Introduction
Neural crest-derived tumors characteristically
express the norepinephrine transporter (NET). This feature can be
used to specifically target tumor cells with agonists of NET. In
this study, we investigated the anticancer effects and underlying
mechanisms of action of mitochondrial inhibitors using
neuroblastoma (NB), a pediatric tumor of neural crest origin, as an
experimental system. NB is unique due to its clinical
heterogeneity. While some tumors (favorable NB) are easily
treatable, almost 50% of tumors (unfavorable NB) exhibit very
aggressive behavior. Unfavorable NBs are also classified as
high-risk NB and are characterized by widespread tumor
dissemination, late relapse and poor long-term survival. Among the
current treatments for high-risk NB,
131I-metaiodobenzylguanidine (MIBG) has been used for
scintigraphic detection and the targeted radiotherapy of NB
(1,2). The use of 131I-MIBG in
clinical practice for the treatment of patients with NB is based on
the fact that MIBG is a norepinephrine analogue and that NBs often
express norepinephrine transporters or NET.
MYC and MYCN are proto-oncogenes that
are members of the MYC gene family. A high MYCN/MYC
expression is associated with the poorest disease outcome in NB.
Approximately half of high-risk NBs exhibit MYCN
amplification, which is associated with older age, rapid tumor
progression and the poorest prognosis (3). A previous study suggested that in
non-MYCN-amplified tumors, MYC expression is responsible for
the aggressive phenotype (4). MYC
and MYCN proteins are stemness factors whose expression, along with
the expression of octamer-binding transcription factor 4 (OCT4),
sex Determining Region Y)-box 2 (SOX2) and Kruppel-like factor 4
(KLF4), help somatic cells regain a stem cell phenotype (5). In a recent study, we suggested that
the high expression of MYC/MYCN is critical to the existence of
stem cell-like tumor-initiating NB cells (6). These observations suggest that MYCN
and/or MYC expression are among the major determining factors of NB
aggressiveness. Previously, we also found that mitochondrial
inhibitors, such as carbonylcyanide
p-trifluoromethoxyphenylhydrazone (FCCP), can destabilize MYC/MYCN
and suppress the growth of NB cells (7), suggesting the potential use of
mitochondrial inhibitors in the treatment of NB.
Studies on genes encoding biomarkers for favorable
outcome in NB (favorable NB genes) have revealed mechanisms
underlying the favorable phenotype of the tumor. To date, several
favorable NB genes have been identified, and include [EPH receptor
B6 (EPHB6), ephrin (EFN)B2, EFNB3,
neurotrophic tyrosine kinase, receptor, type 1 (NTRK1;
TrkA), CD44 and Myc-interacting zinc finger protein
(MIZ-1)] (8–13). High expression levels of favorable
NB genes are associated with a favorable outcome in NB, and the
enforced expression of these genes in cells in unfavorable NB
results in growth suppression. We previously reported that known
favorable NB genes are epigenetically silenced in cells associated
with an unfavorable outcome in NB (9,14).
In the present study, we investigated the effects of three
additional mitochondrial inhibitors, MIBG, metformin and phenformin
on MYC/MYCN. Metformin and phenformin are diabetes medications that
are currently being considered for use as anticancer drugs. Our
data suggest that the destabilization of MYC/MYCN by MIBG,
metformin and phenformin is key to their antitumor effects. In
addition, the effects of these drugs on histone modification, which
is at least in part responsible for a change in the global gene
expression pattern of cells (i.e., upregulated expression of
favorable NB genes and tumor suppressor genes), may provide another
mechanism underlying their anticancer effects.
Materials and methods
Reagents and drug treatments
MIBG and metformin were purchased from
Calbiochem/EMD Chemicals, San Diego, CA, USA. MIBG was dissolved in
5 mM hydrochloric acid (HCl) at the concentration of 10 mM as stock
solution. Metformin was dissolved in H2O at the
concentration of 0.5 M as stock solution. Phenformin was purchased
from Sigma-Aldrich (St. Louis, MO, USA), and the stock solution was
made in 5 mM HCl at 0.25 M. The medium containing fresh drugs was
changed every 48 h.
Cell lines and western blot analysis
SKNBE(2)C, IMR5, Nb69, SY5Y and SKNAS cells were
cultured as previously described (6). SKNBE(2)C and SY5Y cells were
provided by Dr Robert Ross (Fordham University, Bronx, NY, USA).
IMR5 and Nb69 cells were from Dr Roger Kennett and Dr Fred Gilbert
(University of Pennsylvania, Philadelphia, PA, USA). SKNAS cells
were from Dr C. Patrick Reynolds (The Texas Tech University Health
Sciences Center, Lubbock, TX, USA). Western blot analysis was
performed as previously described (6). MYC and MYCN were detected using the
mouse monoclonal antibodies, NCM II 143 and NCM II 100 (15), respectively.
Reverse transcription and TaqMan
real-time polymerase chain reaction (PCR)
RNA was isolated using the Qiagen RNeasy kit
(Qiagen, Valencia, CA, USA). Two micrograms of total RNA were used
to synthesize cDNA. Reverse transcription and TaqMan real-time PCR
were performed as previously described (6).
3-(4,5-Dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium,
inner salt (MTS) assay
An MTS assay (Promega, Madison, WI, USA) was
performed as described in our previous study (6).
Results
Growth suppressive effect of MIBG and its
ability to destabilize MYC/MYCN in NB cells
Non-radiolabeled MIBG has been reported to be
cytotoxic to NB cells in vitro and in vivo(16). However, the mechanisms underlying
its growth suppressive effects are not yet well understood. In this
study, we examined the growth suppressive effects of MIBG on 5 NB
cell lines and investigated the possible mechanisms responsible for
its effects. MIBG suppressed the growth of MYCN-amplified NB
cells [SKNBE(2)C and IMR5 cells] and non-MYCN-amplified NB
cells (SKNAS, Nb69 and SY5Y cells) (Fig. 1A). In addition, SKNBE(2)C cells
were the most susceptible to the effects of MIBG. Fig. 1B shows the NET expression data in
the cell lines examined. SKNAS cells expressed the lowest levels of
NET among the cell lines examined, which was consistent with the
observation that MIBG had the least potent effect in suppressing
the growth of SKNAS cells. Nonetheless, there was no direct
correlation between NET expression and growth suppression mediated
by MIBG in the other four cell lines (Fig. 1B).
We previously demonstrated that FCCP, a known
mitochondrial inhibitor, destabilized MYC and MYCN in NB cells and
induced a growth suppressive effect (7). In this study, to investigate whether
MIBG, which is also a mitochondrial inhibitor, exhibits a similar
effect on MYC/MYCN in NB cells, we examined MYC/MYCN expression in
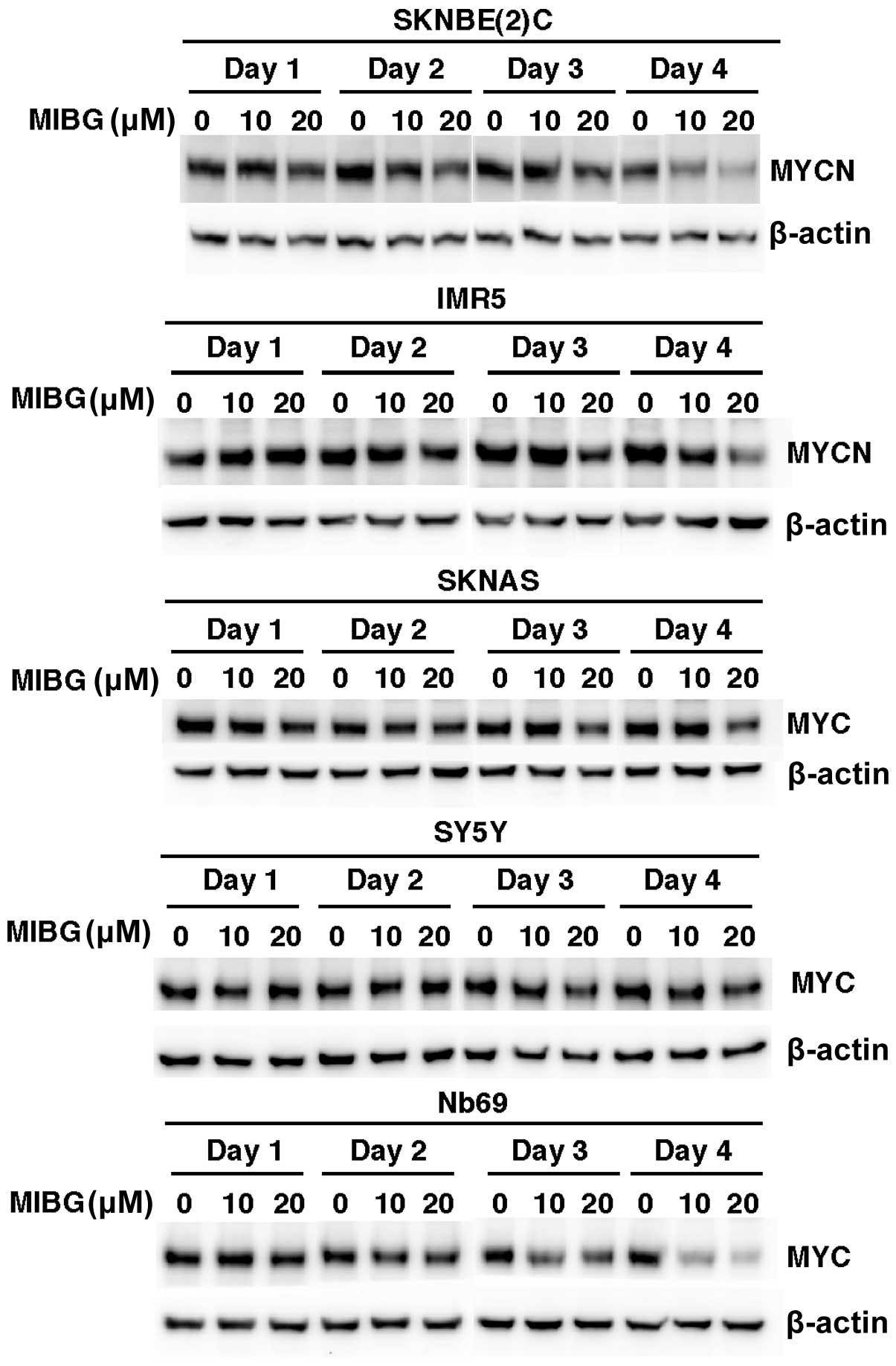
NB cell lines treated with MIBG (10 and 20 μM) for 4 days.
SKNBE(2)C and IMR5 cells expressed high levels of MYCN, whereas
SKNAS, SY5Y and Nb69 cells expressed high levels of MYC. Western
blot analysis revealed that the treatment of these NB cell lines
with MIBG resulted in a reduction in MYCN or MYC expression in a
time-dependent manner (Fig. 2).
The decreased MYC/MYCN expression was detected as early as day 2
[SKNBE(2)C cells] and day 3 (IMR5, SKNAS and Nb69 cells) of the
drug treatment. The destabilization of MYC/MYCN by MIBG also
occurred in a dose-dependent manner in SKNBE(2)C, IMR5, SKNAS and
Nb69 cells. Unlike the other cell lines, the MIBG-treated SY5Y
cells showed only a 15% reduction in MYC expression on day 4 of the
drug-treatment and at the dose of 20 μM (Fig. 2). The effects of MIBG on MYC/MYCN
expression were most prominent in the SKNBE(2)C and Nb69 cells,
followed by the IMR5 cells and, lastly, the SKNAS and SY5Y cells.
Taken together, the data on the growth suppressive effects of MIBG,
shown in Fig. 1A, and the effects
of MIBG on MYC/MYCN, shown in Fig.
2, indicate that with the exception of SY5Y cells, there was a
correlation between the growth suppressive effects of MIBG at 10
and 20 μM and its effects on MYC and MYCN expression among the
other 4 NB cell lines. This observation suggests that the
destabilization of MYC and MYCN is among the important mechanisms
through which MIBG exerts its growth suppressive effects on NB
cells.
Effect of metformin as a single agent or
in combination with MIBG on MYC/MYCN expression and growth of NB
cells
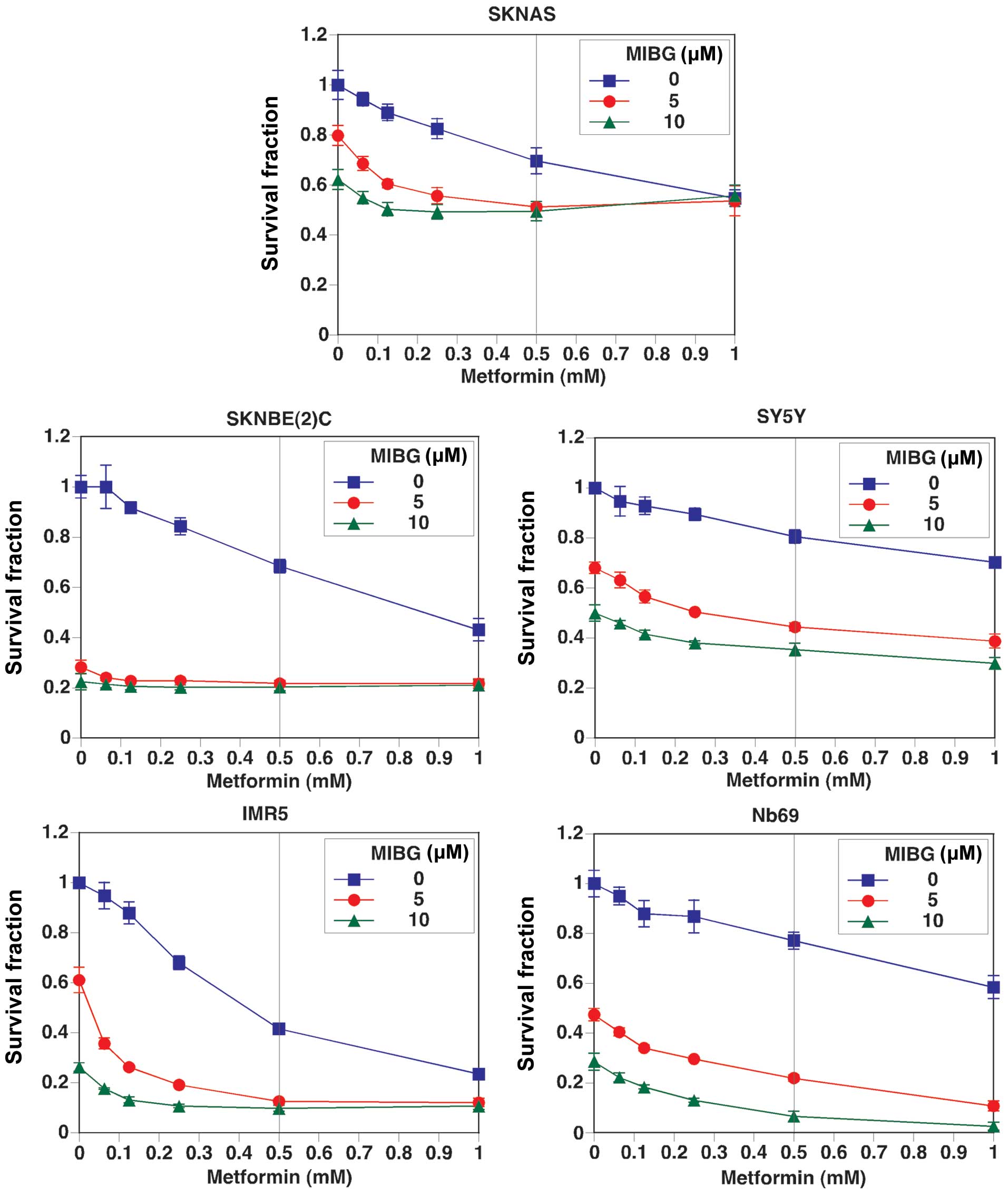
To determine whether the growth suppressive effect
and the MYC/MYCN destabilizing effect are general features of
mitochondrial inhibitors, we extended our investigation to the
effects of metformin on MYC/MYCN expression and the growth of NB
cells. Although metformin is currently being tested in clinical
trials as an anticancer drug, the mechanisms underlying its
anticancer effects are not yet fully understood. The curve with
square data points represents the effects of metformin as a single
agent on NB cell growth, whereas the curves with a solid circle or
solid triangle data points represent the effects of metformin in
combination with MIBG at 5 and 10 μM, respectively (Fig. 3). Our results revealed that
metformin as a single agent suppressed NB cell growth, and there
was an additive growth suppressive effect by the combination
treatment. Among the cell lines examined, the combination treatment
was most effective in suppressing the growth of Nb69, IMR5 and
SKNBE(2)C cells. The growth suppressive effect of metformin as a
single agent was most effective in IMR5 cells and the least
effective in SY5Y cells. Although the effects of metformin on cell
growth were moderate in Nb69 cells, the effects were markedly
enhanced when tested in combination with MIBG. The growth of SKNAS
and SY5Y cells was the least affected under all treatment
conditions.
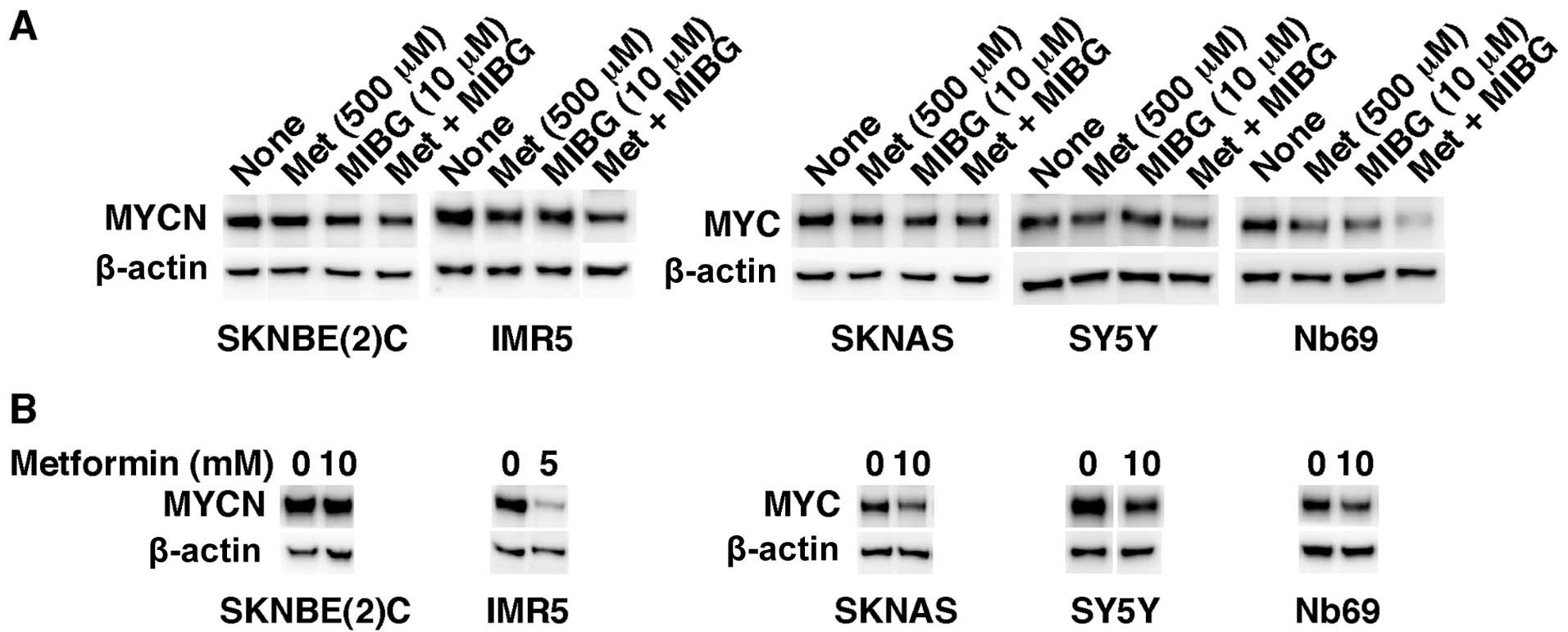
We then investigated the effects of metformin (500
μM) as a single agent and in combination with MIBG (10 μM) on
MYC/MYCN expression in NB cells. The reduction in MYC and MYCN
expression in the cells treated with both drugs at the above
concentrations was the greatest in Nb69 cells followed by IMR5
cells (Fig. 4A). In addition, the
combination drug treatment was the most effective in destabilizing
MYC/MYCN in the treated NB cells (Fig. 4A). The 2 drugs had the least
MYC/MYCN destabilizing effect in SKNBE(2)C, SKNAS and SY5Y cells.
Nonetheless, metformin at much higher concentrations (5 and 10 mM)
was effective in reducing MYC/MYCN expression not only in IMR5 and
Nb69 cells, as expected, but also in SKNAS and SY5Y cells after 1
day of the drug treatment (Fig.
4B). By contrast, SKNBE(2)C cells were resistant to metformin
as regards the destabilization of MYCN even at the higher dose of
the drug (Fig. 4B). The results
presented in Fig. 4A correlate
well with the data points corresponding to the growth suppressive
effects of metformin (500 μM) and MIBG (10 μM) shown in Fig. 3 (see the indicative vertical line
at 500 μM of metformin in Fig.
3). This observation further supports the hypothesis that the
destabilization of MYC/MYCN is an important mechanism responsible
for the ability of the drugs to suppress NB cell growth.
Treatment of NB cells with metformin or
MIBG results in increased expression of genes encoding biomarkers
of favorable outcome in NB (EFNB2, EFNB3, EPHB6, NTRK1, CD44 and
MIZ-1) and tumor suppressor genes [early growth response 1 (EGR1),
EPH receptor A2 (EPHA2), growth arrest and DNA-damage-inducible,
beta (GADD45B), neuregulin 1 (NRG1), TP53 apoptosis effector (PERP)
and sel-1 suppressor of lin-12-like (C. elegans) (SEL1L)]
The data shown in Figs. 2 and 3 suggest that in addition to the
destabilization of MYC/MYCN, other mechanisms may be involved in
the growth suppressive effects on NB cells mediated by MIBG and
metformin, particularly in SY5Y and SKNAS cells. To explore the
possibility that such mechanisms exist, we examined the effects of
MIBG and metformin as single agents and in combination on the
expression of genes encoding biomarkers of favorable outcome in NB
(EFNB2, EFNB3, EPHB6, NTRK1, CD44 and MIZ-1) in NB
cells. The expression of tumor suppressor genes was also examined
in the NB cell lines treated with MIBG and/or metformin. These
genes included EGR1, EPHA2, GADD45B, NRG1, PERP and
SEL1L. Metformin and MIBG upregulated the expression of
favorable NB genes and that of tumor suppressor genes in the
SKNBE(2)C, SKNAS, SY5Y and Nb69 cells (Fig. 5A and B). The drug treatments had
little effect on the expression of these genes in the IMR5
cells.
Metformin and MIBG increase the
expression of acetylated histone H3
We previously demonstrated that the low expression
of favorable NB genes (EFNB2, EFNB3, EPHB6, NTRK1, CD44 and
MIZ-1), as well as that of the tumor growth suppressive
gene, EPHA2, in NB cell lines was due to epigenetic
silencing (9,14,17). We thus examined whether metformin
and MIBG have an effect on chromatin structure, mainly the
alteration of the histone acetylation status, thereby leading to
the increased expression of these genes. With the exception of IMR5
cells, the other NB cell lines treated with metformin and/or MIBG
demonstrated an increased expression of acetylated histone H3
compared with the untreated control (Fig. 5C). These data suggest that
metformin and MIBG function as histone deacetylase (HDAC)
inhibitors, which in turn upregulates the expression of favorable
NB genes and tumor suppressor genes. The inability of the drugs to
augment acetylated histone H3 expression in the IMR5 cells is
consistent with the data shown in Fig. 5A and B, which show that the drugs
had little effect on the expression of the genes examined in IMR5
cells.
Effect of phenformin on MYC/MYCN
expression, acetylation of histone H3 and growth of NB cells
Metformin at 500 μM (Fig. 4A) was less effective than MIBG at
20 μM (Fig. 2) in reducing
MYC/MYCN expression in the NB cells. In addition, high
concentrations of metformin were required to effectively reduce
MYC/MYCN expression in the NB cells (Fig. 4B). We thus examined the effects of
phenformin, another mitochondrial inhibitor and anti-diabetic drug,
on MYC/MYCN expression in the NB cells. It has been reported that
phenformin binds NET (18),
suggesting that NET-positive cells, such as NB cells can
preferentially uptake phenformin. As shown in Fig. 6A, phenformin induced growth
suppressive effects on NB cells in a dose-dependent manner and
destabilized MYC/MYCN at the dose of 250 or 500 μM on days 4 and 6
of the drug treatments (Fig. 6B).
Phenformin was therefore more effective than metformin in reducing
MYC/MYCN expression. Short-term and high-dose treatments of
phenformin (1 day-treatment at the doses of 1 and 2.5 mM)
destabilized MYC/MYCN (Fig. 6C).
In addition, Fig. 6B and C show
that phenformin destabilized MYCN more effectively in the
low-dose/long-term treatment of SKNBE(2)C cells. Finally, the
treatment of NB cells with phenformin resulted in an increased
expression of acetylated histone H3 (Fig. 6D).
Discussion
We investigated the anticancer effects and
underlying mechanisms of action of mitochondrial inhibitors (MIBG,
metformin, and phenformin) using NB cell lines as an experimental
system. MIBG was previously known as a neuroendocrine
tumor-targeting agent. 131I-MIBG has been used for
scintigraphic detection and the targeted radiotherapy of NB
(1,2). Historically, the
radioactive131I residue on 131I-MIBG has been
considered to be the therapeutic effector due to its radiotoxicity
to NB. Our data demonstrated that non-radiolabeled MIBG confers a
growth suppressive effect on NB cells, destabilizes MYC/MYCN and
induces changes in global gene expression. The latter effect is
partly due to the ability of MIBG to increase the acetylation of
histone H3 in the cells.
Metformin and phenformin are anti-diabetic
biguanides that reduce blood glucose levels by inhibiting
gluconeogenesis in the liver. Metformin is one of the first-line
medications for type II diabetes in the United States and other
countries, while there is continued use of phenformin in certain
European and South American countries. Epidemiological evidence
suggests that metformin reduces cancer incidence and mortality in
patients with breast and prostate carcinoma (19–21); however, its exact biochemical
mechanisms are not yet well understood. There are two pre-existing
ideas that need to be re-evaluated in order to gain better insight
into the mechanisms through which biguanides exert their anticancer
effects: i) the involvement of AMP-activated protein kinase (AMPK)
in metformin function (22); and
ii) the Warburg hypothesis (23),
which states that cancer tissues are characterized by their
enhanced glycolysis in oxidative conditions and impaired
mitochondrial oxidative phosphorylation (OXPHOS) functions.
First, the results of several studies are
inconsistent with the hypothesis that the anti-diabetic and growth
inhibitory effects of metformin are linked to the activation of
AMPK: i) studies using AMPK knockout mice have demonstrated that
metformin inhibits mitochondrial OXPHOS Complex I and induces
changes in the energy state, which is solely responsible for the
inhibition of gluconeogenesis (24); ii) it has been suggested that the
growth inhibitory effects of metformin are due to AMPK activation,
leading to the inhibition of the mTOR pathway (25). Nonetheless, recent evidence has
indicated that without functional AMPK, metformin upregulates
REDD1 transcripts and protein, which is involved in the
negative regulation of the mTOR pathway (26). Thus, AMPK activation may be an
epiphenomenon to the direct effects of metformin (mitochondrial
OXPHOS inhibition and transcriptional activation).
Furthermore, several lines of evidence refute the
Warburg hypothesis: i) Weinhouse’s re-evaluation of Warburg’s data
indicated that tumor tissues are in fact active in
mitochondria-driven OXPHOS (27);
ii) several types of cancer cells express high levels of MYC family
proteins that stimulate mitochondrial biogenesis, glycolysis and
glutaminolysis, thus facilitating cancer cell growth (28). These observations suggest that
cancer cells are dependent on mitochondrial OXPHOS augmented by
elevated MYC expression. Thus, the inhibition of mitochondrial
OXPHOS may have a profound effect on cancer cell growth. Our data
revealed that mitochondrial inhibitors induce growth suppressive
effects on NB cells in vitro. We also found that metformin
and phenformin destabilize MYC and MYCN in NB cell lines, although
their effective doses differ (see below).
Finally, the anti-diabetic biguanides interact with
heavy metals (e.g., Zn, Cu and Fe) (29), which can affect the activities of
Zn-dependent histone deacetylases. Our data (Figs. 5C and 6D) are consistent with this observation.
The treatment of NB cells with metformin and phenformin resulted in
an increase in the expression of acetylated histone H3. It remains
to be seen whether the anti-diabetic biguanides have any effect on
histone demethylase.
Our data suggest that the destabilization of MYC and
MYCN is among the important mechanisms through which MIBG,
metformin and phenformin exert their growth suppressive effects on
NB cells. MIBG is more effective than metformin in destabilizing
MYC/MYCN function, and phenformin is more effective than metformin,
but less effective than MIBG in destabilizing MYC/MYCN in NB cell
lines. Structurally, phenformin can be considered a hybrid molecule
between MIBG and metformin. This may account for the observation
that its anti-MYC/MYCN effects are not as effective as those of
MIBG, but more effective than those of metformin. In addition, the
mechanisms through which NB cells uptake drugs may, in part,
explain the difference in the potency of the drugs in destabilizing
MYC/MYCN. Among the 3 drugs examined, it was found that NB cells
uptake MIBG via a receptor-mediated process due to the expression
of NET on their surface. Phenformin may use a similar mechanism as
MIBG since it has been shown to bind NET (18). Phenformin can also easily diffuse
into the cells due to the presence of the phenol ring.
The two new biological and chemical functions of
MIBG, metformin and phenformin reported in this study
(specifically, the destabilizing of MYC/MYCN and the ability to
augment the acetylation of histone H3) provide a better
understanding of the mechanisms through which these drugs function.
Phenformin binds NET, which is also expressed on the NB cell
surface. Therefore, MIBG and phenformin can be tumor-targeting
MYC/MYCN destabilizing agents in NB. The in vitro doses of
metformin and phenformin used in this study seem relatively high.
However, the effective doses in vivo (clinically) are lower
than those used for our in vitro experiments. This is due to
the fact that the positive charge of metformin and phenformin
causes them to accumulate to a very high concentration in the
negatively charged mitochondrial matrix over time (30). In a recent study, we suggested
that MYC/MYCN are important stemness factors that play key roles
during the development of NB stem cells or stem-like cells
(6). Thus, the destabilization of
MYC/MYCN by metformin and phenformin and the ability of metformin
to upregulate tumor suppressor genes may partly explain why these
diabetes drugs protect against cancer incidence and mortality.
Acknowledgements
This study was supported in part by R01 CA127571 and
a grant from St. Baldrick’s Foundation. S.S.W. was supported in
part by the Craig Fellowship, UIC College of Medicine.
References
|
1
|
Treuner J, Feine U, Niethammer D,
Muller-Schaumburg W, Meinke J, Eibach E, Dopfer R, Klingebiel T and
Grumbach S: Scintigraphic imaging of neuroblastoma with
[131-I]iodobenzylguanidine. Lancet. 1:333–334. 1984.
|
|
2
|
Matthay KK, Yanik G, Messina J, Quach A,
Huberty J, Cheng SC, Veatch J, Goldsby R, Brophy P, Kersun LS,
Hawkins RA and Maris JM: Phase II study on the effect of disease
sites, age, and prior therapy on response to
iodine-131-metaiodobenzylguanidine therapy in refractory
neuroblastoma. J Clin Oncol. 25:1054–1060. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Seeger RC, Brodeur GM, Sather H, Dalton A,
Siegel SE, Wong KY and Hammond D: Association of multiple copies of
the N-myc oncogene with rapid progression of neuroblastomas. New
Engl J Med. 313:1111–1116. 1985. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fredlund E, Ringner M, Maris JM and
Pahlman S: High Myc pathway activity and low stage of neuronal
differentiation associate with poor outcome in neuroblastoma. Proc
Natl Acad Sci USA. 105:14094–14099. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Takahashi K, Tanabe K, Ohnuki M, Narita M,
Ichisaka T, Tomoda K and Yamanaka S: Induction of pluripotent stem
cells from adult human fibroblasts by defined factors. Cell.
131:861–872. 2007. View Article : Google Scholar
|
|
6
|
Ikegaki N, Shimada H, Fox AM, Regan PL,
Jacobs JR, Hicks SL, Rappaport EF and Tang XX: Transient treatment
with epigenetic modifiers yields stable neuroblastoma stem cells
resembling aggressive large-cell neuroblastomas. Proc Natl Acad Sci
USA. 110:6097–6102. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ikegaki N, Regan PL, Hicks SL, Maloney N
and Tang XX: Regulation of MYCN stability by reactive oxygen
species in neuroblastoma. Cancer Res. 71(Suppl 1): Abstract LB-142.
2011. View Article : Google Scholar
|
|
8
|
Tang XX, Zhao H, Robinson ME, Cohen B,
Cnaan A, London W, Cohn SL, Cheung NK, Brodeur GM, Evans AE and
Ikegaki N: Implications of EPHB6, EFNB2, and EFNB3 expressions in
human neuroblastoma. Proc Natl Acad Sci USA. 97:10936–10941. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ikegaki N, Gotoh T, Kung B, Riceberg JS,
Kim DY, Zhao H, Rappaport EF, Hicks SL, Seeger RC and Tang XX: De
novo identification of MIZ-1 (ZBTB17) encoding a MYC-interacting
zinc-finger protein as a new favorable neuroblastoma gene. Clin
Cancer Res. 13:6001–6009. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Combaret V, Gross N, Lasset C, Frappaz D,
Peruisseau G, Philip T, Beck D and Favrot MC: Clinical relevance of
CD44 cell-surface expression and N-myc gene amplification in a
multicentric analysis of 121 pediatric neuroblastomas. J Clin
Oncol. 14:25–34. 1996.PubMed/NCBI
|
|
11
|
Nakagawara A, Arima-Nakagawara M, Scavarda
NJ, Azar CG, Cantor AB and Brodeur GM: Association between high
levels of expression of the TRK gene and favorable outcome in human
neuroblastoma. N Engl J Med. 328:847–854. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kogner P, Barbany G, Dominici C, Castello
MA, Raschella G and Persson H: Coexpression of messenger RNA for
TRK protooncogene and low affinity nerve growth factor receptor in
neuroblastoma with favorable prognosis. Cancer Res. 53:2044–2050.
1993.PubMed/NCBI
|
|
13
|
Suzuki T, Bogenmann E, Shimada H, Stram D
and Seeger RC: Lack of high-affinity nerve growth factor receptors
in aggressive neuroblastomas. J Nat Cancer Inst. 85:377–384. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tang XX, Robinson ME, Riceberg JS, Kim DY,
Kung B, Titus TB, Hayashi S, Flake AW, Carpentieri D and Ikegaki N:
Favorable neuroblastoma genes and molecular therapeutics of
neuroblastoma. Clin Cancer Res. 10:5837–5844. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ikegaki N, Bukovsky J and Kennett RH:
Identification and characterization of the NMYC gene product in
human neuroblastoma cells by monoclonal antibodies with defined
specificities. Proc Natl Acad Sci USA. 83:5929–5933. 1986.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Smets LA, Bout B and Wisse J: Cytotoxic
and antitumor effects of the norepinephrine analogue
meta-iodo-benzylguanidine (MIBG). Cancer Chemother Pharmacol.
21:9–13. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kung B, Zhao H, Hicks SL, Tang XX and
Ikegaki N: Biological significance of EPHA2 expression in
neuroblastoma. Int J Oncol. 35:845–850. 2009.
|
|
18
|
Schlessinger A, Geier E, Fan H, Irwin JJ,
Shoichet BK, Giacomini KM and Sali A: Structure-based discovery of
prescription drugs that interact with the norepinephrine
transporter, NET. Proc Natl Acad Sci USA. 108:15810–15815. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Decensi A, Puntoni M, Goodwin P, Cazzaniga
M, Gennari A, Bonanni B and Gandini S: Metformin and cancer risk in
diabetic patients: a systematic review and meta-analysis. Cancer
Prev Res (Phila). 3:1451–1461. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bodmer M, Meier C, Krahenbuhl S, Jick SS
and Meier CR: Long-term metformin use is associated with decreased
risk of breast cancer. Diabetes Care. 33:1304–1308. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wright JL and Stanford JL: Metformin use
and prostate cancer in Caucasian men: results from a
population-based case-control study. Cancer Causes Control.
20:1617–1622. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhou G, Myers R, Li Y, Chen Y, Shen X,
Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, Musi N,
Hirshman MF, Goodyear LJ and Moller DE: Role of AMP-activated
protein kinase in mechanism of metformin action. J Clin Invest.
108:1167–1174. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Warburg OH: Über den Stoffwechsel der
Tumoren. Springer; Berlin: 1926
|
|
24
|
Foretz M, Hebrard S, Leclerc J,
Zarrinpashneh E, Soty M, Mithieux G, Sakamoto K, Andreelli F and
Viollet B: Metformin inhibits hepatic gluconeogenesis in mice
independently of the LKB1/AMPK pathway via a decrease in hepatic
energy state. J Clin Invest. 120:2355–2369. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shackelford DB and Shaw RJ: The LKB1-AMPK
pathway: metabolism and growth control in tumour suppression. Nat
Rev Cancer. 9:563–575. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ben Sahra I, Regazzetti C, Robert G,
Laurent K, Le Marchand-Brustel Y, Auberger P, Tanti JF,
Giorgetti-Peraldi S and Bost F: Metformin, independent of AMPK,
induces mTOR inhibition and cell-cycle arrest through REDD1. Cancer
Res. 71:4366–4372. 2011.PubMed/NCBI
|
|
27
|
Weinhouse S: The Warburg hypothesis fifty
years later. Z Krebsforsch Klin Onkol Cancer Res Clin Oncol.
87:115–126. 1976.PubMed/NCBI
|
|
28
|
Dang CV: Therapeutic targeting of
Myc-reprogrammed cancer cell metabolism. Cold Spring Harb Symp
Quant Biol. 76:369–374. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Sweeney D, Raymer ML and Lockwood TD:
Antidiabetic and antimalarial biguanide drugs are metal-interactive
antiproteolytic agents. Biochem Pharmacol. 66:663–677. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Owen MR, Doran E and Halestrap AP:
Evidence that metformin exerts its anti-diabetic effects through
inhibition of complex 1 of the mitochondrial respiratory chain.
Biochem J. 348:607–614. 2000. View Article : Google Scholar : PubMed/NCBI
|