Introduction
Acute liver failure (ALF) is a syndrome defined by
the sudden onset of severe liver injury, followed by coagulopathy
and encephalopathy. The mortality rates for patients with ALF are
50–70% (1–3). The prognosis of ALF has not improved
significantly over the past decades, despite the development of
treatments, such as plasma exchange, dialysis and antibiotics.
Although liver transplantation is the only promising treatment for
ALF, the rapid progression of the disease and the shortage of
donors limit the success of this treatment option (3). However, the pathogenesis of ALF is
not yet fully understood and therefore prohibits us from
establishing new treatments.
It has been suggested that intrahepatic
microcirculatory disorder is involved in the pathogenesis of ALF.
Intrahepatic macrophage activation has been suggested as an
important factor in the pathogenesis of ALF, causing intrahepatic
microcirculatory disorder, and subsequently, massive levels of
hepatocyte death (4,5). Verification that activated
macrophages disturb intrahepatic microcirculation has not been
achieved due to the difficulties in visualizing the changes
occurring in macrophages and microcapillary structure
simultaneously.
Intravital microscopy, used for the observation of
biological systems with high resolution, is an excellent technique
due to its ability to obtain real-time dynamic microcirculatory
images (6,7). It has often been used in
experimental animal models of liver injury (8–11).
The limitations of this method include difficulty in simultaneously
evaluating blood flow and surrounding pathological changes.
Furthermore, only a limited number of laboratories are able to
utilize this technique as it demands expensive and specialized
equipment and facilities.
In this study, we designed a method for visualizing
hepatic microcapillary structure with the administration of
tetramethylrhodamine isothiocyanate (TRITC)-dextran. Applying this
method to lipopolysaccharide (LPS)- and D-galactosamine
(GalN)-induced liver injury in rats, a widely accepted animal model
of ALF (12), we were able to
obtain detailed findings of intrahepatic microcirculatory
disturbance in relation to intrahepatic macrophage activation.
Given that the massive reduction in the number of viable
hepatocytes is caused by intrahepatic microcirculatory disorder,
there still remains the controversial issue of whether this
reduction in ALF is due to apoptosis or necrosis. A number of
studies have demonstrated that activated macrophages can cause
intrahepatic microcirculatory disorder and consequent parenchymal
necrosis (13–18), while others have indicated that
activated macrophages induce parenchymal apoptosis by secreting
death receptor ligands, such as Fas ligand or tumor necrosis
factor-α (TNF-α) (19,20). Although it is difficult to
determine which manner of cell death is dominant, recent studies
have indicated that apoptosis plays a major role in the reduction
in the number of viable hepatocytes during the development of ALF
(42,43).
Apoptosis is induced through the sequential
activation of caspases that can be activated by death receptors
(extrinsic pathway) also cellular stress (intrinsic pathway)
(21). As regards the intrinsic
pathway, the nicotinamide adenine dinucleotide phosphate oxidase
(NOX) family is a major source of cellular reactive oxygen species
(ROS) (22–24). Among NOX homologues, only NOX4 is
an inducible isoform (25). To
evaluate the mechanisms that trigger hepatocyte apoptosis in ALF,
we conducted an immunohistochemical detection of cleaved caspase-3,
CD68, pimonidazole, hypoxia-inducible factor 1-α (HIF1-α),
4-hydroxy-2-nonenal (4-HNE) and NOX4 in rat livers, and aimed to
determine correlations between their localization and intrahepatic
microcirculatory disorder.
Materials and methods
Chemicals and antibodies
All reagents were purchased from Sigma Chemical Co.
(St. Louis, MO, USA) unless otherwise stated. Monoclonal mouse
anti-CD68 and rabbit anti-NOX4 antibodies, as well as horseradish
peroxidase (HRP)-conjugated polyclonal goat anti-mouse and goat
anti-rabbit antibodies were purchased from Abcam (Cambride, UK).
The polyclonal rabbit anti-cleaved caspase-3 and rabbit anti-HIF1-α
antibodies were from Novus Biologicals (Littleton, CO, USA). A
monoclonal mouse anti-4-HNE antibody was obtained from NOF Corp.
(Tokyo, Japan). Pimonidazole and the mouse anti-pimonidazole
adducts monoclonal antibody (Hypoxyprobe™-1 kit) were purchased
from Hypoxyprobe Inc. (Burlington, MA, USA).
Animals
Eight-week-old male Wistar rats (weighing
approximately 200 g), were purchased from Japan SLC, Inc.
(Hamamatsu, Japan). The animals were provided access to food and
water ad libitum and maintained on a 12-h light-dark cycle
in a temperature (18–21°C) and humidity (55±5%) controlled
environment for 1 week before the experiments. All animals received
humane care, and all experiments in this study were performed
according to the guidelines of the Committee for the Care and Use
of Laboratory Animals at Kyushu University, Fukuoka, Japan.
Experimental protocols
The rats were divided into 2 groups (n=10
rats/group). LPS from Escherichia coli (5 μg/kg body weight)
and GalN (500 mg/kg body weight) were intraperitoneally injected
into the rats in the ALF group, while sterile saline was injected
into the rats in the control group (n=10). At 22 h
post-administration, pimonidazole (60 mg/kg body weight) was
intraperitoneally injected into all the rats. Pimonidazole is
rapidly reduced in hypoxic cells to a highly reactive intermediate
that binds to sulfur on glutathione and proteins. Using an antibody
against these pimonidazole-sulfur adducts, immunohistochemistry can
be employed to detect the hypoxic cells (26–33). Two hours after the pimonidazole
injection, the rats were anesthetized with isoflurane, and the
abdomens were incised and canulated at the inferior vena cava using
a 20 G plastic needle. After a 1-ml blood sample was withdrawn, the
rats were intravenously injected with TRITC-dextran (500 mg/kg body
weight) dissolved in 1 ml of sterile saline and the livers excised.
One lobe of excised liver was fixed in 20% formaldehyde solution
(Mildform 20N; Wako Pure Chemical Industries, Osaka, Japan) for
histological analysis, and another lobe was snap-frozen in liquid
nitrogen for use in western blot analysis.
Preparation of liver sections to assess
TRITC-dextran distribution
The excised livers were fixed in 20% formaldehyde
solution for 1 day, paraffin-embedded and sectioned (5 μm
thickness). The sections were deparaffinized with xylene and
rehydrated by washing through a graded alcohol series and deionized
water. The hydrated tissue sections were washed with
phosphate-buffered saline (PBS). To evaluate TRITC-dextran
distribution, the sections were mounted in aqueous Fluor Mount/plus
(Diagnostic Biosystems Inc., Pleasanton, CA, USA) immediately after
deparaffinization and observed under a fluorescence microscope
(BZ-9000; Keyence, Osaka, Japan).
Hematoxylin and eosin (H&E)
staining
Some sections were deparaffinized, rehydrated and
stained with H&E to evaluate the general histological state.
Some sections were deparaffinized, rehydrated and stained with
H&E according to a standard procedure (66) to evaluate the general histological
state.
Immunohistochemistry
The liver sections were deparaffinized, rehydrated
and exposed to 3% (v/v) H2O2 for 15 min at
room temperature to quench endogenous peroxidase activity. The
sections were incubated with antigen retrieval solution (Target
Retrieval Solution; Dako, Tokyo, Japan) for 30 min at 95°C.
Following 2 washes in PBS, the sections were incubated with
blocking buffer (Blocking One Histo; Nakalai Tesque, Inc., Kyoto,
Japan) for 30 min. The sections were then incubated with antibodies
dissolved in Can Get Signal Immunostain solution A (Toyobo, Osaka,
Japan) for 1 h at room temperature, followed by incubation with the
appropriate secondary antibody conjugated to HRP in Can Get Signal
Immunostain solution B for 30 min at room temperature. Following 2
washes in PBS, the sections were incubated with
3,3′-diaminobenzidine tetrahydrochloride (DAB) solution for 5
min.
For double immunohistochemical staining, the same
steps from antigen retrieval to incubation with a chromogen
described above were repeated, except that acetonitrile solution
(Vector SG; Vector Laboratories, Burlingame, CA, USA) instead of
DAB was used as a chromogen. The sections were washed twice with
PBS and mounted in aqueous Fluor Mount/plus. Hematoxylin
counterstaining was only conducted for the samples incubated with a
single antibody. For CD68 and pimonidazole immunohistochemistry, we
obtained bright-field and fluorescent images from the same liver
sections and merged them. For double immunohistochemistry and
HIF1-α, 4-HNE and NOX4 immunohistochemistry, staining was carried
out on serial sections. The pathological findings assessed by one
of the authors blinded to the group allocations.
Western blot analysis
Liver tissue samples (20 mg) were homogenized in
lysis buffer and protein levels were quantified using a commercial
protein assay kit (Bio-Rad, Hercules, CA, USA). Liver extracts (30
μg protein/lane) were electrophoresed and subjected to SDS-PAGE
under reducing conditions using 10% polyacrylamide gels. Proteins
were transferred to Immobilon-P polyvinylidene difluoride transfer
membranes (Millipore, Billerica, MA, USA) and probed with an
anti-NOX4 rabbit monoclonal antibody. Detection was carried out
using an anti-rabbit IgG goat HRP-conjugated antibody in
conjunction with ECL Prime Western Blotting Detection reagent (GE
Healthcare, UK Ltd., Buckinghamshire, UK). The quantification of
enhanced chemiluminescence (ECL) was performed using an ImageQuant
LAS 4000 imaging system (GE Healthcare, UK Ltd.).
Statistical analysis
Data are expressed using box and whisker plots. The
Steel test was applied to analyze results between 2 groups, and the
Steel-Dwass test was used to analyze results between 3 or more
groups. A P-value ≤0.05 was considered to indicate a statistically
significant difference.
Results
Histology of livers of rats with
LPS/GalN-induced ALF
The liver sections obtained from the control rats
had an integrated structure of hepatic lobules (Fig. 1A). The intraperitoneal injection
of LPS/GalN induced marked hepatic injuries, accompanied by
hemorrhaging and inflammatory cell infiltration (Fig. 1B).
 | Figure 1Hematoxylin and eosin
(H&E)-stained sections and fluorescent images from
tetramethylrhodamine isothiocyanate (TRITC)-dextran-injected
livers. Liver sections from (A) control rats, and (B) rats with
acute liver failure (ALF) induced by
lipopolysaccharide/D-galactosamine (LPS/GalN) stained with H&E.
Fluorescent images of TRITC-dextran-injected livers from (C and E)
the control rats and (D and F) rats with ALF. (G) The TRITC-dextran
distribution ratio for each zone in the livers from the control
rats and rats with ALF. (C and E) In the livers of the control
rats, TRITC-dextran was evenly distributed in the portal veins (P),
sinusoids and central veins (V). (D and F) In the livers of rats
with ALF, TRITC-dextran was distributed in the portal veins (P) and
zone 1 sinusoids, but not in the central veins and zone 3
sinusoids. In zone 2, several spotty signals of TRITC-dextran can
be obseved (*). Original magnification, ×200 (A, B, E and F), and
×40 (C and D). The TRITC-dextran distribution ratio in zone 3 of
the livers of rats with ALF was significantly decreased compared
with ALF zones 1 and 2, and zones 1–3 in the controls
(*P≤0.05). Zone 1, periportal zone; zone 2, intermediate
zone; zone 3, pericentral zone. |
TRITC-dextran distribution in liver
sections
We varied the interval from TRITC-dextran injection
to liver excision (30 sec, 1, 2, 3, 5 and 10 min), and concluded
that clear and strong signals of TRITC-dextran could be obtained
after 1 min (data not shown). The interval was therefore fixed to 1
min in subsequent experiments. In the liver sections from the
control rats, TRITC-dextran was distributed evenly across the liver
acinus (portal veins, sinusoids and central veins) (Fig. 1C and E). In the liver sections
from the rats with ALF, TRITC-dextran was clearly distributed in
the portal veins and zone 1 (periportal zone) sinusoids, and could
be faintly observed in the central veins and zone 3 (pericentral
zone) sinusoids. Additionally, several spotty signals corresponding
to TRITC-dextran appeared in zone 2 (intermediate zone) (Fig. 1D and F). We measured the ratio of
TRITC-dextran-positive areas to the parenchymal area in each zone
(Fig. 1G). TRITC-dextran
distribution in zone 3 of the livers of rats with ALF was markedly
decreased when compared with that in zones 1 and 2 of the livers of
rats wtih ALF, and all zones in the livers of the control rats.
Correlation between intrahepatic
microcirculation and infiltration of CD68+ positive
cells
In the control rats, only some cells were positive
for CD68 (Fig. 2C). In the rats
with ALF, there was a marked infiltration of CD68+ cells
distributed among the liver acinus (Fig. 2D). We observed that
CD68+ cells dominantly localized around the spotty
signals of TRITC-dextran in zone 2 (Fig. 2F). The ratio of CD68+
cells to the parenchymal area in each zone indicated that the
CD68+ cells in all zones in the livers of rats with ALF
were significantly increased in comparison to those in the control
rats; the CD68+ cells were most abundant in zone 2 in
the livers of rats with ALF (Fig.
2G).
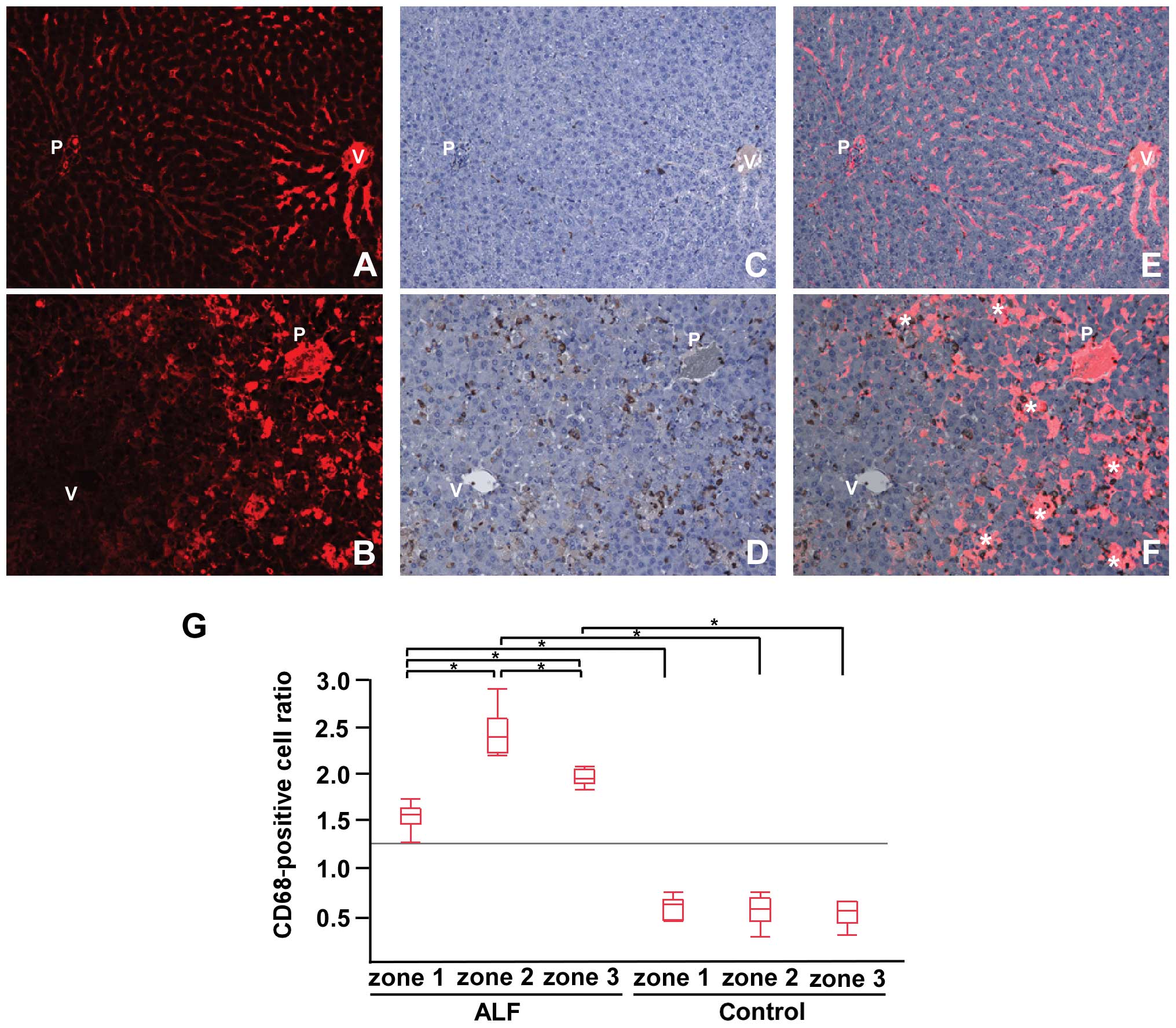 | Figure 2Tetramethylrhodamine isothiocyanate
(TRITC)-dextran and CD68 immunohistochemistry. (A and B)
TRITC-dextran distribution. (C and D) Immunohistochemical detection
of CD68 and (E and F) merge images for controls (upper panel) and
acute liver failure (ALF) (lower panel) samples. (G) The
CD68+ cell ratio in each zone for the control and ALF
samples. In the controls, TRITC-dextran was evenly distributed in
the liver acinus (A) with some CD68+ cells (C). In ALF
livers, marked infiltration of CD68+ was observed in the
liver acinus (D). Merge images (B and D) showed that
CD68+ cells were predominantly localized around the
spotty signals of TRITC-dextran in zone 2 (*) (F) (original
magnification, ×200). CD68+ cells in the ALF samples
were significantly increased compared with the controls. (G) In the
ALF samples, CD68+ cells were most abundant in zone 2,
followed by zone 3 and then zone 1 (*P≤0.05). P, portal
vein; V, central vein. Zone 1, periportal zone; zone 2,
intermediate zone; zone 3, pericentral zone. |
Apoptotic hepatocytes and their
localization
In the control rats, a few cells were positive for
cleaved caspase-3 (Fig. 3A). In
the rats with ALF, high numbers of cleaved caspase-3+
hepatocytes were observed (Fig.
3B) and were mainly localized in zones 2 and 3 (Fig. 3C and D). In the rats with ALF, we
evaluated the ratio of cleaved caspase-3+ hepatocytes to
the parenchymal area in each zone (Fig. 3E). Cleaved caspase-3+
hepatocytes were most abundant in zone 3.
 | Figure 3Immunohistochemical detection of
cleaved caspase-3 and the apoptotic hepatocyte ratio.
Immunohistochemistry for cleaved caspase-3 in livers from (A)
control rats and (B–D) rats with acute liver failure (ALF). (E) The
cleaved caspase-3+ cell ratio in each ALF zone. (A) In
the controls, few cells were positive for cleaved caspase-3. (B) In
the livers of mice with ALF, high numbers of cleaved
caspase-3+ hepatocytes were observed, particularly in
zones 2 (C) and 3 (D). Original magnification, ×200 (A and B), ×400
(C and D). Cleaved caspase-3+ cell ratios in the livers
of rats with ALF were greatest in zone 3, followed by zones 2 and 1
(E) (*P≤0.05). P, portal vein; V, central vein. Zone 1,
periportal zone; zone 2, intermediate zone; zone 3, pericentral
zone. |
Localization of parenchymal hypoxia,
HIF1-α, oxidative stress and NOX4
On the basis of the results presented above, we
hypothesized that hypoxia due to intrahepatic microcirculatory
disorder may result in parenchymal oxidative stress and consequent
apoptosis in zone 3 via an intrinsic pathway. In the control rats,
whole liver tissue was negative for pimonidazole (normoxic
conditions) (Fig. 4C). In the
rats with ALF, the zone 1 parenchyma was predominantly negative for
pimonidazole staining, whereas the zone 3 parenchyma was strongly
positive (hypoxic conditions) (Fig.
4D). Hypoxia in zone 3 appeared to be induced by impaired
intrahepatic microcirculation (Fig.
4F). An immunohistochemical detection of HIF1-α (Fig. 4G and H), 4-HNE (Fig. 4I and J) and NOX4 (Fig. 4K and L) was conducted. The whole
liver tissue from the control rats was mostly negative, whereas the
zone 3 parenchyma in the rats with ALF was positive for all
proteins. The localization of HIF1-α, 4-HNE and NOX4 corresponded
with that of pimonidazole. NOX4 protein expression was markedly
increased in the rats with ALF compared with the control rats
(Fig. 4M).
 | Figure 4Immunohistochemical detection of
pimonidazole, hypoxia inducible factor 1-α (HIF1-α),
4-hydroxy-2-nonenal (4-HNE) and nicotinamide adenine dinucleotide
phosphate oxidase (NOX4), and NOX4 western blot analysis results.
(A and B) Tetramethylrhodamine isothiocyanate (TRITC)-dextran
distribution. (C and D) Immunohistochemical detection of
pimonidazole and (E and F) merge images from the livers of (A, C
and E) control rats and (B, D and F) rats with acute liver failure
(ALF). Immunohistochemistry for (G and H) HIF1-α, (I and J) 4-HNE
and (K and L) NOX4 in livers of (G, I and K) control rats and (H, J
and L) rats with ALF. (M) Western blot analysis of NOX4. In the
controls, TRITC-dextran was evenly distributed in the liver acinus
lacking hypoxic areas (A and C). In the livers of rats with ALF,
zone 1 was normoxic with sustained sinusoidal flow, but zone 3 was
hypoxic with reduced sinusoidal flow (B, D and F). In the controls,
all zones were predominantly negative for HIF1-α (G), 4-HNE (I) and
NOX4 (K). In the ALF samples, zone 1 was mainly negative, and zone
3 positive for HIF1-α (H), 4-HNE (J) and NOX4 (L) (original
magnification, ×200). In the livers of rats with ALF, NOX4 protein
expression was increased compared with the controls (M)
(*P≤0.05). P, portal vein; V, central vein. Zone 1,
periportal zone; zone 2, intermediate zone; zone 3, pericentral
zone. |
Analysis of zonal differences in
hepatocyte apoptosis
The simultaneous immunohistochemical detection of
cleaved caspase-3 and CD68 (Fig.
5A–C) revealed that the majority of the apoptotic hepatocytes
in zone 2 were localized adjacent to infiltrated macrophages. Few
apoptotic hepatocytes in zone 3 were similarly localized. The
simultaneous immunohistochemical detection of cleaved caspase-3 and
NOX4 (Fig. 5D–F) revealed that
few apoptotic hepatocytes in zone 2 colocalized with NOX4, whereas
the majority of apoptotic hepatocytes in zone 3 colocalized with
NOX4.
 | Figure 5Simultaneous immunohistochemical
detection of cleaved caspase-3 and CD68, and cleaved caspase-3 and
nicotinamide adenine dinucleotide phosphate oxidase (NOX4) in
livers of rats with acute liver failure (ALF). Immunohistochemical
detection of cleaved caspase-3 (brown color) and CD68 (dark blue
color) (A–C), and cleaved-caspase-3 (brown) and NOX4 (dark blue)
(D–F) in livers of rats with ALF. The majority of apoptotic
hepatocytes in zone 2 (B), and a few apoptotic hepatocytes in zone
3 (C) were localized adjacent to macrophages. A few apoptotic
hepatocytes in zone 2 colocalized with NOX4 (E), and the majority
of apoptotic hepatocytes in zone 3 were colocalized with NOX4 (F).
Original magnification, ×200 (A and D), ×400 (B, C, E and F). P,
portal vein; V, central vein. Zone 1, periportal zone; zone 2,
intermediate zone; zone 3, pericentral zone. |
Discussion
In this study, we demonstrate that the intrahepatic
microcirculatory structure can be visualized in formaldehyde-fixed
paraffin-embedded (FFPE) liver sections from rats treated with
TRITC-dextran. This method can be applied to both healthy and ALF
livers stimulated with LPS/GalN. Previously, the administration of
fluorescent dextran has been used to analyze tissue
microcirculation under various conditions with the aim of
demonstrating exceptional blood flow or extravasation. Vollmar
et al (34) administered
fluorescent dextran intravenously to rats with liver cirrhosis and
observed its excretion to the lymph vessels. Sun et al
(35) administered fluorescent
dextran to rats with ischemia/reperfusion injury via the superior
mesenteric artery to visualize extravasation at the intestinal
villi. To our knowledge, no preceding study, however, has used this
method to assess sinusoidal blood flow. This technically convenient
method could be applied to evaluate other animal models mimicking
liver diseases, such as graft-versus-host disease, sinusoidal
obstruction syndrome and ischemia/reperfusion injury, which
encompass intrahepatic microcirculatory disorder during their
progression.
In our study, in the livers of rats with ALF,
several spotty signals of TRITC-dextran were observed in zone 2
(Fig. 1D and F). These spotty
signals seemed to represent a pooling of sinusoidal blood flow as a
result of extravasation into the parenchyma. This likely indicates
the disrupted integrity of sinusoidal endothelial cells (SECs),
which is consistently described in previous studies involving
animal models of ALF (36) and
patients (37). Additionally,
signals of TRITC-dextran in zone 3, the downstream region of
sinusoidal flow, were markedly reduced (Fig. 1D, F and G). This is a reasonable
result due to the destruction of the microcirculatory structure
upstream of zone 3. To confirm whether these sinusoidal findings in
ALF were connected to hepatic macrophage activation, we generated
merged images comprising CD68 immunohistochemistry and
TRITC-dextran fluorescent images.
Macrophage activation has been observed in patients
with ALF (19,38) and is thought to be an important
aspect of the pathogenesis of ALF (4,5).
Nonetheless, the pathway from macrophage activation to massive
hepatocyte death remains unclear. As sinusoidal fibrin deposition
has been observed in patients with ALF (39,40), activated macrophages are thought
to induce sinusoidal fibrin deposition and cause hepatic
microcirculatory disorder, thereby resulting in parenchymal hypoxia
and necrosis (13–18). An essential role of activated
macrophages has been reported in parenchymal apoptosis through the
secretion of death receptor ligands, such as Fas ligand and TNF-α
(19,20). Evidence indicates that the same
stimuli can result in either the necrosis or the apoptosis of
hepatocytes (21). Bantel et
al (41) showed that serum
M30, which selectively recognizes a caspase cleaved neoepitope of
cytokeratin 18, reflected hepatocyte apoptosis in patients with
chronic hepatitis C. Rutherford et al (42,43) reported the findings of Bantel
et al (41) as an
effective predictor for the prognosis of ALF. Although the relative
contribution of necrosis and apoptosis to ALF remains controversial
(44), it is acceptable that a
pathway triggered by macrophage activation that results in
widespread hepatic hypoxia and subsequent massive hepatocyte death
would exist, and that apoptosis would be involved in this process.
Therefore, an investigation of the mechanisms of apoptosis in ALF
may prove useful in the development of novel treatment strategies
to protect liver cells from uncontrollable cell destruction.
In our rat model of ALF, CD68+ cells were
localized around pooling TRITC-dextran in zone 2, indicating that
infiltrated macrophages directly caused SEC destruction in that
area (Fig. 2F). The number of
infiltrated macrophages was most abundant in zone 2, followed by
zone 3, and then zone 1 (Fig.
2G). The number of apoptotic hepatocytes was most abundant in
zone 3, followed by zone 2 then zone 1 (Fig. 3E). The difference in the
distribution of macrophages and apoptotic cells strongly suggests
that the activation of an apoptotic pathway in zone 3 may be
promoted by factors other than macrophages.
Apoptosis is induced by the sequential activation of
caspases. This activation can be triggered by the activation of
death receptors located on cell membranes (extrinsic pathway), and
by cellular stress, i.e., endoplasmic reticulum stress or oxidative
stress (intrinsic pathway) (21).
Among the possible enzymatic systems contributing to the oxidative
stress of hepatocytes (xanthine oxidoreductase, cyclooxygenase,
endothelial nitric oxide synthases, cytochrome P450 monoxygenases
and NOX) (45), NOX is a major
source of cellular ROS (22–24). ROS generation by NOX has been
considered to occur only in phagocytes. Six homologues of phagocyte
catalytic NOX (NOX2) have been isolated: NOX1, NOX3, NOX4, NOX5,
DUOX1 and DUOX2 (46). NOX4 has
been shown to have unique characteristics compared with other NOX
homologues. NOX4 requires no cytosolic component for ROS-producing
activity and produces large amounts of ROS constitutively (47,48). NOX4 is expressed in fairly
ubiquitous organs (49) and its
expression levels are generally higher than those of other NOX
homologues. NOX4 has therefore been suggested to be an inducible
NOX (25). NOX4 has been shown to
play a crucial role in the pathophysiology of cardiovascular
diseases (23), diabetic
complications (50) and fibrosis
(51–53). In the liver, NOX4 is primarily
expressed in hepatocytes, stellate cells and endothelial cells
(54), and plays a pivotal role
in the cellular senescence of hepatocytes (55), hepatitis C virus-induced ROS
generation (56,57) and liver fibrosis (58–62).
Recently, NOX4 was found to be a target of HIF-1α, a
master regulator of the cellular response to hypoxia (63), and therefore was suggested to be
involved in cellular destruction in ALF. We hypothesized that
intrahepatic microcirculatory disorder in the livers of rats with
ALF may cause parenchymal hypoxia, and that the consequent
activation of HIF1-α would upregulate NOX4, leading to the
accumulation of oxidative stress and resultant hepatocyte apoptosis
via an intrinsic pathway. In this study, zone 1 was almost normoxic
and zone 3 was highly hypoxic (Fig.
4D). The hypoxic area was under impaired intrahepatic
microcirculation (Fig. 4F) in the
livers of rats with ALF. The results from immunohistochemistry for
HIF1-α (Fig. 4G and H), 4-HNE
(Fig. 4I and J) and NOX4
(Fig. 4K and L) suggested that
their distributions were in accordance with those for pimonidazole.
This indicates that oxidative stress may be induced by
hypoxia-dependent NOX4 activation.
For a more precise evaluation of the localization of
macrophages and NOX4-expressing cells in conjunction with that of
apoptotic hepatocytes, we used simultaneous immunohistochemical
staining. The majority of the apoptotic hepatocytes in zone 2 were
localized adjacent to macrophages (Fig. 5B), but were not colocalized with
NOX4-positive cells (Fig. 5E).
The greatest portion of apoptotic hepatocytes in zone 3 was not
localized adjacent to macrophages (Fig. 5C), but colocalized with
NOX4-positive cells (Fig. 5F).
These results indicated that hepatocyte apoptosis in zones 2 and 3
of our rat model of ALF was triggered mainly by activated
macrophages and hypoxia, respectively.
To the best of our knowledge, ours is the first
study to suggest that NOX4 upregulation may be involved in
hepatocyte apoptosis during the progression of ALF. We recognize,
however, that this study is based on histological observations only
in one experimental animal model with no intervention. To date,
NOX4-specific small-molecule inhibitors are not readily available
(64,65), and further studies involving
genetic interventions of NOX4 in an ALF model are required.
In conclusion, the intravenous injection of
TRITC-dextran clearly revealed intrahepatic microcirculation in
FFPE sections, and demonstrated intrahepatic microcirculatory
disorder in the livers of rats with ALF. This method enables the
simultaneous examination of microcirculation and
immunohistochemistry to be conducted on the same liver sections,
and is useful for investigating correlations between sinusoidal
blood flow and other physiological factors. Zonal differences in
hepatocyte apoptosis were revealed. The data from the present study
suggest that hypoxia in zone 3 and intrahepatic microcirculatory
disorder followed by HIF1-α upregulation may induce NOX4
upregulation, thus resulting in cellular oxidative stress.
Acknowledgements
We appreciate the technical support from the
Research Support Center, Graduate School of Medical Sciences,
Kyushu University.
Abbreviations:
|
ALF
|
acute liver failure
|
|
TRITC
|
tetramethylrhodamine
isothiocyanate
|
|
LPS
|
lipopolysaccharide
|
|
GalN
|
D-galactosamine
|
|
TNF-α
|
tumor necrosis factor-α
|
|
NOX
|
nicotinamide adenine dinucleotide
phosphate oxidase
|
|
ROS
|
reactive oxygen species
|
|
HIF1-α
|
hypoxia-inducible factor 1-α
|
|
4-HNE
|
4-hydroxy-2-nonenal
|
|
HRP
|
horseradish peroxidase
|
|
PBS
|
phosphate-buffered saline
|
|
H&E
|
hematoxylin and eosin
|
|
DAB
|
3,3′-diaminobenzidine
tetrahydrochloride
|
|
ECL
|
enhanced chemiluminescence
|
|
FFPE
|
formaldehyde-fixed
paraffin-embedded
|
|
SEC
|
sinusoidal endothelial cell
|
References
|
1
|
Lee WM, Squires RH, Nyberg SL, Doo E and
Hoofnagle JH: Acute liver failure: Summary of a workshop.
Hepatology. 47:1401–1415. 2008.PubMed/NCBI
|
|
2
|
Marudanayagam R, Shanmugam V, Gunson B, et
al: Aetiology and outcome of acute liver failure. HPB (Oxford).
11:429–434. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Oketani M, Ido A, Nakayama N, et al:
Etiology and prognosis of fulminant hepatitis and late-onset
hepatic failure in japan: Summary of the annual nationwide survey
between 2004 and 2009. Hepatol Res. 43:97–105. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Han DW: Intestinal endotoxemia as a
pathogenetic mechanism in liver failure. World J Gastroenterol.
8:961–965. 2002.PubMed/NCBI
|
|
5
|
Antoniades CG, Berry PA, Wendon JA and
Vergani D: The importance of immune dysfunction in determining
outcome in acute liver failure. J Hepatol. 49:845–861. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
McCuskey RS, Urbaschek R, McCuskey PA and
Urbaschek B: In vivo microscopic studies of the responses of the
liver to endotoxin. Klin Wochenschr. 60:749–751. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Uhlmann S, Uhlmann D and Spiegel HU:
Evaluation of hepatic microcirculation by in vivo microscopy. J
Invest Surg. 12:179–193. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Klintman D, Schramm R, Menger MD and
Thorlacius H: Leukocyte recruitment in hepatic injury:
selectin-mediated leukocyte rolling is a prerequisite for
CD18-dependent firm adhesion. J Hepatol. 36:53–59. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Eipel C, Kidess E, Abshagen K, et al:
Antileukoproteinase protects against hepatic inflammation, but not
apoptosis in the response of D-galactosamine-sensitized mice to
lipopolysaccharide. Br J Pharmacol. 151:406–413. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roller J, Laschke MW, Scheuer C and Menger
MD: Heme oxygenase (HO)-1 protects from lipopolysaccharide
(LPS)-mediated liver injury by inhibition of hepatic leukocyte
accumulation and improvement of microvascular perfusion.
Langenbecks Arch Surg. 395:387–394. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ito Y, Bethea NW, Abril ER and McCuskey
RS: Early hepatic microvascular injury in response to acetaminophen
toxicity. Microcirculation. 10:391–400. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lawson JA, Fisher MA, Simmons CA, Farhood
A and Jaeschke H: Parenchymal cell apoptosis as a signal for
sinusoidal sequestration and transendothelial migration of
neutrophils in murine models of endotoxin and Fas-antibody-induced
liver injury. Hepatology. 28:761–767. 1998. View Article : Google Scholar
|
|
13
|
Fujiwara K, Ogata I, Ohta Y, et al:
Intravascular coagulation in acute liver failure in rats and its
treatment with antithrombin III. Gut. 29:1103–1108. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yamada S, Ogata I, Hirata K, Mochida S,
Tomiya T and Fujiwara K: Intravascular coagulation in the
development of massive hepatic necrosis induced by
Corynebacterium parvum and endotoxin in rats. Scand J
Gastroenterol. 24:293–298. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Mochida S, Ohno A, Arai M, Tamatani T,
Miyasaka M and Fujiwara K: Role of adhesion molecules in the
development of massive hepatic necrosis in rats. Hepatology.
23:320–328. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Miyazaki M, Kato M, Tanaka M, et al:
Antithrombin III injection via the portal vein suppresses liver
damage. World J Gastroenterol. 18:1884–1891. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hirata K, Ogata I, Ohta Y and Fujiwara K:
Hepatic sinusoidal cell destruction in the development of
intravascular coagulation in acute liver failure of rats. J Pathol.
158:157–165. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ding JW, Ning Q, Liu MF, et al: Fulminant
hepatic failure in murine hepatitis virus strain 3 infection:
tissue-specific expression of a novel fgl2 prothrombinase. J Virol.
71:9223–9230. 1997.PubMed/NCBI
|
|
19
|
Mita A, Hashikura Y, Tagawa Y, Nakayama J,
Kawakubo M and Miyagawa S: Expression of Fas ligand by hepatic
macrophages in patients with fulminant hepatic failure. Am J
Gastroenterol. 100:2551–2559. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Streetz K, Leifeld L, Grundmann D, et al:
Tumor necrosis factor α in the pathogenesis of human and murine
fulminant hepatic failure. Gastroenterology. 119:446–460. 2000.
|
|
21
|
Malhi H and Gores GJ: Cellular and
molecular mechanisms of liver injury. Gastroenterology.
134:1641–1654. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Touyz RM, Briones AM, Sedeek M, Burger D
and Montezano AC: NOX isoforms and reactive oxygen species in
vascular health. Mol Interv. 11:27–35. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen F, Haigh S, Barman S and Fulton DJ:
From form to function: the role of Nox4 in the cardiovascular
system. Front Physiol. 3:4122012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Katsuyama M: NOX/NADPH oxidase, the
superoxide-generating enzyme: its transcriptional regulation and
physiological roles. J Pharmacol Sci. 114:134–146. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Brown DI and Griendling KK: Nox proteins
in signal transduction. Free Radic Biol Med. 47:1239–1253. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Arteel GE, Thurman RG, Yates JM and
Raleigh JA: Evidence that hypoxia markers detect oxygen gradients
in liver: pimonidazole and retrograde perfusion of rat liver. Br J
Cancer. 72:889–895. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Arteel GE, Iimuro Y, Yin M, Raleigh JA and
Thurman RG: Chronic enteral ethanol treatment causes hypoxia in rat
liver tissue in vivo. Hepatology. 25:920–926. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhong Z, Arteel GE, Connor HD, et al:
Binge drinking disturbs hepatic microcirculation after
transplantation: prevention with free radical scavengers. J
Pharmacol Exp Ther. 290:611–620. 1999.
|
|
29
|
Rosmorduc O, Wendum D, Corpechot C, et al:
Hepatocellular hypoxia-induced vascular endothelial growth factor
expression and angiogenesis in experimental biliary cirrhosis. Am J
Pathol. 155:1065–1073. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bardag-Gorce F, French BA, Li J, et al:
The importance of cycling of blood alcohol levels in the
pathogenesis of experimental alcoholic liver disease in rats.
Gastroenterology. 123:325–335. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Corpechot C, Barbu V, Wendum D, et al:
Hepatocyte growth factor and c-Met inhibition by hepatic cell
hypoxia: a potential mechanism for liver regeneration failure in
experimental cirrhosis. Am J Pathol. 160:613–620. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Deng X, Luyendyk JP, Zou W, et al:
Neutrophil interaction with the hemostatic system contributes to
liver injury in rats cotreated with lipopolysaccharide and
ranitidine. J Pharmacol Exp Ther. 322:852–861. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chaudhuri S, McCullough SS, Hennings L, et
al: Acetaminophen hepatotoxicity and HIF-1α induction in
acetaminophen toxicity in mice occurs without hypoxia. Toxicol Appl
Pharmacol. 252:211–220. 2011.
|
|
34
|
Vollmar B, Wolf B, Siegmund S, Katsen AD
and Menger MD: Lymph vessel expansion and function in the
development of hepatic fibrosis and cirrhosis. Am J Pathol.
151:169–175. 1997.PubMed/NCBI
|
|
35
|
Sun Z, Wang X, Deng X, et al: Beneficial
effects of lexipafant, a PAF antagonist on gut barrier dysfunction
caused by intestinal ischemia and reperfusion in rats. Dig Surg.
17:57–65. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Takenaka K, Sakaida I, Yasunaga M and
Okita K: Ultrastructural study of development of hepatic necrosis
induced by TNF-alpha and D-galactosamine. Dig Dis Sci. 43:887–892.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Le Bail B, Bioulac-Sage P, Senuita R,
Quinton A, Saric J and Balabaud C: Fine structure of hepatic
sinusoids and sinusoidal cells in disease. J Electron Microsc Tech.
14:257–282. 1990.PubMed/NCBI
|
|
38
|
Kotoh K, Kato M, Kohjima M, Nakamuta M and
Enjoji M: A new treatment strategy for acute liver failure. World J
Hepatol. 2:395–400. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Levy GA, Liu M, Ding J, et al: Molecular
and functional analysis of the human prothrombinase gene (HFGL2)
and its role in viral hepatitis. Am J Pathol. 156:1217–1225. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Marsden PA, Ning Q, Fung LS, et al: The
Fgl2/fibroleukin prothrombinase contributes to immunologically
mediated thrombosis in experimental and human viral hepatitis. J
Clin Invest. 112:58–66. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Bantel H, Lugering A, Heidemann J, et al:
Detection of apoptotic caspase activation in sera from patients
with chronic HCV infection is associated with fibrotic liver
injury. Hepatology. 40:1078–1087. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rutherford AE, Hynan LS, Borges CBS, et
al: Serum apoptosis markers in acute liver failure: a pilot study.
Clin Gastroenterol Hepatol. 5:1477–1483. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Rutherford A, King LY, Hynan LS, et al:
Development of an accurate index for predicting outcomes of
patients with acute liver failure. Gastroenterology. 143:1237–1243.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Malhi H, Gores GJ and Lemasters JJ:
Apoptosis and necrosis in the liver: A tale of two deaths?
Hepatology. 43(Suppl 1): S31–S44. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
De Minicis S, Bataller R and Brenner DA:
NADPH oxidase in the liver: defensive, offensive, or fibrogenic?
Gastroenterology. 131:272–275. 2006.
|
|
46
|
Bedard K and Krause KH: The NOX family of
ROS-generating NADPH oxidases: physiology and pathophysiology.
Physiol Rev. 87:245–313. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Martyn KD, Frederick LM, von Loehneysen K,
Dinauer MC and Knaus UG: Functional analysis of Nox4 reveals unique
characteristics compared to other NADPH oxidases. Cell Signal.
18:69–82. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Serrander L, Cartier L, Bedard K, et al:
NOX4 activity is determined by mRNA levels and reveals a unique
pattern of ROS generation. Biochem J. 406:105–114. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Krause KH: Tissue distribution and
putative physiological function of NOX family NADPH oxidases. Jpn J
Infect Dis. 57:S28–S29. 2004.PubMed/NCBI
|
|
50
|
Meng D, Mei A, Liu J, et al: NADPH oxidase
4 mediates insulin-stimulated HIF-1α and VEGF expression, and
angiogenesis in vitro. PLoS One. 7:e483932012.PubMed/NCBI
|
|
51
|
Hecker L, Vittal R, Jones T, et al: NADPH
oxidase-4 mediates myofibroblast activation and fibrogenic
responses to lung injury. Nat Med. 15:1077–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Barnes JL and Gorin Y: Myofibroblast
differentiation during fibrosis: role of NAD(P)H oxidases. Kidney
Int. 79:944–956. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sedeek M, Callera G, Montezano A, et al:
Critical role of Nox4-based NADPH oxidase in glucose-induced
oxidative stress in the kidney: implications in type 2 diabetic
nephropathy. Am J Physiol Renal Physiol. 299:F1348–F1358. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Reinehr R, Becker S, Eberle A,
Grether-Beck S and Haussinger D: Involvement of NADPH oxidase
isoforms and Src family kinases in CD95-dependent hepatocyte
apoptosis. J Biol Chem. 280:27179–27194. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Senturk S, Mumcuoglu M, Gursoy-Yuzugullu
O, Cingoz B, Akcali KC and Ozturk M: Transforming growth
factor-beta induces senescence in hepatocellular carcinoma cells
and inhibits tumor growth. Hepatology. 52:966–974. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Boudreau HE, Emerson SU, Korzeniowska A,
Jendrysik MA and Leto TL: Hepatitis C virus (HCV) proteins induce
NADPH oxidase 4 expression in a transforming growth factor
beta-dependent manner: a new contributor to HCV-induced oxidative
stress. J Virol. 83:12934–12946. 2009. View Article : Google Scholar
|
|
57
|
de Mochel NS, Seronello S, Wang SH, et al:
Hepatocyte NAD(P)H oxidases as an endogenous source of reactive
oxygen species during hepatitis C virus infection. Hepatology.
52:47–59. 2010.PubMed/NCBI
|
|
58
|
Carmona-Cuenca I, Roncero C, Sancho P, et
al: Upregulation of the NADPH oxidase NOX4 by TGF-beta in
hepatocytes is required for its pro-apoptotic activity. J Hepatol.
49:965–976. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Caja L, Sancho P, Bertran E,
Iglesias-Serret D, Gil J and Fabregat I: Overactivation of the
MEK/ERK pathway in liver tumor cells confers resistance to
TGF-{beta}-induced cell death through impairing up-regulation of
the NADPH oxidase NOX4. Cancer Res. 69:7595–7602. 2009.PubMed/NCBI
|
|
60
|
Carmona-Cuenca I, Herrera B, Ventura JJ,
Roncero C, Fernandez M and Fabregat I: EGF blocks NADPH oxidase
activation by TGF-beta in fetal rat hepatocytes, impairing
oxidative stress, and cell death. J Cell Physiol. 207:322–330.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Sancho P, Bertran E, Caja L,
Carmona-Cuenca I, Murillo MM and Fabregat I: The inhibition of the
epidermal growth factor (EGF) pathway enhances TGF-beta-induced
apoptosis in rat hepatoma cells through inducing oxidative stress
coincident with a change in the expression pattern of the NADPH
oxidases (NOX) isoforms. Biochim Biophys Acta. 1793:253–263. 2009.
View Article : Google Scholar
|
|
62
|
Sancho P, Mainez J, Crosas-Molist E, et
al: NADPH oxidase NOX4 mediates stellate cell activation and
hepatocyte cell death during liver fibrosis development. PLoS One.
7:e452852012. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Diebold I, Petry A, Hess J and Görlach A:
The NADPH oxidase subunit NOX4 is a new target gene of the
hypoxia-inducible factor-1. Mol Biol Cell. 21:2087–2096. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Borbely G, Szabadkai I, Horvath Z, et al:
Small-molecule inhibitors of NADPH oxidase 4. J Med Chem.
53:6758–6762. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Cifuentes-Pagano E, Csanyi G and Pagano
PJ: NADPH oxidase inhibitors: a decade of discovery from Nox2ds to
HTS. Cell Mol Life Sci. 69:2315–2325. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Henriques U: Histological technique in
routine histopathology. An opinion. Pathol Res Pract. 171:417–422.
1981. View Article : Google Scholar : PubMed/NCBI
|














