β-dependent HO-1. J Leukoc Biol. 79:277–284.
2006.PubMed/NCBI
|
|
22
|
Liu FC, Hwang TL, Lau YT and Yu HP:
Mechanism of salutary effects of astringinin on rodent hepatic
injury following trauma-hemorrhage: Akt-dependent hemeoxygenase-1
signaling pathways. PLoS One. 6:e259072011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Devey L, Mohr E, Bellamy C, et al: c-Jun
terminal kinase-2 gene deleted mice overexpress hemeoxygenase-1 and
are protected from hepatic ischemia reperfusion injury.
Transplantation. 88:308–316. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Yu HP, Yang SC, Lau YT and Hwang TL: Role
of Akt-dependent up-regulation of hemeoxygenase-1 in
resveratrol-mediated attenuation of hepatic injury after trauma
hemorrhage. Surgery. 148:103–109. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Elmarakby AA, Faulkner J, Baban B and
Sullivan JC: Induction of hemeoxygenase-1 reduces renal oxidative
stress and inflammation in diabetic spontaneously hypertensive
rats. Int J Hypertens. 2012:9572352012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hou HS, Liao CL, Sytwu HK, et al:
Deficiency of interleukin-15 enhances susceptibility to
acetaminophen-induced liver injury in mice. PLoS One. 7:e448802012.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Zhou YC, Chen S, Cao JJ, Chen SY, Xie YF
and Niu QX: Adenovirus-mediated viral interleukin-10 gene transfer
prevents concanavalin A-induced liver injury. Dig Liver Dis.
44:398–405. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhang LJ, Zheng WD, Shi MN and Wang XZ:
Effects of interleukin-10 on activation and apoptosis of hepatic
stellate cells in fibrotic rat liver. World J Gastroenterol.
12:1918–1923. 2006.PubMed/NCBI
|
|
29
|
Lee TS and Chau LY: Heme oxygenase-1
mediates the anti-inflammatory effect of interleukin-10 in mice.
Nat Med. 8:240–246. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Gunnett CA, Heistad DD and Faraci FM:
Interleukin-10 protects nitric oxide-dependent relaxation during
diabetes: role of superoxide. Diabetes. 51:1931–1937. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang LJ, Zheng WD, Chen YX, et al:
Antifibrotic effects of interleukin-10 on experimental hepatic
fibrosis. Hepatogastroenterology. 54:2092–2098. 2007.PubMed/NCBI
|
|
32
|
Hamaguchi M, Kojima T, Takeda N, et al:
Nonalcoholic fatty liver disease is a novel predictor of
cardiovascular disease. World J Gastroenterol. 13:1579–1584. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Bedogni G, Miglioli L, Masutti F,
Tiribelli C, Marchesini G and Bellentani S: Prevalence of and risk
factors for nonalcoholic fatty liver disease: the Dionysos
nutrition and liver study. Hepatology. 42:44–52. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Donati G, Stagni B, Piscaglia F, et al:
Increased prevalence of fatty liver in arterial hypertensive
patients with normal liver enzymes: role of insulin resistance.
Gut. 53:1020–1023. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Pollare T, Lithell H and Berne C: Insulin
resistance is a characteristic feature of primary hypertension
independent of obesity. Metabolism. 39:167–174. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Raitakari OT, Porkka KV, Rönnemaa T, et
al: The role of insulin in clustering of serum lipids and blood
pressure in children and adolescents. The cardiovascular risk in
young finns study. Diabetologia. 38:1042–1050. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Allemann Y, Horber FF, Colombo M, et al:
Insulin sensitivity and body fat distribution in normotensive
offspring of hypertensive parents. Lancet. 341:327–331. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ogihara T, Asano T, Ando K, et al:
High-salt diet enhances insulin signaling and induces insulin
resistance in Dahl salt-sensitive rats. Hypertension. 40:83–89.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ogihara T, Asano T and Fujita T:
Contribution of salt intake to insulin resistance associated with
hypertension. Life Sci. 73:509–523. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Petta S, Macaluso FS, Barcellona MR, et
al: Serum γ-glutamyl transferase levels, insulin resistance and
liver fibrosis in patients with chronic liver diseases. PLoS One.
7:e511652012.
|
|
41
|
Aithal GP, Thomas JA, Kaye PV, et al:
Randomized, placebo-controlled trial of pioglitazone in nondiabetic
subjects with nonalcoholic steatohepatitis. Gastroenterology.
135:1176–1184. 2008. View Article : Google Scholar
|
|
42
|
Cohen JC, Horton JD and Hobbs HH: Human
fatty liver disease: old questions and new insights. Science.
332:1519–1523. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Day CP and James OF: Steatohepatitis: a
tale of two ‘hits’? Gastroenterology. 114:842–845. 1998.
|