Introduction
Breast cancer is the leading diagnosed cancer and
cause of cancer mortality in females worldwide. Breast cancer is
responsible for ~23% of total new cancer cases and 14% of total
cancer deaths in recent years in women (1).
DNA methylation in cancer has received attention as
it has been shown to participate in the complex multistage process
of malignant tumor emergence. Aberrant promoter methylation in CpG
islands involves DNA methyltransferases (Dnmts) transfer of methyl
groups from S-adenosyl-L-methionine to the fifth carbon position of
the cytosines in 5′-CpG-3′ dinucleotides (2). Hypermethylation may suppress gene
transcription and reduce the stability of the genome by recruiting
a complex containing transcriptional corepressors and histone
deacetylases. This likely plays a crucial role in the inactivation
of tumor suppressor genes, which is a step in tumorigenesis
(3,4).
As a candidate tumor suppressor gene that is
associated with 90% of sporadic breast cancers (5), RhoBTB2 was cloned by Hamaguchi et
al using representational difference analysis (RDA) (6). RhoBTB2 is located on chromosome 8p,
which is a hotspot region where many breast cancer tumor suppressor
genes, including NGR1, FEZ1/LZTS1 and 14-3-3σ (7–10),
become inactivated by hypermethylation and their silencing
contributes to breast tumor development. Aberrant methylation of
RhoBTB2 was also found to reduce RhoBTB2 expression in bladder
cancers (11). Results of the
study by Fu et al revealed that mutations in the RhoBTB2
promoter and the seventh exon were seldom identified in Chinese
patients, and are not associated with the risk of breast cancer as
it is not a frequent mechanism of inactivation (12). Findings by those authors are
consistent with results by Knowles et al suggesting other
mechanisms may be involved, such as methylation, which are more
common than mutations in RhoBTB2 (4), which are responsible for the loss of
expression. CpGplot (http://www.ebi.ac.uk/emboss/cpgplot) was used to
examine the RhoBTB2 promoter region and CpG islands were
identified.
In this study, we determined the methylation status
of RhoBTB2 and the mRNA expression in breast cancer tissues by nMSP
and quantitative reverse transcription PCR (qRT)-PCR. We also
correlated the methylation changes with the transcript expression
and correlated results with clinicopathological characteristics to
investigate CpG methylation and the role of RhoBTB2 in breast
cancer.
Materials and methods
Tissues
Samples were collected after obtaining informed
consent from 50 female patients, who were between 33 and 78 years
of age (average age, 51.3±11.4 years), and who underwent surgery
due to breast cancer at the Union Hospital, Fuzhou, China, between
September 2010 and April 2011. All of the cases were definitively
diagnosed by pathology; including 46 infiltrating ductal carcinomas
(IDC), two infiltrating lobular carcinoma (ILC), and two ductal
carcinoma in situ (DCIS). None of the patients had received
neoadjuvant therapy. Primary tumor samples and corresponding normal
breast tissues, taken 5 cm from the cancer margin, were obtained.
Samples were immediately snap-frozen in liquid nitrogen after
resection and stored at −80°C overnight. The rest of the tumor was
examined by routine histopathology and immunohistochemistry in the
Pathology Department of Union Hospital, Fuzhou, China and the
clinical data, including age of onset, tumor size, lymph node
metastasis status of the patient, the histology grade, TNM staging,
sex hormone and Her-2 levels, and the p53 gene status of the tumor
were cataloged. The study was approved by the Ethics Committee of
the Union Hospital, Fuzhou, China.
qRT-PCR and semiquantitative RT-PCR
Total RNA from tissues was extracted by TRIzol
reagent (Takara, Shiga, Japan), and quantified with GeneQuant Pro
(Amersham Biosciences, Pittsburg, PA, USA). Samples with a ratio of
OD280/260 between 1.9 and 2.0 were processed further after total
RNA was confirmed to be without degradation by agarose gel
electrophoresis. The samples were stored at −80°C. Reverse
transcription of RNA was performed in a total volume of 20 μl
reaction mixture containing 2 μg RNA, in accordance with the
protocol for reverse transcriptase reagent M-MLV (Bioteke, Beijing,
China). cDNA products were amplified using primers specific for
RhoBTB2 and β-actin. β-actin was used for normalization of the
quantity of cDNA.
RhoBTB2 primers for quantitative PCR were designed
by Sangon Biotech (Shanghai, China), while the primers for PCR were
obtained from previous studies (11,13); all primers were synthesized by
Sangon. The primer sequences are presented in Table I, and quantitative PCR reaction
systems are presented in Table
II. The amplification conditions were as follows: 50°C for 2
min, 95°C for 2 min, then 30 cycles of 95°C for 15 sec, 60°C for 30
sec and 72°C for 30 sec.
 | Table ISummary of primer sequences for
PCR. |
Table I
Summary of primer sequences for
PCR.
| Gene name | Primer sequence
(5′→3′) | Product size
(bp) |
|---|
| β-actin (for both
real-time PCR and semiquantitative PCR) | F:
TCACCCACACTGTGCCCATCTACGA | 295 |
| R:
CAGCGGAACCGCTCATTGCCAATGG | 295 |
| RhoBTB2 (for
real-time PCR) | F:
ATGTGGTGGTTCTGTGCTTCT | 209 |
| R:
GGGCAGGATTTCATTAGGTTT | 209 |
| RhoBTB2 (for
semiquantitative PCR) | F:
TGTGGGCTCAGAGCTCAGGAGT | 316 |
| R:
CTGTAGAGGGCAGCATACGCGT | 316 |
| Table IIQuantitative PCR reaction system. |
Table II
Quantitative PCR reaction system.
| Component | Volume |
|---|
| 2X PCR mix | 7.5 μl |
| Forward primer (10
pmol/μl) | 0.5 μl |
| Reverse primer (10
pmol/μl) | 0.5 μl |
| ROX | 0.05 μl |
| cDNA template | 0.5 μl |
| ddH2O | up to 15 μl |
qRT-PCR was performed using a Platinum®
SYBR®-Green qPCR SuperMix UDG (Invitrogen, Carlsbad, CA,
USA) and the ABI 7500 Sequence Detection System (Applied
Biosystems, USA). Optimization of amplification conditions was
carried out according to the manufacturer’s instructions. Each
experimental reaction was performed in triplicate and the relative
expression was calculated using the ΔΔCt method (14). The relative levels of RhoBTB2 mRNA
in the breast cancer tissues that were normalized to the internal
control β-actin by subtraction were calculated as ΔCt (cancer
tissue), and the levels in normal tissues as ΔCt (normal tissue).
The RhoBTB2 mRNA score in each tissue pair was defined as follows:
ΔCt of cancer tissue - the ΔCt of normal tissue yielded a ΔΔCt
value, which was used to calculate the result of
2(−ΔΔCt). RhoBTB2 mRNA was upregulated when the score
was >1.0 and downregulated when the score was <1.0.
Semiquantitative PCR was carried out in a PTC-200
thermal cycler (MJ Research, Watertown, MA, USA). Amplification was
carried out in a 10 μl reaction mixture containing 0.5 μl of cDNA
sample, 5 μl of 2X PCR mix (Bioteke), and 0.5 μl of the primers
(the final concentration of each pair was 10 pmol/μl), with 3.5 μl
of deionized water. The amplification program was: 94°C for 5 min,
then 30 cycles of 94°C for 30 sec, 64°C for 30 sec, 72°C for 45
sec, followed by a final extension at 72°C for 10 min. PCR products
were examined with Marker (MBI Fermentas, Vilnius, Lithuania) on 2%
agarose gel electrophoresis (100 V, 70 mA) and visualized under UV
illumination (Syngene, Cambridge, UK). RhoBTB2 mRNA quantity in
each sample was represented and analyzed in the form of Gray
Intensity of RhoBTB2/β-actin, then the relative level of RhoBTB2
mRNA in normal breast tissue was subtracted from the relative level
in the tumor. RhoBTB2 mRNA was upregulated when the score was >0
and downregulated when the score was <0.
DNA preparation and bisulfate
modification
Genomic DNA was extracted from ~50–100 mg of fresh
tissue, using an animal tissue genomic DNA isolation kit (Bioteke)
according to the manufacturer’s instructions. DNA was quantified
with GeneQuant Pro (Amersham Biosciences). The samples with a ratio
of OD280/260 between 1.7 and 1.9 were accepted, then bisulfate
modification of 200 ng DNA was performed using protocols from the
Methylamp™ One-Step DNA Modification kit (Epigentek, Brooklyn, NY,
USA).
Nested methylation-specific PCR
analysis
Nested primers for RhoBTB2 were synthesized by
Invitrogen using primer sequences obtained from a previous study
(11). The nMSP primer sequences
are presented in Table III.
| Table IIISummary of primer sequences for
nMSP. |
Table III
Summary of primer sequences for
nMSP.
| Gene name | Primer sequence
(5′→3′) | Product size
(bp) |
|---|
| RhoBTB2 (outside
primer) | F:
GGTGGTTTATTTGGTGATATTG | 439 |
| R:
CCTACAACCTTACCTCCTAACAC | 439 |
| RhoBTB2 (M, inside
primer) | F:
GCGAGTTGGTATGTTATGTC | 144 |
| R:
TAATCTTACCCACGACGTTA | 144 |
| RhoBTB2 (U, inside
primer) | F:
GGTGAGTTGGTATGTTATGTT | 144 |
| R: CTAATCTTACCCAC
AACATTA | 144 |
First round amplifications in 15 μl reactions
included: 7.5 μl of 2X PCR mix (Bioteke, Beijing, China), 10 pmol
of outsider primer and 0.5 μl of 200 ng modified DNA, with
deionized water to make up the volume, using the following cycle
parameters: 94°C for 5 min, then 35 cycles at 94°C for 30 sec, 56°C
for 45 sec and 72°C for 45 sec, followed by a final extension at
72°C for 5 min. Aliquots of 0.5 μl PCR products were subjected to a
second round of amplification.
Second round amplification was carried out using
methylated and unmethylated primers in 15 μl reactions, with the
following cycle parameters: 94°C for 5 min, then 30 cycles at 94°C
for 30 sec, 50°C for 45 sec and 72°C for 45 sec, followed by a
final extension at 72°C for 5 min.
Then, 15 μl of the final PCR products were confirmed
by 2% agarose gel electrophoresis at 100 V for 30 min, and
visualized under UV illumination. DNA from the peripheral blood of
healthy adults, treated and untreated with DNA methyltransferase,
was used as a positive control for methylated and unmethylated DNA.
H2O instead of DNA was used in the negative control in
each set of PCR experiments.
Statistical analysis
Calculations were carried out using SPSS11.0
statistical software (SPSS, Inc., Chicago, IL, USA). The numerical
data correlation among mRNA expression and epigenetic events was
analyzed by the Chi-Square (χ2) test. Fisher’s exact
test or continuity correction was used to test the statistical
significance of the observed differences between the methylation
status or mRNA expression and clinical parameters as appropriate. A
comparison of mRNA expression in cancer and normal tissues was
performed using the Paired-samples t-test. P-values presented were
two-sided, and P<0.05 was considered statistically
significant.
Results
RhoBTB2 mRNA expression in breast
cancer
In the 50 pairs of tumor and control tissues,
RhoBTB2 mRNA in 29/50 breast cancer tumor samples (58%) was
reduced, compared to the corresponding normal tissues. The average
RhoBTB2 expression in breast carcinoma tissues was significantly
lower than that in control tissues. The gray intensity value was
0.19±0.01 in tumors vs. 0.25±0.01 in normal tissues and the ΔCt
value was 5.74±0.45 in tumors vs. 3.07±0.12 in normal tissues
(P<0.05). No significant relationship was observed between
RhoBTB2 mRNA expression and the patient age of onset, tumor size,
lymph node involvement, TNM staging, tumor grade, sex hormones or
Her-2 levels, or p53 protein expression (P>0.05) (Table IV).
| Table IVCorrelation between
clinicopathological factors and RhoBTB2 mRNA levels. |
Table IV
Correlation between
clinicopathological factors and RhoBTB2 mRNA levels.
| | RhoBTB2 mRNA | |
|---|
| |
| |
|---|
| Variables | N | Down Up | P-value | |
|---|
| Age (years) | | | | |
| <50 | 24 | 13 | 11 | 0.598 |
| ≥50 | 26 | 16 | 10 | |
| Tumor size (cm) | | | | |
| ≤2 | 27 | 16 | 11 | 0.845 |
| >2 | 23 | 13 | 10 | |
| Lymph node
involvement | | | | |
| No | 26 | 15 | 11 | 0.441 |
| Yes | 24 | 14 | 10 | |
| TNM staging | | | | |
| I–II | 39 | 24 | 15 | 0.543 |
| III–IV | 11 | 5 | 6 | |
| Tumor gradea | | | | |
| I–II | 36 | 19 | 17 | 0.230 |
| III | 14 | 10 | 4 | |
| ER status | | | | |
| Negative | 14 | 10 | 4 | 0.230 |
| Positive | 36 | 19 | 17 | |
| PR status | | | | |
| Negative | 16 | 12 | 4 | 0.095 |
| Positive | 34 | 17 | 17 | |
| Her-2 status | | | | |
| Negative | 33 | 18 | 15 | 0.479 |
| Positive | 11 | 8 | 3 | |
| p53 status | | | | |
| Negative | 18 | 10 | 8 | 0.354 |
| Positive | 26 | 18 | 8 | |
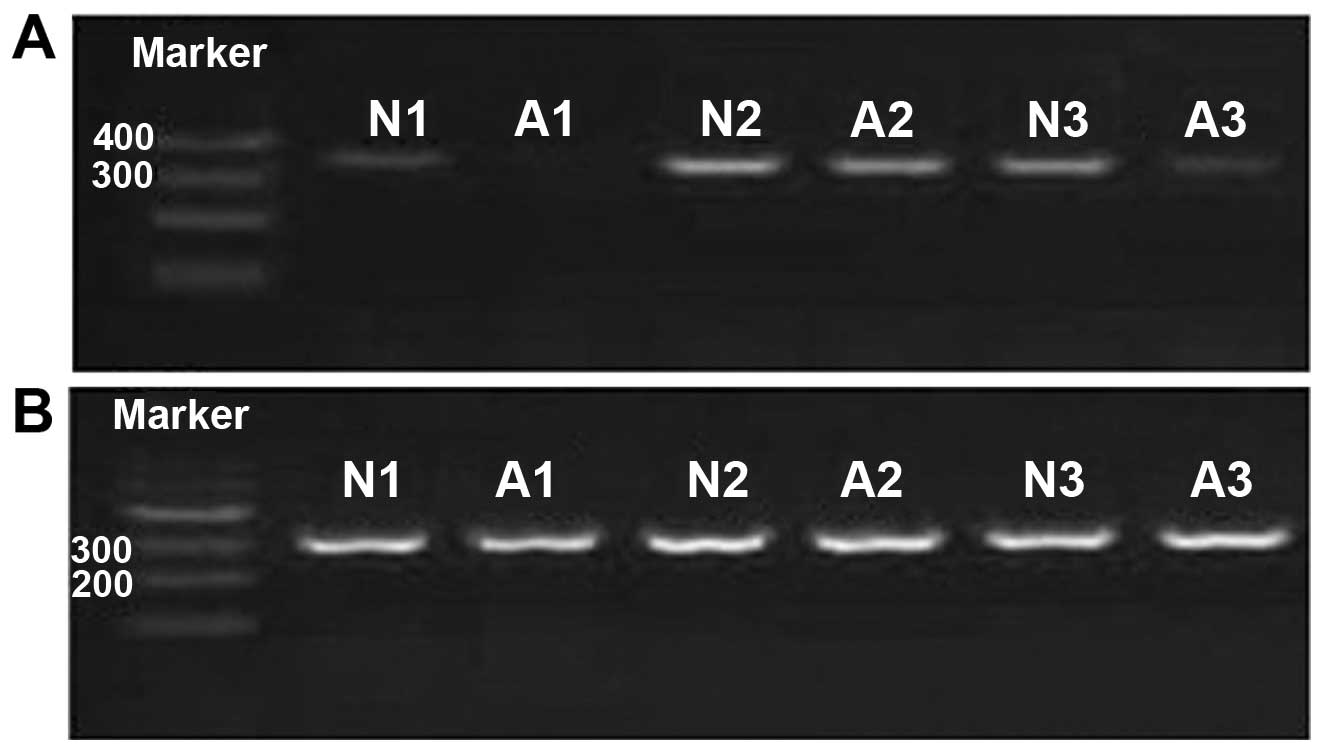
All the samples were assayed in triplicate,
including three assays for RhoBTB2 and three for β-actin. The
analysis of solubility curves demonstrated that the curves of
RhoBTB2 and β-actin presented a single peak, with the same DNA
melting temperature (Tm) and a sharp peak. No abnormal reaction
wave form was observed in other locations, which shows that the PCR
products were specific. The DNA amplification curves from the
target gene of the same sample were smooth, full, and repeatable.
In addition, 2% agarose gel electrophoresis verification (Fig. 1) showed the PCR products were pure
and consistent with the results of quantitative PCR.
Methylation profile of RhoBTB2 in breast
cancer
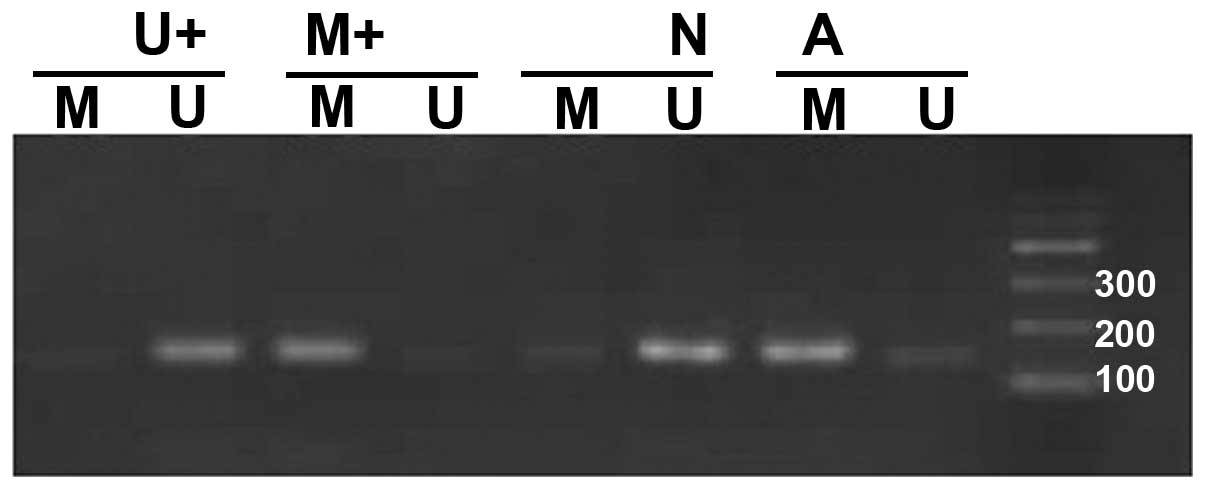
The determining standards for the methylated and
unmethylated primers were: in one sample the amplification with the
methylated and unmethylated primers was considered partial
methylation, amplifications with the methylated primer yielding
correct bands were considered full methylation, amplifications with
the unmethylated primer yielding correct bands were considered
methylation-negative. Both partial and full methylation were
considered methylation-positive.
Fifty pairs of samples with the best quality of
genomic DNA which had been previously examined for RhoBTB2 mRNA
expression were selected for methylation analysis.
Methylation-specific bands were detected in 26 tumor samples
(26/50, 52%). When we compared the methylation status in the tumors
(52% positive methylation) and corresponding adjacent normal
samples (0% positive methylation), the difference in the frequency
of methylation between them was significant (P<0.05) (Fig. 2).
We analyzed the relationship between the RhoBTB2
gene methylation status and clinicopathological characteristics of
patients (Table V). There was no
notable association between RhoBTB2 methylation and patient age of
onset, tumor size, lymph node involvement, TNM staging, tumor
grade, ER status, Her-2 expression, or p53 gene status (P>0.05).
There was a significant correlation of RhoBTB2 methylation with PR
status (P=0.026) and the ratio of RhoBTB2 methylation was higher in
PR− than in PR+ tissues, 75.0 vs. 41.2%,
indicating that the patients who were PR− were prone to
have RhoBTB2 methylation.
| Table VCorrelation between
clinicopathological factors and methylation status of RhoBTB2 in
breast cancer. |
Table V
Correlation between
clinicopathological factors and methylation status of RhoBTB2 in
breast cancer.
| | Methylation | |
|---|
| |
| |
|---|
| Variables | N | Yes | No | P-value |
|---|
| Age (years) | | | | |
| <50 | 24 | 12 | 12 | 0.786 |
| ≥50 | 26 | 14 | 12 | |
| Tumor size
(cm) | | | | |
| ≤2 | 27 | 16 | 11 | 0.266 |
| >2 | 23 | 10 | 13 | |
| Lymph node
involvement | | | | |
| No | 26 | 15 | 11 | 0.402 |
| Yes | 24 | 11 | 13 | |
| TNM staging | | | | |
| I–II | 39 | 22 | 17 | 0.240 |
| III–IV | 11 | 4 | 7 | |
| Tumor gradea | | | | |
| I–II | 36 | 17 | 19 | 0.278 |
| III | 14 | 9 | 5 | |
| ER status | | | | |
| Negative | 14 | 10 | 4 | 0.086 |
| Positive | 36 | 16 | 20 | |
| PR status | | | | |
| Negative | 16 | 12 | 4 | 0.026 |
| Positive | 34 | 14 | 20 | |
| Her-2 status | | | | |
| Negative | 33 | 16 | 17 | 0.162 |
| Positive | 11 | 8 | 3 | |
| p53 status | | | | |
| Negative | 18 | 7 | 11 | 0.139 |
| Positive | 26 | 16 | 10 | |
Relationship between RhoBTB2 methylation
and mRNA expression
The correlation of RhoBTB2 methylation with mRNA
expression is shown in Table VI.
There are 22 cases that were methylation-positive from the 29 cases
which had suppressed RhoBTB2 mRNA expression (22/29, 75.9%), while
only 4/21 cases were methylation-positive from the cases without
suppressed mRNA expression (19.0%). Compared with the normal
RhoBTB2 mRNA expression group, methylation was more frequent in the
low RhoBTB2 mRNA expression group, and the difference was
statistically significant (χ2 = 15.751, P<0.001).
Tumor tissue with hypermethylation of the gene tended to have lower
RhoBTB2 mRNA expression, and the suppression of RhoBTB2 expression
was observed in samples with gene methylation. This finding
suggests that methylation of RhoBTB2 significantly contributes to
the inhibition of transcription of the gene in breast cancer
tumors.
| Table VIRelationship between RhoBTB2
methylation and mRNA expression (n=50). |
Table VI
Relationship between RhoBTB2
methylation and mRNA expression (n=50).
| Promoter
methylation | |
|---|
|
| |
|---|
| mRNA
expression | Yes | No | Total |
|---|
| Low | 22 | 7 | 29 |
| Normal | 4 | 17 | 21 |
| Total | 26 | 24 | 50 |
Discussion
Aberrant DNA methylation plays a crucial role in the
pathogenesis of human cancer since it may cause epigenetic
inactivation in tumor suppressor genes in order to promote
tumorigenesis. In this study, we detected mRNA expression of
RhoBTB2 and the methylation status in breast cancer tissues and
correlated the transcript expression and methylation with
clinicopathological characteristics of the patients. RhoBTB2 mRNA
expression in breast carcinoma was significantly depressed compared
to the corresponding normal tissues (P<0.001), and the loss of
mRNA was found in significantly more samples with methylation of
the gene (χ2 = 15.751, P<0.001). Additionally, there
was a statistical correlation of the aberrant methylation changes
with PR-negative cancer tissues (P<0.05). To the best of our
knowledge, this is the first study on CpG methylation-mediated
transcriptional silencing of RhoBTB2 in breast cancer, which is a
significant event during the genesis of breast cancer.
RhoBTB2 is located on chromosome 8p, which is a
region of the chromosome with frequent abnormality in various types
of cancer. As a member of the RhoGTPases family, RhoBTB2 plays a
critical role in preventing the invasion and metastasis of
malignant tumor cells through cytoskeleton remodeling in order to
influence cell division, motility, contraction, and cytokinesis. It
is also involved in nerve growth, mitogenesis, membrane
trafficking, transcriptional activation, and cell growth control
such as proliferation and apoptosis through upregulation of the
E2F1 protein (15–18). Thus, inactivation of RhoBTB2 is
important in the development of breast cancer, and the study of its
methylation status is useful in the clarification of the causes of
breast cancer.
In the analysis of RhoBTB2 mRNA expression from 50
cases, there were 29 breast tumor samples that had low or loss of
expression, as the RhoBTB2 transcription in breast cancer was
greatly suppressed compared with normal tissues (P<0.001). The
result of semiquantitative PCR was consistent with the quantitative
PCR and the results were repeatable. According to the analysis of
ISH and semiquantitative PCR reported by Mao et al (19), the mRNA and protein levels of
RhoBTB2 in 60 breast cancer tissues was lower than that in the 30
benign breast lesions. In addition, follow-up observations of these
cases revealed that the survival rate was significantly higher in
RhoBTB2-positive patients than in RhoBTB2-negative patients,
suggesting that the expression of RhoBTB2 may be regarded as an
independent prognostic factor in breast carcinoma patients. As a
breast cancer tumor suppressor gene, the downregulation of RhoBTB2
expression is a biological index of poor prognosis for breast
cancer patients. In the RNA expression analysis in the study by
Hamaguchi et al, absence of RhoBTB2 was found in 58% (11/19)
of breast cancer tissues and 50% (7/14) of lung cancer tissues, but
normal expression was found in the corresponding normal tissues
(6). Knowles et al
(4) found that, RhoBTB2 mRNA was
decreased in 75% of bladder cancer cell lines. Compared with the
corresponding normal tissues, bladder cancer tissues had
significantly less RhoBTB2 mRNA, which was correlated with the
clinical TNM stage and histological grade, and a low RhoBTB2
expression was regarded as a poor prognostic indicator for bladder
cancer patients (11). However,
the expression of RhoBTB2 was not decreased in colon tumors or
other types of cancer (4),
suggesting that RhoBTB2 is a tissue-specific tumor suppressor
gene.
MSP results show that the total frequency of the
RhoBTB2 promoter methylation in tumor tissues is 52%, which was
significantly higher than that in the corresponding normal tissues
(P<0.05). In our breast cancer tissue samples, there were more
methylation-positive samples with downregulation of RhoBTB2 mRNA
than there were cases without decreased RhoBTB2 mRNA (75.9 vs.
19.0%, χ2 = 15.751, P<0.001). This suggests that
RhoBTB2 mRNA expression is associated with its methylation status,
and that the RhoBTB2 gene can be silenced by promoter methylation
in breast cancer, which may affect the development of breast
cancer. Jones and Takai (20)
reported that the hypermethylation of CpG islands in the DNA
promoter is the third mechanism of deactivation of an anti-oncogene
along with mutation and deletion, and even a unique one in some
cases. Aberrant methylation inhibits the transcription of genes and
abrogates gene expression, but does not alter the DNA sequence or
the gene product. This results in tumor suppressor gene silencing
and the stability of the genome decreases (21,22), promoting tumorigenesis and tumor
development. The RhoBTB2 gene is located on chromosome 8p21.3. In
this same region of 8p, several breast cancer-related tumor
suppressor genes, such as NGR1, FEZ1/LZTS1, and 14-3-3σ, also
exhibit expression abrogation by promoter methylation (7–10).
Previous investigations into mutations in the RhoBTB2 gene have
demonstrated that the occurrence of mutations was less than the
occurrence of reduced mRNA expression (12). Knowles et al (4) hypothesized that there was another
mechanism for RhoBTB2 silencing, for example, promoter methylation,
that was noted more frequently than mutations in the gene.
Recently, Shi et al (11)
used MSP and RT-PCR and found that the frequency of methylation in
CpG islands of the RhoBTB2 promoter is much higher in bladder
cancer tissues than in normal tissues. The RhoBTB2 mRNA level in
the tumor tissues with methylation is much lower than that in the
tissues without methylation. Hypermethylation of the RhoBTB2
promoter is therefore a significant mechanism of RhoBTB2
deactivation in bladder cancer. Findings of Mao et al
(19) demonstrated that the mRNA
levels of RhoBTB2 were consistent with the protein levels,
suggesting that RhoBTB2 expression was blocked at the transcription
level. Downregulation of these transcripts and silencing of the
promoter may be the primary mechanism of gene suppression, and our
results are consistent with the abovementioned studies.
Both methylated and unmethylated genes were
identified in several tumor samples, known as partly methylated,
and the samples were considered methylation-positive. In the study
by Herman et al on the sensitivity of MSP for detecting the
methylated alleles in lung cancer samples (23), 0.1% of P16 DNA had methylated
alleles, although the cells were always associated with normal
cells that masked the presence of methylated sites. Tumor tissues
consist of many normal cells, such as stromal, endothelial and
inflammatory cells that do not have CpG methylation and their DNA
may affect the results of the analysis. We suggest three reasons to
explain partly methylated samples: first, we cannot avoid sampling
normal interstitial cells within these tumor tissues. Second, DNA
methylation may be important in early stages of tumor development
(22) such as precancerous
lesions, and the tumor tissues contain cells at various stages of
development with methylation only occurring in the cells during the
early stages of development. Third, since human genes are diploid,
we cannot exclude the existence of different methylation status of
the two alleles. Fan et al (24) suggested that, there is decreased
and unstable aberrant methylation in part of the tumor, and less
methylation in an early tumor may represent a cancer cell subset
with an unstable methylation status of tumor suppressor gene CpG
islands. Methylation of CpG islands is a progressive process, with
every pyrimidine base in CpG islands becoming gradually methylated.
Some CpG islands are methylated early and then methylation expands
to more CpG islands. Thus, key genes evolve into a silent steady
state, and the gradual loss of function of these genes may be a
mechanism that avoids early death of abnormal cells. This allows
the evolution of an immortal tumor cell with its altered germplasm
(25).
In the analysis of clinicopathological
characteristics, RhoBTB2 methylation was observed preferentially in
samples that were PR-negative (P<0.05), but there was no notable
association between RhoBTB2 methylation and other
clinicopathological characteristics, such as age of onset, tumor
size, lymph node involvement, TNM staging, tumor grade, ER status,
Her-2 expression, or p53 gene status. This observation suggests
that hypermethylation of the RhoBTB2 promoter may be involved in
the initial stages of tumorigenesis. Moreover, the PR levels may be
affected by hypermethylation of RhoBTB2 and aberrant methylation of
RhoBTB2 is also important in the pathogenesis of certain types of
breast cancer. These findings suggest that RhoBTB2 methylation may
function as an early risk event for breast cancer in a
phase-specific and tissue-specific manner. In addition, the
clinicopathological characteristics analysis also revealed that
there was no significant correlation between loss of RhoBTB2
expression and the clinicopathological characteristics. Mao et
al (19) reported that the
loss of RhoBTB2 mRNA expression was associated with age of onset,
PR status and histopathological types but not with TNM staging,
lymph node involvement, or ER and Her-2 status in breast cancer.
Although our results are different from those of Mao et al,
there were differences in the studies. The samples investigated by
Mao et al were obtained from unpaired breast cancer tissues,
whereas, we examined paired samples. Patients included in this
study were from the Fujian region and there may be a difference in
the genetic character of populations from different regions.
Additionally, differences in laboratory and statistical methods may
also have influenced the results. Moreover, Bi et al
(5) compared differences in the
age at onset of breast cancer, lymph node involvement, TNM staging,
ER/PR, Her-2 status and survival time between groups, and showed no
marked difference in RhoBTB2 expression. However, data from a
larger sample may aid in obtaining better information on the
distribution of RhoBTB2 expression in breast cancer patients.
In summary, we provide new evidence that
hypermethylation of the RhoBTB2 promoter was an important mechanism
associated with inactivation of RhoBTB2 transcription in breast
cancer, which is important in the tumorigenesis and progression of
breast cancer. There was also a correlation between RhoBTB2
methylation and PR downregulation, and a detailed explanation of
the connection requires further investigation. At the present time,
testing for DNA methylation is a feasible assay for determining the
prognosis and for diagnoses by DNA-based biomarkers. Thus, our
study revealed that the risk of breast cancer was connected with
the expression of the RhoBTB2 gene, which is a new molecular target
for breast cancer diagnosis, therapy and prognosis. As DNA
methylation is reversible, the feasibility of a clinical
application of modifying methylation of the RhoBTB2 gene in order
to restore its antitumor function merits further investigation.
Acknowledgements
This study was supported by the Fujian Provincial
Natural Science Foundation of China (grant no. 2013J01313). We
gratefully thank Limin Chen, Lili Chen, Wenhui Guo, Jie Zhang,
Xueying Wu, Yuanjing Chen and Zhao Hu for providing invaluable
technical assistance and Stephen Brooks for English language
supervision.
References
|
1
|
Jemal A, Bray F, Center MM, Ferlay J, Ward
E and Forman D: Global cancer statistics. CA Cancer J Clin.
61:69–90. 2011. View Article : Google Scholar
|
|
2
|
Jeltsch A: Beyond Watson and Crick: DNA
methylation and molecular enzymology of DNA methyltransferases.
Chembiochem. 3:274–293. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baylin SB, Esteller M, Rountree MR,
Bachman KE, Schuebel K and Herman JG: Aberrant patterns of DNA
methylation, chromatin formation and gene expression in cancer. Hum
Mol Genet. 10:687–692. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Knowles MA, Aveyard JS, Taylor CF, Harnden
P and Bass S: Mutation analysis of the 8p candidate tumour
suppressor genes DBC2 (RHOBTB2) and LZTS1 in bladder cancer. Cancer
Lett. 225:121–130. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bi Y, Zheng G, Mao HT and Zhuo WS:
Anti-oncogene RhoBTB2 and breast cancer. Int J Surg. 34:177–181.
2007.
|
|
6
|
Hamaguchi M, Meth JL, von Klitzing C, et
al: DBC2, a candidate for a tumor suppressor gene involved in
breast cancer. Proc Natl Acad Sci USA. 99:13647–13652. 2002.
View Article : Google Scholar
|
|
7
|
Chua YL, Ito Y, Pole JC, et al: The NRG1
gene is frequently silenced by methylation in breast cancers and is
a strong candidate for the 8p tumour suppressor gene. Oncogene.
28:4041–4052. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Chen L, Zhu Z, Sun X, et al:
Down-regulation of tumor suppressor gene FEZ1/LZTS1 in breast
carcinoma involves promoter methylation and associates with
metastasis. Breast Cancer Res Treat. 116:471–478. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Umbricht CB, Evron E, Gabrielson E,
Ferguson A, Marks J and Sukumar S: Hypermethylation of 14-3-3 sigma
(stratifin) is an early event in breast cancer. Oncogene.
20:3348–3353. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ferguson AT, Evron E, Umbricht CB, et al:
High frequency of hypermethylation at the 14-3-3 sigma locus leads
to gene silencing in breast cancer. Proc Natl Acad Sci USA.
97:6049–6054. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Shi Y, Chen JY, Yang J, Li B, Chen ZH and
Xiao CG: DBC2 gene is silenced by promoter methylation in bladder
cancer. Urol Oncol. 26:465–469. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Fu GP, Liao SG, Wang N and Wang YJ: The
association between DBC2 gene mutation and breast cancer in Chinese
population. Tumor Clin Res. 29:575–577. 2009.
|
|
13
|
Huang J, Zheng DL, Qin FS, et al: Genetic
and epigenetic silencing of SCARA5 may contribute to human
hepatocellular carcinoma by activating FAK signaling. J Clin
Invest. 120:223–241. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
|
|
15
|
Van Aelst L and D’Souza-Schorey C: Rho
GTPases and signaling networks. Genes Dev. 11:2295–2322.
1997.PubMed/NCBI
|
|
16
|
Nielsen R, Bustamante C, Clark AG, et al:
A scan for positively selected genes in the genomes of humans and
chimpanzees. PLoS Biol. 3:e1702005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Stanelle J, Stiewe T, Theseling CC, Peter
M and Putzer BM: Gene expression changes in response to E2F1
activation. Nucleic Acids Res. 30:1859–1867. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
D’Souza SJ, Vespa A, Murkherjee S, Maher
A, Pajak A and Dagnino L: E2F-1 is essential for normal epidermal
wound repair. J Biol Chem. 277:10626–10632. 2002.
|
|
19
|
Mao H, Qu X, Yang Y, Zuo W, Bi Y, Zhou C,
Yin H, Deng B, Sun J and Zhang L: A novel tumor suppressor gene
RhoBTB2 (RHOBTB2): Frequent loss of expression in sporadic breast
cancer. Mol Carcinog. 49:283–289. 2010.PubMed/NCBI
|
|
20
|
Jones PA and Takai D: The role of DNA
methylation in mammalian epigenetics. Science. 293:1068–1070. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Singal R and Ginder GD: DNA methylation.
Blood. 93:4059–4070. 1999.PubMed/NCBI
|
|
22
|
Jones PA and Laird PW: Cancer-epigenetics
comes of age. Nat Genet. 21:163–167. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Herman JG, Graff JR, Myohanen S, Nelkin BD
and Baylin SB: Methylation-specific PCR: a novel PCR assay for
methylation status of CpG islands. Proc Natl Acad Sci USA.
93:9821–9826. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Fan X, Inda M, Tunon T and Castresana J:
Improvement of the methylation specific PCR technical conditions
for the detection of p16 promoter hypermethylation in small amounts
of tumor DNA. Oncol Rep. 9:181–184. 2002.PubMed/NCBI
|
|
25
|
Belinsky SA, Nikula KJ, Palmisano WA, et
al: Aberrant methylation of p16(INK4a) is an early event in lung
cancer and a potential biomarker for early diagnosis. Proc Natl
Acad Sci USA. 95:11891–11896. 1998. View Article : Google Scholar : PubMed/NCBI
|