Introduction
Atherosclerosis is a chronic immuno-inflammatory
disease with high morbidity and atherosclerosis-related
cardiovascular diseases are the leading cause of mortality
worldwide (1). The
destabilization and rupture of atherosclerotic plaques is the main
pathological basis of acute cardiovascular disease events without
effective treatments. Macrophages play a key role in each stage of
atherosclerosis (2). Stimulation
of high levels of oxidized low-density lipoprotein (ox-LDL), led to
the monocyte-derived macrophages become lipid-laden and are
eventually transformed into foam cells. A central feature of
atherosclerosis is the accumulation of foam cells in the lesion
and, notably macrophage recruitment into plaques is critical for,
and increases with, disease progression (3–5).
ox-LDL is also a potential inducer of cell apoptosis in
atherosclerosis. Previous studies have demonstrated that ox-LDL
induced apoptosis in a variety of tissues and cells, including
endothelial cells (ECs), vascular smooth muscle cells (VSMCs) and
macrophages (6–9). Apoptosis of macrophages and VSMCs in
atherosclerotic plaques is thought to lead to increased necrotic
core formation, inflammation, plaque rupture and atherothrombosis
(10,11). In human atherosclerotic plaques,
apoptosis of macrophages is detected during all stages, which
occurs more frequently compared with apoptosis of the VSMCs.
Accumulating evidence has shown that the phagocytic clearance of
apoptotic cells, or ‘efferocytosis’ in macrophages is effective in
the early stage of atherosclerosis, whereas efferocytosis in
advanced atherosclerosis becomes defective, which is causally
linked to the progression of atherosclerosis (12). Therefore, the enhancement in
efferocytosis by drugs or other methods in macrophages potentially
contributes to the inhibition of atherosclerotic plaques
progression and reduction of acute coronary events.
Results of recent studies on macrophage autophagy
have shown a novel pathway through which these cells contribute to
vascular disease (13–16). Autophagy may be a new target for
therapeutic utility in atherosclerosis. Originally described as
‘self-eating’ in the 1960s, autophagy is an evolutionarily
conserved controlled cellular catabolic process that mediates the
degradation of altered and damaged proteins and organelles. The
cellular symbol of autophagy is the formation of characteristic
double-membrane vesicles, known as autophagosomes. The origins of
this structure remain to be elucidated, although it may be
generated from multiple sources including the endoplasmic reticulum
(17,18), the outer mitochondrial membrane
(17,19), and the plasma membrane (20,21). The autophagosomes are targeted to
lysosomes to form single-membraned submicroscopic vesicles termed
autolysosomes with degradative capacity. The altered and damaged
proteins and organelles were contained in autolysosomes and were
eliminated by a series of lysosomal enzymes. Autophagy exerts a
protective effect in nutrients generating and maintaining survival
(22). Recent evidence suggested
that maintenance of basal autophagy in macrophages was useful in
the clearance of apoptotic and necrotic cells, which may enhance
the efferocytosis of apoptotic macrophages (23–25).
The sirtuins are a family of nicotinamide adenine
dinucleotide (NAD)-dependent deacetylases that have been linked to
the regulation of life span initially found in yeast cells.
Sirtuin1 (Sirt1) is the closest relative of yeast Sir2 in mammalian
cells which play important roles in multiple disease-related
pathways such as cell cycle regulation, cell apoptosis and
migration (26). Resveratrol
(3,4′,5-trihydroxy-trans-stilbene, RSV), a polyphenolic
phytoalexin, is a potent activator of Sirt1. Nicotinamide (NAM),
the precursor for the synthesis of NAD+, has been
recognized as an inhibitor of Sirt1. Previous results indicated
that RSV suppressed atherosclerosis in hypercholesterolemic rabbits
and endothelium-specific overexpression of Sirt1 decreased
atherosclerosis in apolipoprotein E-deficient (apoE−/−)
mice. Moreover, Sirt1 modulated neointima formation following
vascular injury in mice (27–30). Sirt1 is considered a novel target
in the prevention of atherosclerosis by regulating lipid
metabolism, promoting endothelial survival and improving
endothelial function, repressing vascular smooth muscle cell
migration and proliferation and most importantly, inducing cellular
autophagy (31–34). Sirt1 was reported to prevent
atherosclerosis by potentially regulating the degree of autophagy
to match current cellular needs with real-time metabolic status
(35).
Focus on VSMCs and ECs has been given in previous
investigations on autophagy (23,36,37). Transmission electron microscopy
(TEM) of VSMCs in the fibrous cap of experimental or human plaques
reveals features of autophagy such as the formation of myelin
figures (36). Moreover,
experimental exposure of endothelial cells directly to ox-LDL
strongly enhanced autophagy compared with exposure to LDL only,
indicating that autophagy may contribute to the degradation of
ox-LDL (37,38). Autophagy has also been reported to
regulate intracellular lipid metabolism to avoid the formation of
foam cells (39,40). As recently reviewed (41,42), lipophagy, a special type of
autophagy, contributes to cholesterol egress from lipid-laden cells
to high-density lipoprotein (HDL) via lysosomal lipases. Autophagy
can play a role in the hydrolysis of stored cholesterol droplets in
macrophages, thus facilitating cholesterol efflux (43). The function of macrophage
autophagy in atherosclerosis has been complicated by the strong
phagocytic activity of these cells. Currently, few studies have
focused on the enhancement in efferocytosis of apoptotic
macrophages through Sirt1-mediated autophagy. However, autophagy is
a ‘double-edged sword’. The general consensus is that basal
autophagy can protect plaque cells against oxidative stress by
degrading damaged intracellular material and promoting cell
survival, and excessive stimulation of autophagy may cause
autophagic cell death, leading to reduced synthesis of collagen,
thinning of the fibrous cap, plaque destabilization, lesional
thrombosis, and acute clinical events (44,23,24). Thus the aim of this study was to
determine whether there was a connection between enhancement in
efferocytosis of apoptotic macrophages and autophagy mediated by
Sirt1.
Materials and methods
Reagents and antibodies
A Cell Counting Kit-8 (CCK-8) was purchased from
Dojindo Laboratories (Kumamoto, Japan). ox-LDL was purchased from
AbD Serotec (Kidlington, Oxford, UK). Oil red O staining,
5(6)-carboxyfluorescein diacetate
N-succinimidyl ester (CFSE) staining, Sirt1-activator RSV,
Sirt1-inhibitor NAM and autophagy-inhibitor 3-methyladenine (3-MA)
were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). The
anti-Sirt1 antibody was obtained from Cell Signaling (Beverly, MA,
USA). The anti-light chain (LC) 3, anti-Atg5 and anti-Atg7
antibodies were obtained from Epitomics (Burlingame, CA, USA).
Horseradish peroxidase-linked anti-rabbit, anti-mouse secondary
antibodies and RIPA buffer were obtained from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA).
Cell culture and experimental design
A mouse macrophage-like RAW264.7 cell line was
purchased from the Cell Bank of the Shanghai Institutes for
Biological Sciences, Chinese Academy of Sciences. The cells were
cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco, NY,
USA), which contained 5 mM glucose, and were supplemented with 10%
fetal bovine serum (FBS) (Gibco) and 1% penicillin/streptomycin at
37°C with 5% CO2 in a humidified atmosphere. The cells
were passaged every 2–3 days and then placed into 6-well plates
with slides at a density of 4×105 cells/cm2
and made apoptotic by incubation with ox-LDL. The optimal
concentration and time point of ox-LDL were analyzed using western
blotting of Sirt1 and the autophagy marker proteins. The apoptotic
cells were randomly divided into the following groups: i) control
group, only apoptotic cells, ii) low concentration RSV group:
apoptotic cells incubated with RSV of low concentration, iii) high
concentration RSV group: apoptotic cells incubated with RSV of high
concentration, iv) low concentration NAM group: apoptotic cells
incubated with NAM of low concentration, v) high concentration NAM
group: apoptotic cells incubated with NAM of high concentration,
vi) 3-MA + low concentration RSV group: the apoptotic cells were
pretreated with 10 mM 3-MA for 2 h, and then incubated with low
concentration RSV. The low and high concentration of RSV and NAM
were assayed from the cell proliferation of RAW264.7 incubation
with RSV and NAM for 24 h.
Protein extraction and western blot
analysis
Total proteins were obtained by rinsing treated
cells with ice-cold phosphate-buffered saline (PBS), and lysing in
lysis buffer (10 mM Tris pH 7.4, 20 mM NaCl, 5 mM MgCl2,
0.5% NP-40 and 0.1 mM PMSF). The extracts were then centrifuged at
12,000 × g for 10 min at 4°C, and the clear supernatants containing
total protein were collected. The protein concentration was
measured with the Bio-Rad protein assay (Hercules, CA, USA), an
equal amount of protein was separated on SDS-polyacrylamide gel
electrophoresis and transferred to nitrocellulose membranes. After
blocking with 5% non-fat milk, the membranes were incubated
overnight with the previously mentioned first antibodies at 4°C.
The membranes were then incubated with the appropriate secondary
antibodies, and the bands were detected by enhanced
chemiluminescence. The band density value was quantified using the
ImageJ image processing program. Since the extent of conversion of
LC3-I to LC3-II is correlated with the level of autophagy, LC3-I
and LC3-II were detected by western blot analysis, and the
conversion of LC3 was demonstrated by LC3-II/LC3-I ratio. The
LC3-II/LC3-I ratio was calculated as follows: LC3-II/LC3-I ratio =
the band density of LC3-II/the band density of LC3-I. We also
detected the expression of two separate autophagy proteins, Atg5
and Atg7, which were ultimately required for the formation of the
autophagosome and the subsequent induction of autophagy (45–47).
Cell Counting Kit-8 assay
Cell proliferation was assayed using a CCK-8 assay.
Cell suspension (100 μl) of RAW264.7 cells seeded in 96-well plates
at a density of 1×105 cells/well was performed. RAW264.7
cells were then treated with RSV or NAM. The final concentrations
of RSV were 50, 25, 12.5, 5 and 0 μM, respectively. The final
concentrations of NAM were 80, 40, 20, 10, 5 and 0 mM,
respectively. Each group was prepared with five parallel wells and
incubated at 37°C, 5% CO2, for 24 h. At the end of the
culture period, CCK-8 was added to each well according to the
manufacturer’s instructions at a final concentration of 0.5 mg/ml
for 3 h. The absorbance was measured with an enzyme calibrator at
450 and 650 nm and optical density (OD) values were measured.
Experiments were repeated three times.
Oil red O staining
The RAW264.7 cells, measured for lipid accumulation
through staining of ox-LDL with oil red O, were placed in 6-well
plates with slides at a density of 4×105
cells/cm2 and followed with the aforementioned
treatments. At the end of the treatment period, the cells were
rinsed with PBS and fixed with 10% formalin for 5 min at room
temperature. The cells were then rinsed again with 60% isopropanol
and incubated with fresh-filtered oil red O solution (60% saturated
oil red O/40% deionized water) for 20 min. For analysis, the slides
were washed in isopropanol for 10 min, rinsed in distilled water,
counterstained with hematoxylin and mounted in glycerol/gelatin
solution. Images of cells were captured using a fluorescence
microscope.
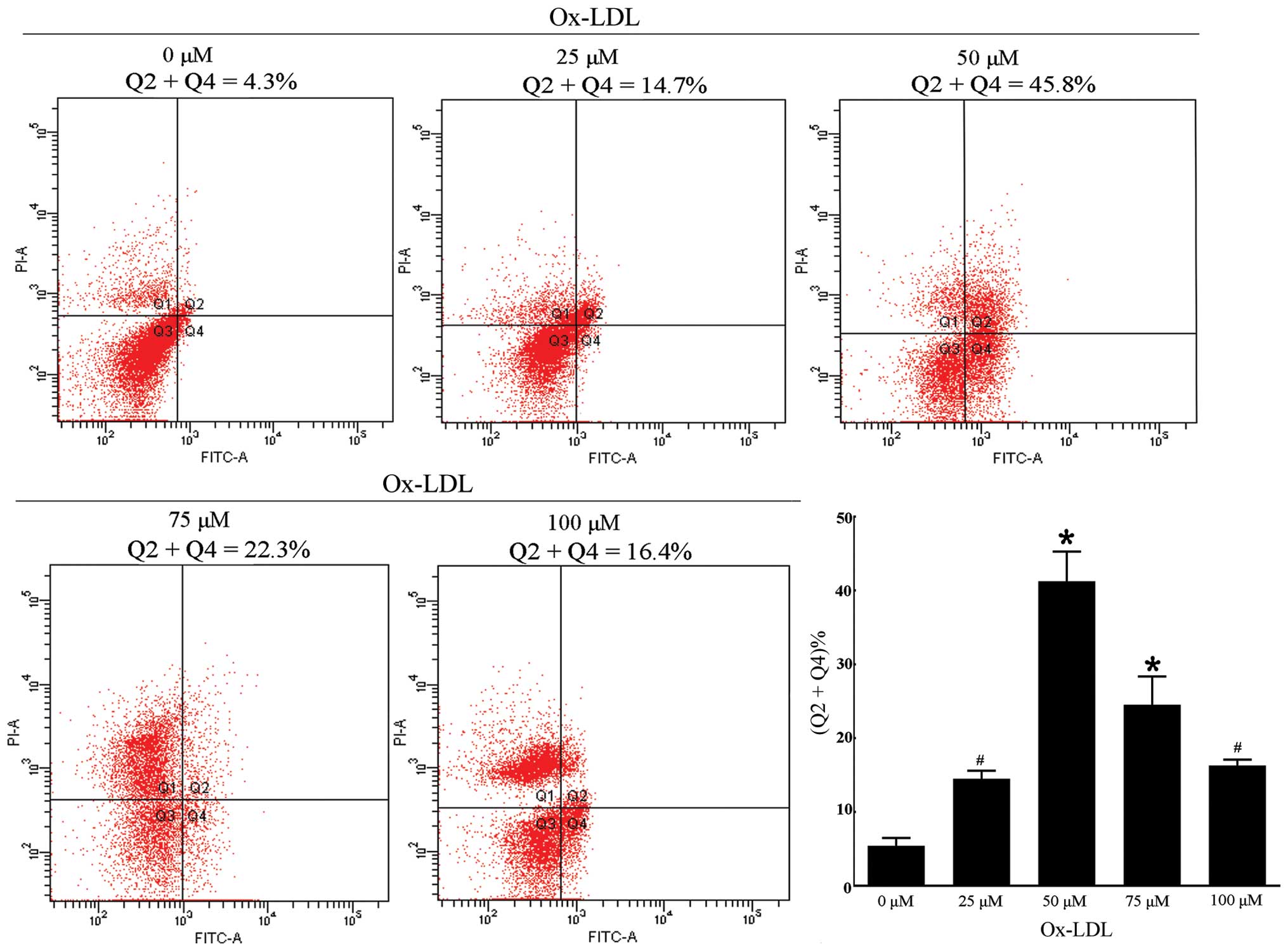
Analysis of apoptosis by flow cytometry
(FCM) of Annexin-V/propidium iodide (AV/PI) dual staining
The RAW264.7 cells were incubated with ox-LDL of
designed concentrations and were then processed with an AV-FITC kit
(Keygene, KGA108) according to the manufacturer’s instructions. The
samples were analyzed by FACScan flow cytometer (Becton-Dickinson,
Franklin Lakes, NJ, USA) in order to quantify the apoptotic rate.
Different subpopulations were distinguishable: Q1, Annexin
V-negative but PI-positive, i.e., necrotic cells; Q2, Annexin
V/PI-double positive, i.e., late apoptotic cells; Q3, Annexin
V/PI-double negative, i.e., live cells; Q4, Annexin V-positive but
PI-negative, i.e., early apoptotic cells. The apoptotic rate was
determined as the percentage of Q2 + Q4.
Detection of autophagosomes by TEM
analysis
The treated cells were rinsed with ice-cold PBS and
centrifuged at 1,000 × g for 5 min at room temperature, after which
the clear supernatants were removed. Cell pellets were fixed with
2.5% glutaraldehyde in 0.1 M cacodylate buffer, pH 7.4 for at least
30 min at 4°C. After fixation, the specimens were thoroughly washed
in 0.1 M cacodylate buffer and then post-fixed with 1% osmium
tetroxide in the same buffer for 1 h at room temperature. The
specimens were dehydrated in a graded series of ethanol, and
embedded in Epon, then 0.1 μm thin sections were stained with
uranyl-acetate/lead citrate and viewed in a Hitachi H-300 TEM.
Measurement of the efferocytosis of
apoptotic RAW264.7 cells
The measurement of the efferocytosis of RAW264.7
cells was performed as described by Li et al, Yancey et
al and Jehle et al (48–50). The RAW264.7 cells were made
apoptotic by incubation with ox-LDL followed by treatments in the
aforementioned 6 groups. After vigorous washing with PBS, the cells
were fixed in 4% paraformaldehyde and counterstained with PI. The
cells in 6 groups were incubated for 2 h with fresh RAW264.7 cells
which were labeled with CFSE cell tracer. The efferocytosis of
apoptotic RAW264.7 cells was visualized using fluorescence
microscopy. PI red-labeled apoptotic RAW264.7 cells merged into
CFSE cell tracer green-labeled fresh RAW264.7 cells, which was
considered as phagocytosis of the apoptotic cells by fresh RAW264.7
cells. The phagocytic index was used to evaluate the efferocytosis
of apoptotic RAW264.7 cells. The phagocytic index was calculated
using the formula: Phagocytic index = (number of phagocytized
RAW264.7 cells/number of total cells) × 100. Experiments were
repeated five times and the analysis was performed in a blinded
fashion by two independent observers.
Statistical analysis
Data are expressed as mean ± SD. Statistical
analysis of data was performed by applying the Student’s t-test to
determine the significance between two groups. Statistical
significance of pairwise differences among three or more groups
were determined using one-way analysis of variance (ANOVA) followed
by post-hoc test. P<0.05 was considered statistically
significant. Analysis was performed using SPSS for Windows (SPSS
Inc., Version 16.0, Chicago, IL, USA).
Results
Expression of Sirt1 and autophagy marker
proteins was elevated at optimal concentrations and time point of
ox-LDL
The effects of ox-LDL (25, 50 and 100 μM) on the
expression of Sirt1 and autophagy marker proteins at different time
points (12, 24 and 48 h) were examined. Our results showed that
ox-LDL of appropriate concentration elevated the levels of Sirt1
and autophagy marker proteins such as Atg5, Atg7 and LC3-II/LC3-I
at optimal time points. Results of the western blot analysis shown
in Figs. 1 and 2 revealed that the expression of Sirt1
and autophagy marker proteins was increased at 24 h (all P<0.05
vs. 12 h), and then decreased at 48 h (all P<0.05 vs. 24 h). The
expression of Sirt1 and autophagy marker proteins was significantly
higher at 50 μM ox-LDL (all P<0.05 vs. 0 μM), but was reduced
when the cells were treated with 75 and 100 μM ox-LDL (all
P<0.05 vs. 0 μM). Thus, cells treated with 50 μM ox-LDL for 24 h
may be considered optimal for the expression of Sirt1 and autophagy
marker proteins. Moreover, the results suggested that autophagy was
induced concomitantly with the induction of expression of Sirt1 by
a moderate stimulus of ox-LDL, suggesting that Sirt1 is involved in
autophagy under treatment of ox-LDL to some extent.
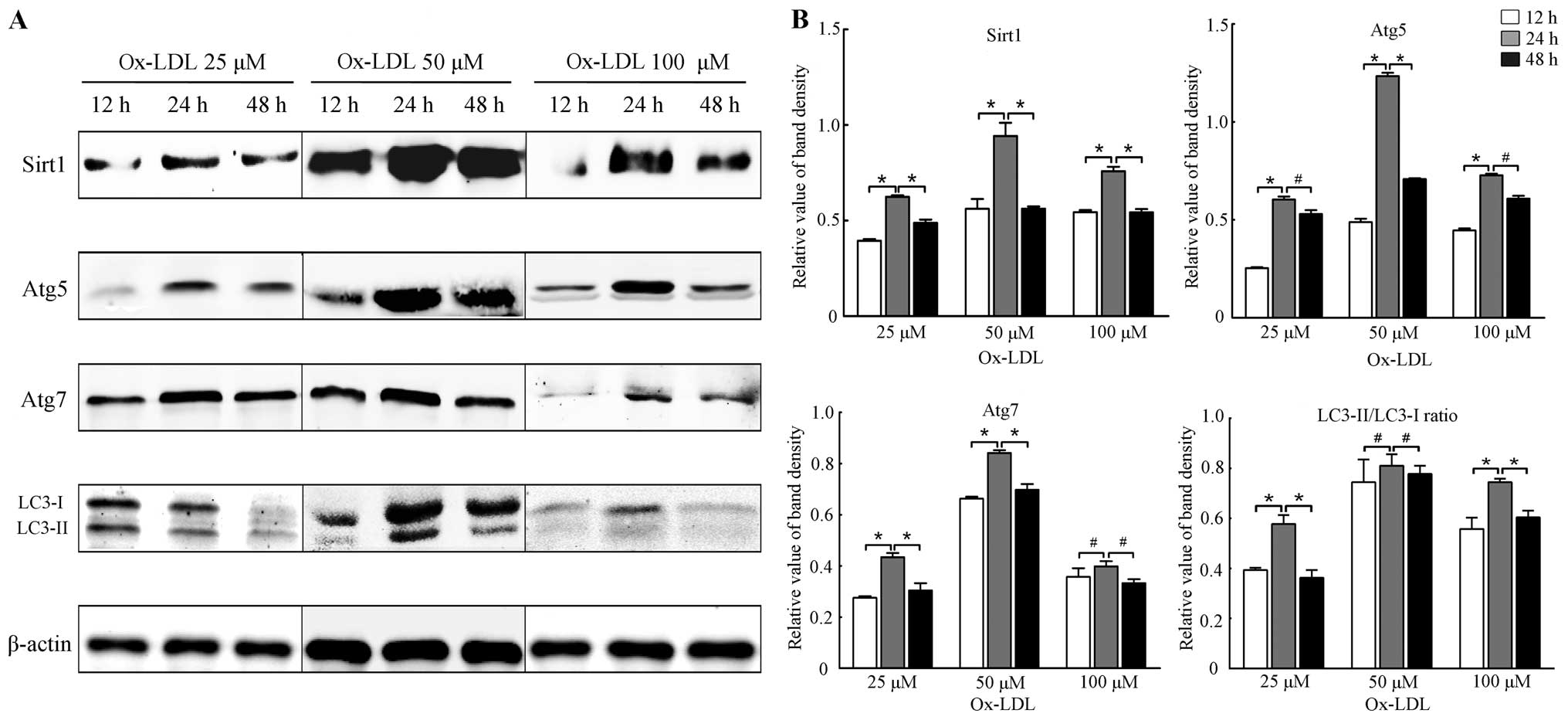 | Figure 1Expression of Sirtuin1 (Sirt1) and
autophagy marker proteins in RAW264.7 cells treated with ox-LDL at
different concentrations and time points. (A) Representative
results of assays of Sirt1, Atg5, Atg7, LC3-I and LC3-II and
β-actin abundances in RAW264.7 cells with ox-LDL of designated
concentrations (25, 50 and 100 μM) and time points (12, 24 and 48
h) using western blot analysis. (B) Protein expression levels of
Sirt1 were analyzed by western blotting by using polyclonal
antibodies to Sirt1, Atg5, Atg7, LC3-I and LC3-II, respectively, in
order to quantify the expression in RAW264.7 cells. LC3-II/LC3-I
ratio was calculated. β-actin was used as an equal loading control.
The band value was quantified by densitometric analysis. The
expression of all the proteins was increased most at 24 h compared
with 12 or 48 h (all P<0.05). Experiments were repeated at least
three times. Data are expressed as the mean ± SD in the
corresponding bar graph and statistical significance was determined
by the Student’s t-test. Columns, mean; error bars, ±SD;
#P<0.05, *P<0.01. |
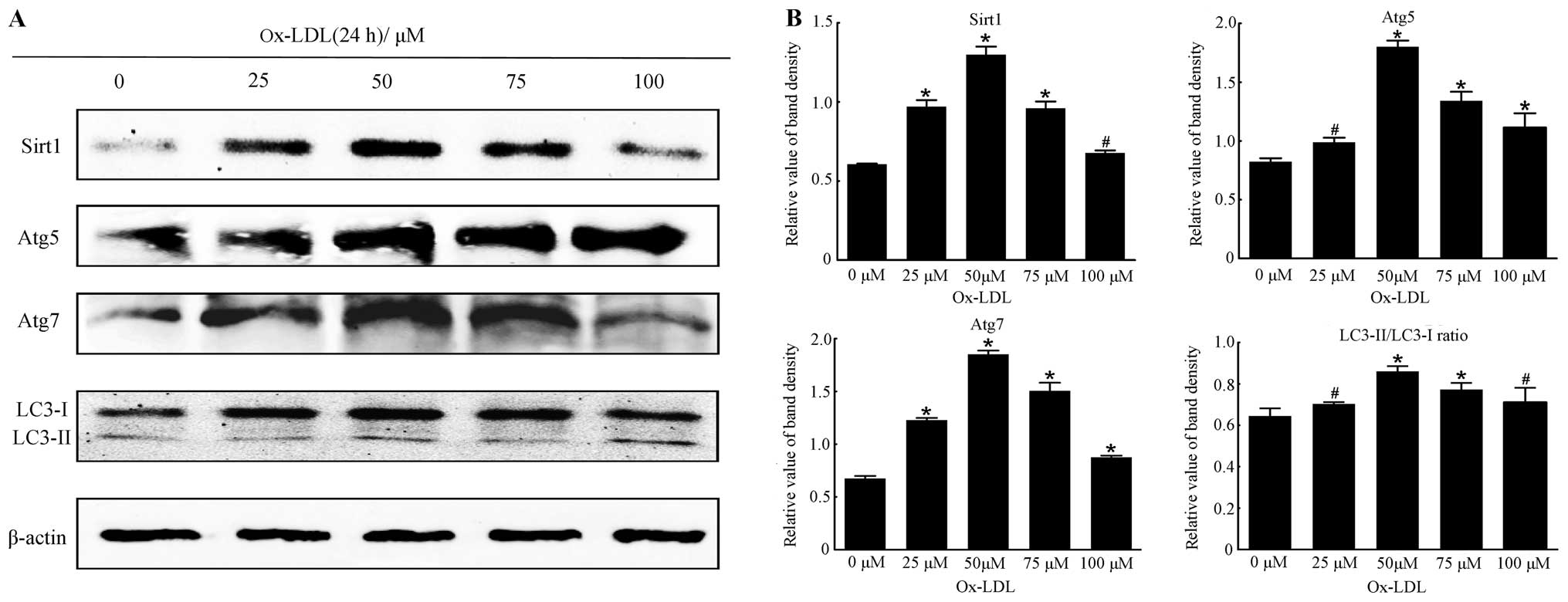 | Figure 2The optimal concentration of ox-LDL
for the expression of Sirtuin1 (Sirt1) and autophagy marker
proteins in RAW264.7 cells after incubation for 24 h. (A)
Representative western blot analysis results of Sirt1, Atg5, Atg7,
LC3-I and LC3-II and β-actin abundances in RAW264.7 cells following
stimulation with different concentrations of ox-LDL (0, 25, 50, 75
and 100 μM) for 24 h. (B) Protein levels of Sirt1, Atg5, Atg7,
LC3-I and LC3-II were analyzed by western blotting in RAW264.7
cells, respectively. The LC3-II/LC3-I ratio was calculated. β-actin
was used as an equal loading control. The band value was quantified
by densitometric analysis. The expression of all the proteins was
increased after incubation with ox-LDL at the designated
concentrations compared with 0 μM ox-LDL (all P<0.05). The
optimal concentration of ox-LDL was 75 μM compared with the other
groups. Experiments were repeated at least three times. Data are
expressed as the mean ± SD in the corresponding bar graph and
statistical significance was determined by the Student’s t-test.
Columns, mean; error bars, ±SD; #P<0.05 vs. 0 μM
group, *P<0.01 vs. 0 μM group. |
Ox-LDL treatment simultaneously induces
autophagy and apoptosis in RAW264.7 cells
We investigated cell apoptosis when the cells were
incubated with different concentrations of ox-LDL for 24 h. An
appropriate concentration of ox-LDL for further measurement of
efferocytosis was required according to the apoptotic rate.
Additionally, the quantitative analysis of apoptosis by FCM of
AV/PI dual staining at designated concentrations showed that the
apoptotic rate increased at 25 μM (14.52±1.08% vs. 0 μM,
5.43±1.04%) and 50 μM (41.23±4.02%) in a dose-dependent manner and
markedly accelerated when treated with 50 μM ox-LDL (Fig. 3). The apoptotic rate decreased
when the cells were incubated with 75 μM (24.53±3.82%) and 100 μM
(16.30±0.79%) ox-LDL. By comparing the apoptotic rate among these
concentrations, the data suggested that the apoptotic rate was
appropriate when the cells were treated with 50 μM ox-LDL
(P<0.001 vs. 0 μM). Furthermore, the previous results showed
that, cells treated with 50 μM ox-LDL for 24 h would be optimal for
the expression of autophagy marker proteins, which is similar to
the condition of cell apoptosis. The evidence suggested that
autophagy and apoptosis of RAW264.7 cells were triggered by
incubation with 50 μM ox-LDL for 24 h.
Different doses of RSV and NAM exert dual
effects on cell proliferation and the expression of Sirt1 in
RAW264.7 cells
Low concentration RSV exerted a protective effect on
cells. However, high concentration RSV may induce cell necrosis and
apoptosis of RAW264.7 cells (51). NAM of different concentrations was
also able to promote or inhibit cell proliferation (52,53). The dual effects of RSV and NAM on
cell proliferation and the expression of Sirt1 in RAW264.7 cells
were examined. The appropriate high and low concentrations of RSV
or NAM for cell proliferation, respectively, were identified. The
results of CCK-8 assay indicated that the cell proliferation rates
increased significantly when incubated with 12.5 μM RSV (0.69±0.01
OD), compared with that of the 0 μM group (0.45±0.03 OD), 5 μM
group (0.49±0.02 OD), 25 μM group (0.62±0.02 OD), 50 μM group
(0.61±0.04 OD) and 100 μM group (0.34±0.01 OD). The proliferation
rates decreased significantly when the cells incubated with 100 μM
RSV (0.34±0.01 OD vs. 50 μM: 0.61±0.04 OD) (Fig. 4A). Similarly, the cell
proliferation rates remained appropriate when incubated with 5 mM
NAM (0.52±0.03 OD) compared with 0 mM NAM (0.52±0.04 OD). The
proliferation rates decreased in the 10 mM NAM group (0.41±0.02
OD), 20 mM NAM group (0.40±0.02 OD), and most significantly, the 40
mM NAM (0.25±0.03 OD) and 80 mM NAM groups (0.14±0.02 OD) (Fig. 4B). These results suggested that
the cell status was extremely poor when incubated with 100 μM RSV,
40 and 80 mM NAM. Thus, the high and low concentrations of RSV were
50 and 12.5 μM, while the corresponding high and low concentrations
of NAM were 20 and 5 mM. Furthermore, after the RAW264.7 cells were
made apoptotic, the results of western blotting revealed that Sirt1
expression was increased when cells were incubated with 12.5 μM RSV
and 5 mM NAM compared with that of 50 μM RSV and 20 mM NAM,
respectively (both P<0.05 vs. control) (Fig. 5A and B).
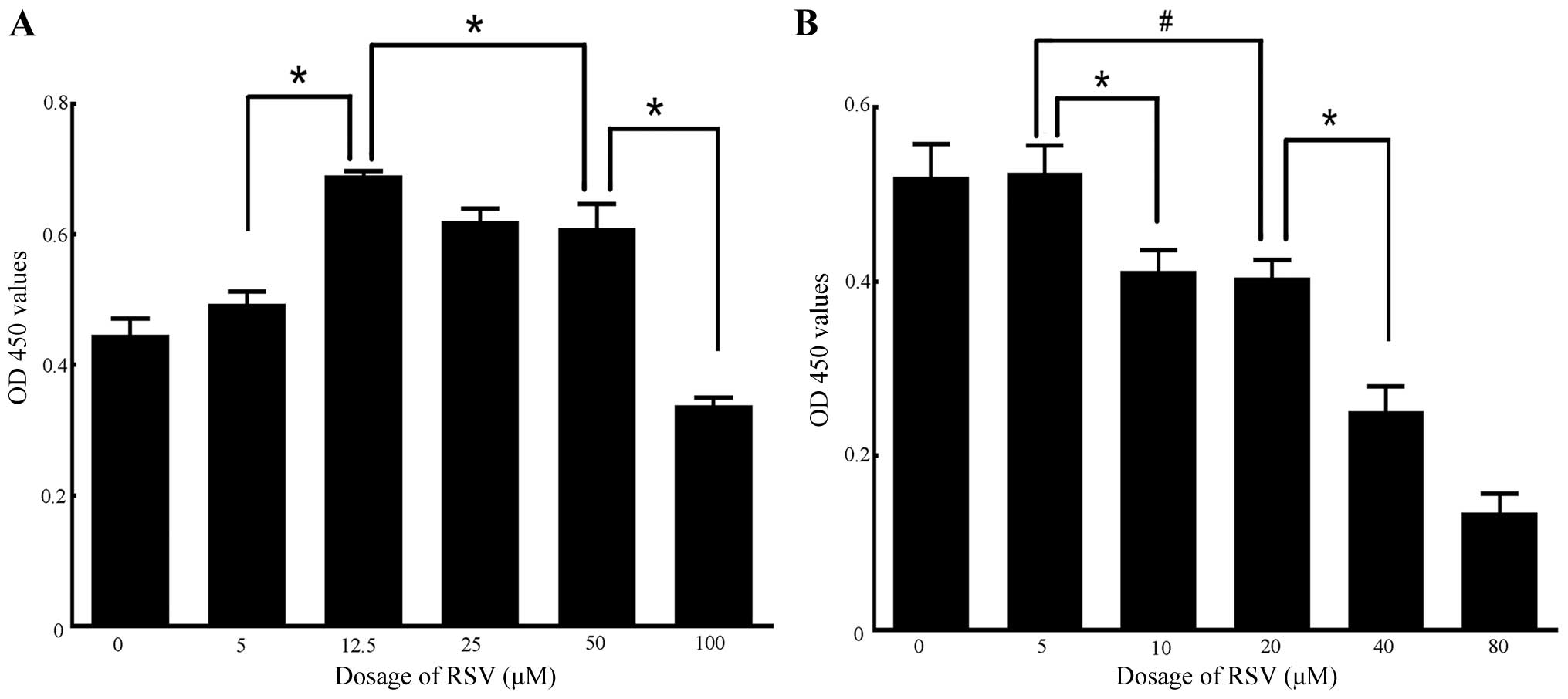 | Figure 4Determination of high and low
concentrations of resveratrol (RSV) or nicotinamide (NAM) by
analysis of cell proliferation rates using the Cell Counting Kit-8
(CCK-8) assay. (A) Results of CCK-8 assay on RAW264.7 cells
incubated with different doses of RSV (0, 5, 12.5, 25, 50 and 100
μM). No significant differences were observed in comparisons
between 0 and 5 μM, and 25 and 50 μM groups, respectively. A
significant difference was observed between the 5 and 12.5 μM, 12.5
and 50 μM group, and 50 and 100 μM groups, respectively (all
P<0.01). (B) Results of CCK-8 assay on RAW264.7 cells incubated
with different doses of NAM (0, 5, 10, 20, 40 and 80 mM). No
significant differences were shown in comparisons between the 0 and
5 mM, and 10 and 20 mM groups, respectively. A significant
difference was observed between the 5 and 10 mM, 5 and 20 mM, and
20 and 40 mM groups, respectively (all P<0.05). Experiments were
repeated three times. Data are expressed as the mean ± SD in the
corresponding bar graph and statistical significance was determined
by the Student’s t-test. Columns, mean; error bars, ±SD;
#P<0.05, *P<0.01. |
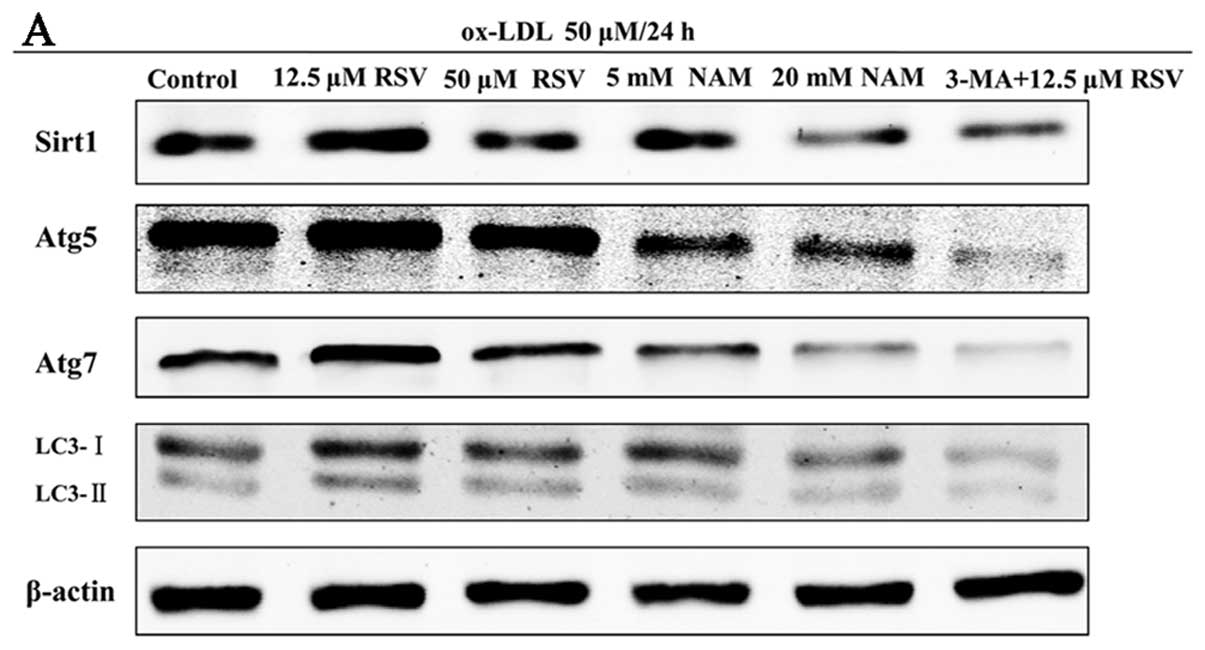 | Figure 5Expression of Sirtuin1 (Sirt1) and
autophagy marker proteins in ox-LDL-induced apoptotic RAW264.7
cells of different treatments. The apoptotic cells were randomly
divided into 6 groups: i) control, ii) 12.5 μM resveratrol (RSV),
iii) 50 μM RSV, iv) 5 mM nicotinamide (NAM), v) 20 mM NAM, vi)
3-methyladenine (3-MA) + 12.5 μM RSV. (A) Representative results of
assays of Sirt1, Atg5, Atg7, LC3-I, LC3-II and β-actin abundances
of 6 groups using western blot analysis. (B) The levels of Sirt1,
Atg5, Atg7, LC3-I and LC3-II protein expression were analyzed by
western blot analysis by using polyclonal antibodies to Sirt1,
Atg5, Atg7, LC3-I and LC3-II to quantify the expression in these
groups. LC3-II/LC3-I ratio was calculated. Sirt1 expression was
significantly elevated in 12.5 μM RSV group, compared with control
group (P=0.002). No statistical significance was observed between
the 3-MA + 12.5 μM RSV and control groups following inhibition of
autophagy. The levels of all autophagy marker proteins were
significantly increased in 12.5 μM RSV group simultaneously,
compared with the control group (all P<0.05). β-actin was used
as an equal loading control. The band value was quantified by
densitometric analysis. Experiments were repeated at least three
times. Data are expressed as the mean ± SD in the corresponding bar
graph and statistical significance was determined by the Student’s
t-test. Columns, mean; error bars, ±SD; #P<0.05 vs.
control group, *P<0.01 vs. control group. |
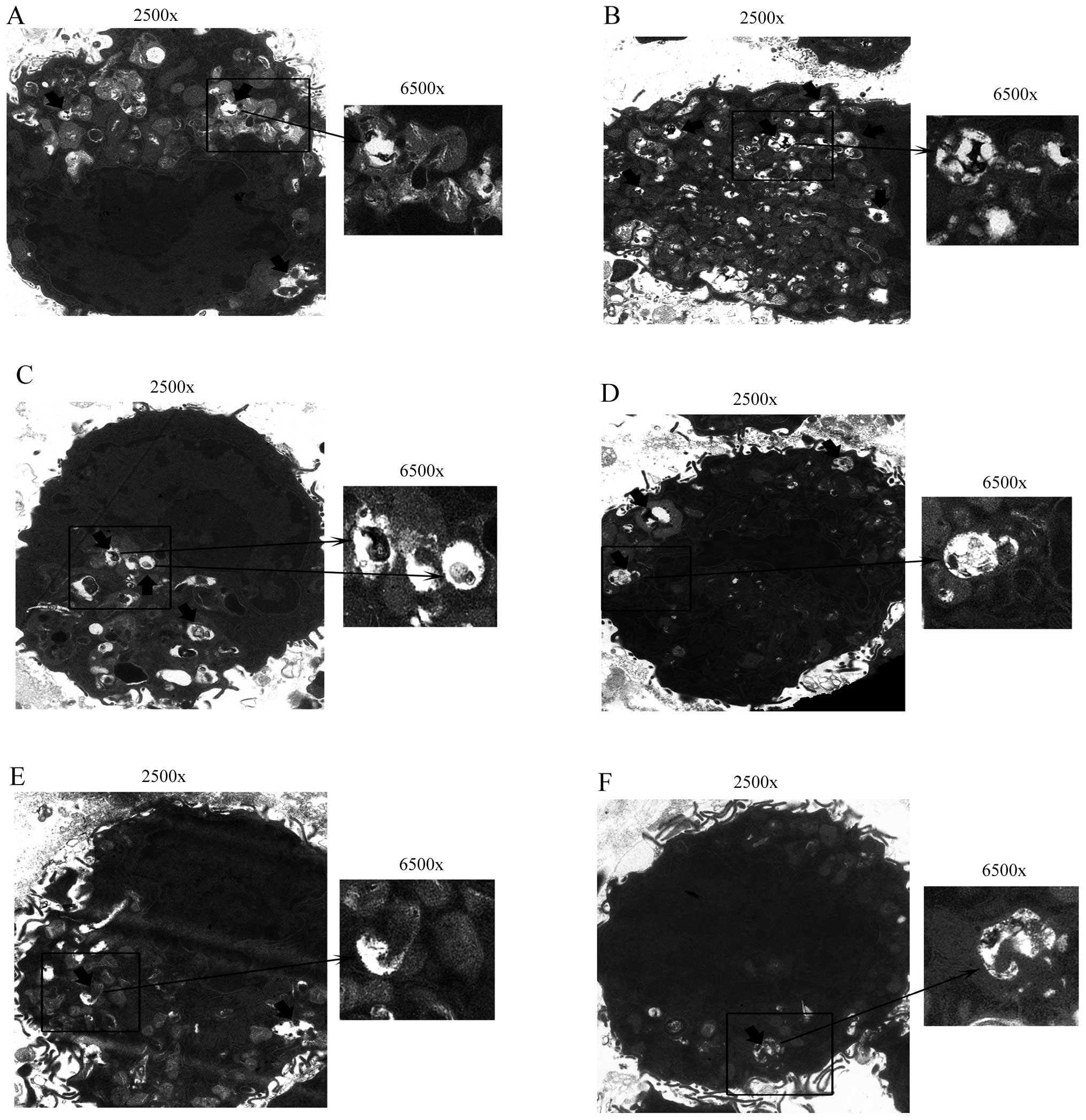
Sirt1 possibly contributes to autophagy
in apoptotic RAW264.7 cells following treatment of ox-LDL
To define the potential role of Sirt1 in autophagy,
we examined the expression of Sirt1 and autophagy marker proteins
in apoptotic RAW264.7 cells following the treatment of 50 μM ox-LDL
for 24 h, using high and low concentration RSV or NAM. We also
investigated the expression of these proteins when the cells were
incubated with 3-MA, the chemical inhibitor of autophagy. Treatment
with 12.5 μM RSV significantly increased the expression of
autophagy marker proteins such as Atg5, Atg7 and LC3-II/LC3-I,
which was accompanied by the activation of Sirt1 (all P<0.05 vs.
control group). The expression of Sirt1 and autophagy marker
proteins in the 5 and 20 mM NAM groups were simultaneously
decreased as compared to the control group (all P<0.05)
(Fig. 5). Moreover, the
expression of Sirt1 in the 3-MA + 12.5 μM RSV group showed no
significant difference compared with the control group (P=0.07)
subsequent to inhibition of autophagy (Fig. 5A and B). We also detected the
autophagosomes of these groups via TEM analysis. Significantly more
autophagosomes were identified in the 12.5 μM RSV group compared
with the other groups, while there was hardly any formation of
autophagosomes in the 3-MA + 12.5 μM RSV group (Fig. 6). These results showed that Sirt1
was able to regulate the expression of autophagy marker proteins in
RAW264.7 cells.
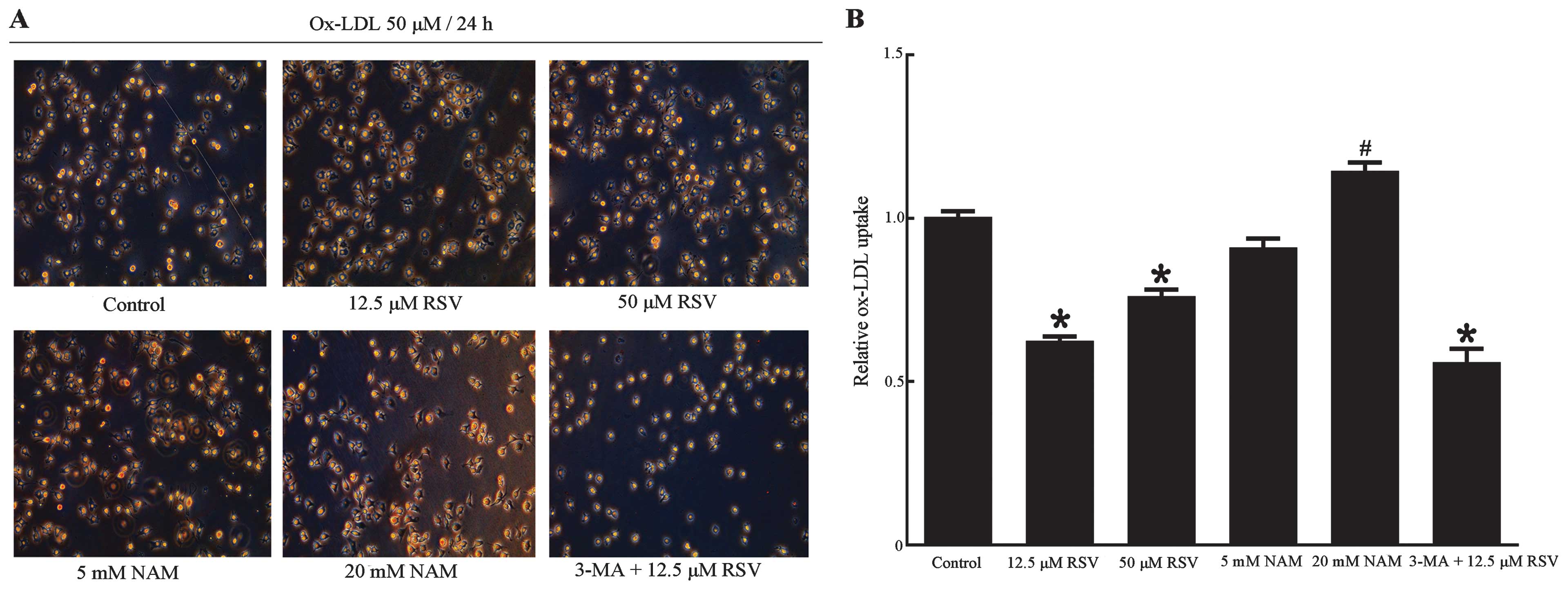
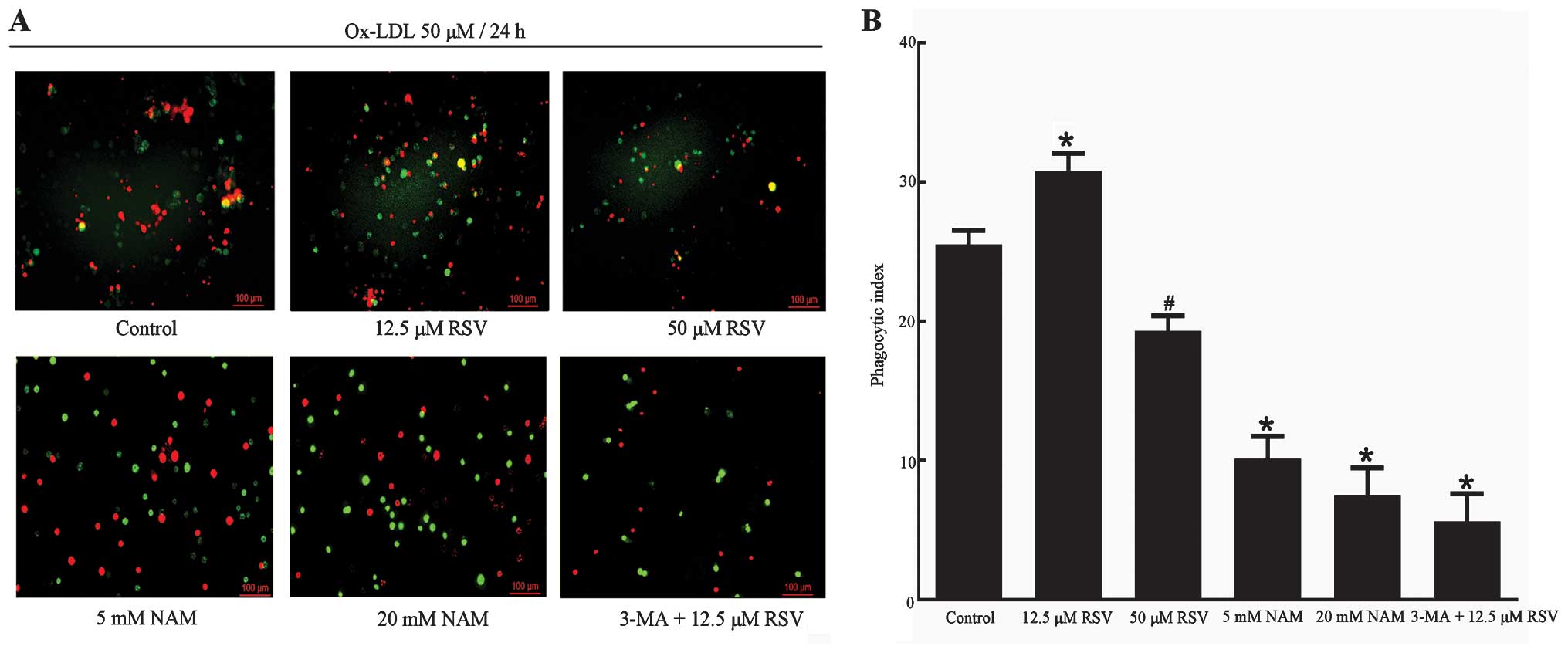
Upregulation of autophagy enhanced
efferocytosis of apoptotic RAW264.7 cells
The relationship between Sir1-mediated autophagy and
efferocytosis in apoptotic RAW264.7 cells was investigated. ox-LDL
uptake was detected, which may be useful in the elevation of the
phagocytosis of RAW264.7 cells regulated by Sirt1 and autophagy. As
determined by oil red O staining (Fig. 7), ox-LDL uptake in the 12.5 and 50
μM RSV groups were decreased compared with the control group
(P<0.001, P=0.008 respectively), which was accompanied by the
upregulation of autophagy. By contrast, ox-LDL uptake in the 20 mM
group NAM showed a significant increase (P=0.03 vs. control group),
whereas a decrease was identified in the 3-MA + 12.5 μM RSV group
following the inhibition of autophagy (P<0.001 vs. control
group). This finding may be attributed to the expression of Sirt1
since Sirt1 has been reported to decrease ox-LDL uptake and prevent
macrophage foam cell formation through suppression of the
expression of the scavenger receptor Lox-1 in macrophages (54). Another possible reason is the poor
status of the cells in this group, since there were significant
fewer cells compared with the other groups, as noted in the images
obtained via fluorescence microscopy (Figs. 7A and 8A). The results from the measurement of
the efferocytosis demonstrated that 12.5 μM RSV caused a marked
increase in the efferocytosis of apoptotic RAW264.7 cells compared
with the control group (P<0.001) (Fig. 8), suggesting that upregulation of
autophagy contributes to the phagocytic clearance of apoptotic
cells. Similarly, the efferocytosis of apoptotic RAW264.7 cells was
decreased in the 5 and 20 mM NAM groups (P=0.001, P<0.001
respectively vs. the control group). It is also likely that
inhibition of autophagy by 3-MA contributes to defective
efferocytosis, although Sirt1 was expressed. All our results showed
that upregulation of autophagy was capable of enhancing
efferocytosis of apoptotic RAW264.7 cells. Since the expression of
Sirt1 was able to regulate autophagy and the improvement of
efferocytosis was accompanied by an increase in the expression of
Sirt1, the enhancement in efferocytosis of apoptotic RAW264.7 cells
could be Sirt1-mediated. However, only the expression of Sirt1 did
not induce enhancement in efferocytosis following inhibition of
autophagy.
Discussion
The main aim of this study was to determine whether
the upregulation of autophagy mediated by Sirt1 could enhance
efferocytosis of apoptotic macrophages induced by ox-LDL. The
expression of Sirt1 and autophagy marker proteins was investigated,
using Sirt1 activator RSV and inhibitor NAM. The findings in our
study suggest that with the increase in the expression of Sirt1
activated by an appropriate dose of RSV, the expression of
autophagy marker proteins was also increased. Furthermore, the
efferocytosis of apoptotic RAW264.7 cells was also improved
simultaneously with this increase.
Previous studies (13–16) have shown that macrophage autophagy
in atherosclerosis becomes dysfunctional in atherosclerosis and its
deficiency promotes vascular inflammation, oxidative stress, and
plaque necrosis, suggesting a mechanism-based strategy to
therapeutically suppress atherosclerosis progression. It is
difficult to determine whether the vacuoles in their cytoplasm
result from autophagocytosis or heterophagocytosis through
conventional electron microscopy analysis. In addition, LC3 is
poorly expressed in macrophages and overexpression of other
lysosomal marker proteins may give rise to false-positive signals
in immunoelectron microscopy (23,24). Notably, pharmacological modulation
of macrophage autophagy has been shown to affect vascular
inflammation. Stent-based delivery of everolimus (a well-known
autophagy inducer) in atherosclerotic plaques of high-fat diet-fed
rabbits leads to a marked reduction of macrophages via autophagic
cell death without altering the VSMC plaque content (13). Two recent studies have provided
new dimensions to the understanding of the role of macrophage
autophagy in regulating atherosclerosis. Razani and colleagues
(15) demonstrated that initially
autophagy is functional and becomes severely compromised with
disease progression. This deficiency of macrophage autophagy may
induce inflammosome hyperactivation through lysosomal leakage,
generation of reactive oxygen species (ROS), and impaired
mitophagy, thus increased vascular inflammation and plaque
formation in apoE−/− mice. Findings of another study
provided evidence that inhibition of macrophage autophagy enhanced
apoptosis and riphosphopyridine nucleotide (NADPH) oxidase-mediated
oxidative stress, rendering the apoptotic cells less recognizable
to efferocytosis in low-density lipoprotein receptor
(LDLr)−/− mice (16).
The defective efferocytosis could promote plaque necrosis in
advanced atherosclerosis. These data indicated a protective role
played by macrophage autophagy in the two most widely used mouse
models of atherosclerosis.
Sirt1 has been recently regarded as a new factor in
the regulation of autophagy. The decrease in Sirt1 protein
expression may lead to inflammation through dysregulation of
autophagy and increased levels of acetylated nuclear factor-κB
(NF-κB) (55,56). Sirt1 regulated autophagy by
promoting the formation of autophagosome in a cellular model of
oxidative stress (57). Moreover,
RSV exerted protective effects on cells through the activation of
the adenosine 5′-monophosphate-activated protein kinase
(AMPK)-SIRT1-autophagy pathway. Inhibition of Sirt1 contributes to
the dysregulation of other nutrient-sensing pathways including
mammalian target of rapamycin (mTOR) and AMPK, thereby leading to
the impairment of autophagy in human macrophage, which may induce
inflammation through NF-κB activation and the accumulation of
autophagy marker proteins (58,59). However, few studies have explored
the relationship between efferocytosis of apoptotic macrophage
cells and autophagy mediated by Sirt1. Our results mainly provide
preliminary evidence that Sirt1 potentially affects the
efferocytosis of apoptotic macrophages via the activation of
autophagy, which is useful in understanding the molecular mechanism
and pathogenesis of atherosclerosis. Our findings may have
implications regarding the influence of defective autophagy on cell
survival and the expression of Sirt1 in atherosclerosis, since the
cell status was poor and the level of Sirt1 was not elevated when
the cells were incubated with 3-MA + 12.5 μM RSV.
Limitations to our study should be noted. The study
was performed using in vivo experimental systems.
Additionally, the relationship between efferocytosis of apoptotic
macrophages and Sirt1 should be confirmed in the atherosclerotic
plaques through in vitro experiments. We only detected the
efferocytosis of apoptotic macrophages enhanced by Sirt1-mediated
moderate autophagy. Thus, the effect of Sirt1-mediated excessive
autophagy on efferocytosis of apoptotic macrophages should also be
determined. Studies are also required to elucidate the signaling
pathways underlying this mechanism.
In conclusion, results of the present study showed
that autophagy was upregulated by an appropriate dose of Sirt1
activator RSV, and that the efferocytosis of apoptotic RAW264.7 was
significantly improved when incubated with the appropriate dose of
RSV compared with Sirt1 inhibitor NAM and autophagy inhibitor 3-MA.
This enhancement in efferocytosis may be associated with
Sirt1-mediated autophagy.
Ackowledgements
This study was supported by National Natural Science
Foundation of China (No. 81200198) and Shanghai Municipal Health
Bureau research projects (Grant No. 20124224), P.R. China to Buchun
Zhang. We completed this study at the Central Laboratory of the
Shanghai Tenth People’s Hospital of Tongji University. The authors
would like to thank Jianhui Zhuang, Ke Wang and Hailing Li from
Tongji University School of Medicine for their helpful
discussions.
References
|
1
|
Gersh BJ, Sliwa K, Mayosi BM and Yusuf S:
Novel therapeutic concepts: the epidemic of cardiovascular disease
in the developing world: global implications. Eur Heart J.
31:642–648. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Moore KJ and Tabas I: Macrophages in the
pathogenesis of atherosclerosis. Cell. 145:341–355. 2011.
View Article : Google Scholar
|
|
3
|
Gautier EL, Jakubzick C and Randolph GJ:
Regulation of the migration and survival of monocyte subsets by
chemokine receptors and its relevance to atherosclerosis.
Arterioscler Thromb Vasc Biol. 29:1412–1418. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gerrity RG: The role of the monocyte in
atherogenesis: I. Transition of blood-borne monocytes into foam
cells in fatty lesions. Am J Pathol. 103:181–190. 1981.PubMed/NCBI
|
|
5
|
Swirski FK, Pittet MJ, Kircher MF, Aikawa
E, Jaffer FA, Libby P and Weissleder R: Monocyte accumulation in
mouse atherogenesis is progressive and proportional to extent of
disease. Proc Natl Acad Sci USA. 103:10340–10345. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ylä-Herttuala S, Palinski W, Rosenfeld ME,
Parthasarathy S, Carew TE, Butler S, Witztum JL and Steinberg D:
Evidence for the presence of oxidatively modified low density
lipoprotein in atherosclerotic lesions of rabbit and man. J Clin
Invest. 84:1086–1095. 1989.PubMed/NCBI
|
|
7
|
Colles SM, Maxson JM, Carlson SG and
Chisolm GM: Oxidized LDL-induced injury and apoptosis in
atherosclerosis. Potential roles for oxysterols. Trends Cardiovasc
Med. 11:131–138. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Salvayre R, Auge N, Benoist H and
Negre-Salvayre A: Oxidized low-density lipoprotein-induced
apoptosis. Biochim Biophys Acta. 1585:213–221. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Napoli C: Oxidation of LDL, atherogenesis,
and apoptosis. Ann NY Acad Sci. 1010:698–709. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Stoneman VE and Bennett MR: Role of
apoptosis in atherosclerosis and its therapeutic implications. Clin
Sci (Lond). 107:343–354. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tabas I: Apoptosis and efferocytosis in
mouse models of atherosclerosis. Curr Drug Targets. 8:1288–1296.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Seimon T and Tabas I: Mechanisms and
consequences of macrophage apoptosis in atherosclerosis. J Lipid
Res. 50:S382–S387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Verheye S, Martinet W, Kockx MM, Knaapen
MW, Salu K, Timmermans JP, Ellis JT, Kilpatrick DL and De Meyer GR:
Selective clearance of macrophages in atherosclerotic plaques by
autophagy. J Am Coll Cardiol. 49:706–715. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
De Meyer I, Martinet W, Schrijvers DM,
Timmermans JP, Bult H and De Meyer GR: Toll-like receptor 7
stimulation by imiquimod induces macrophage autophagy and
inflammation in atherosclerotic plaques. Basic Res Cardiol.
107:2692012.PubMed/NCBI
|
|
15
|
Razani B, Feng C, Coleman T, Emanuel R,
Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB and Semenkovich CF:
Autophagy links inflammasomes to atherosclerotic progression. Cell
Metab. 15:534–544. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Liao X, Sluimer JC, Wang Y, Subramanian M,
Brown K, Pattison JS, Robbins J, Martinez J and Tabas I: Macrophage
autophagy plays a protective role in advanced atherosclerosis. Cell
Metab. 15:545–553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Axe EL, Walker SA, Manifava M, Chandra P,
Roderick HL, Habermann A, Griffiths G and Ktistakis NT:
Autophagosome formation from membrane compartments enriched in
phosphatidylinositol 3-phosphate and dynamically connected to the
endoplasmic reticulum. J Cell Biol. 182:685–701. 2008. View Article : Google Scholar
|
|
18
|
Hayashi-Nishino M, Fujita N, Noda T,
Yamaguchi A, Yoshimori T and Yamamoto A: A subdomain of the
endoplasmic reticulum forms a cradle for autophagosome formation.
Nat Cell Biol. 11:1433–1437. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
19
|
Hailey DW, Rambold AS, Satpute-Krishnan P,
Mitra K, Sougrat R, Kim PK and Lippincott-Schwartz J: Mitochondria
supply membranes for autophagosome biogenesis during starvation.
Cell. 141:656–667. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tooze SA and Yoshimori T: The origin of
the autophagosomal membrane. Nat Cell Biol. 12:831–835. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Ravikumar B, Moreau K, Jahreiss L, Puri C
and Rubinsztein DC: Plasma membrane contributes to the formation of
pre-autophagosomal structures. Nat Cell Biol. 12:747–757. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Goligorsky MS: SIRTing out the link
between autophagy and ageing. Nephrol Dial Transplant.
25:2434–2436. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Schrijvers DM, De Meyer GR and Martinet W:
Autophagy in atherosclerosis: a potential drug target for plaque
stabilization. Arterioscler Thromb Vasc Biol. 31:2787–2791. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Martinet W and De Meyer GR: Autophagy in
atherosclerosis: a cell survival and death phenomenon with
therapeutic potential. Circ Res. 104:304–317. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Martinez J, Almendinger J, Oberst A, Ness
R, Dillon CP, Fitzgerald P, Hengartner MO and Green DR:
Microtubule-associated protein 1 light chain 3 alpha
(LC3)-associated phagocytosis is required for the efficient
clearance of dead cells. Proc Natl Acad Sci USA. 108:17396–17401.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yu W, Fu YC, Chen CJ, Wang X and Wang W:
SIRT1: a novel target to prevent atherosclerosis. J Cell Biochem.
108:10–13. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Norata GD, Marchesi P, Passamonti S,
Pirillo A, Violi F and Catapano AL: Anti-inflammatory and
anti-atherogenic effects of cathechin, caffeic acid and
trans-resveratrol in apolipoprotein E deficient mice.
Atherosclerosis. 191:265–271. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang Z, Zou J, Cao K, Hsieh TC, Huang Y
and Wu JM: Dealcoholized red wine containing known amounts of
resveratrol suppresses atherosclerosis in hypercholesterolemic
rabbits without affecting plasma lipid levels. Int J Mol Med.
16:533–540. 2005.
|
|
29
|
Zhang QJ, Wang Z, Chen HZ, Zhou S, Zheng
W, Liu G, Wei YS, Cai H and Liu DP: Endothelium-specific
overexpression of class III deacetylase SIRT1 decreases
atherosclerosis in apolipoprotein E-deficient mice. Cardiovasc Res.
80:191–199. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Li L, Zhang HN, Chen HZ, Gao P, Zhu LH, Li
HL, Lv X, Zhang QJ, Zhang R, Wang Z, She ZG, Zhang R, Wei YS, Du
GH, Liu DP and Liang CC: SIRT1 acts as a modulator of neointima
formation following vascular injury in mice. Circ Res.
108:1180–1189. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Houtkooper RH, Pirinen E and Auwerx J:
Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol
Cell Biol. 13:225–238. 2012.PubMed/NCBI
|
|
32
|
Mattagajasingh I, Kim CS, Naqvi A,
Yamamori T, Hoffman TA, Jung SB, DeRicco J, Kasuno K and Irani K:
SIRT1 promotes endothelium-dependent vascular relaxation by
activating endothelial nitric oxide synthase. Proc Natl Acad Sci
USA. 104:14855–14860. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Brunet A, Sweeney LB, Sturgill JF, Chua
KF, Greer PL, Lin Y, Tran H, Ross SE, Mostoslavsky R, Cohen HY, Hu
LS, Cheng HL, Jedrychowski MP, Gygi SP, Sinclair DA, Alt FW and
Greenberg ME: Stress-dependent regulation of FOXO transcription
factors by the SIRT1 deacetylase. Science. 303:2011–2015. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stein S and Matter CM: Protective roles of
SIRT1 in atherosclerosis. Cell Cycle. 10:640–647. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lee IH, Cao L, Mostoslavsky R, Lombard DB,
Liu J, Bruns NE, Tsokos M, Alt FW and Finkel T: A role for the
NAD-dependent deacetylase Sirt1 in the regulation of autophagy.
Proc Natl Acad Sci USA. 105:3374–3379. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kockx MM, De Meyer GR, Buyssens N, Knaapen
MW, Bult H and Herman AG: Cell composition, replication, and
apoptosis in atherosclerotic plaques after 6 months of cholesterol
withdrawal. Circ Res. 83:378–387. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nowicki M, Zabirnyk O, Duerrschmidt N,
Borlak J and Spanel-Borowski K: No upregulation of lectin-like
oxidized low-density lipoprotein receptor-l in serum-deprived
EAhy926 endothelial cells under oxLDL exposure, but increase in
autophagy. Eur J Cell Biol. 86:605–615. 2007. View Article : Google Scholar
|
|
38
|
Yoshimori T: Autophage: a regulated bulk
degradation process inside cells. Biochem Biophys Res Commm.
313:453–458. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Singh R and Cuervo AM: Autophagy in the
cellular energetic balance. Cell Metab. 13:495–504. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Singh R and Cuervo AM: Lipophagy:
connecting autophagy and lipid metabolism. Int J Cell Biol.
2012:2820412012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Liu K and Czaja MJ: Regulation of lipid
stores and metabolism by lipophagy. Cell Death Differ. 20:3–11.
2013. View Article : Google Scholar
|
|
43
|
Ouimet M, Franklin V, Mak E, Liao X, Tabas
I and Marcel YL: Autophagy regulates cholesterol efflux from
macrophage foam cells via lysosomal acid lipase. Cell Metab.
13:655–667. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kiffin R, Bandyopadhyay U and Cuervo AM:
Oxidative stress and autophagy. Antioxid Redox Signal. 8:152–162.
2006. View Article : Google Scholar
|
|
45
|
Ohsumi Y: Molecular dissection of
autophagy: two ubiquitin-like systems. Nat Rev Mol Cell Biol.
2:211–216. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Shintani T and Klionsky DJ: Autophagy in
health and disease: a double-edged sword. Science. 306:990–995.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lum JJ, DeBerardinis RJ and Thompson CB:
Autophagy in metazoans: cell survival in the land of plenty. Nat
Rev Mol Cell Biol. 6:439–448. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Li S, Sun Y, Liang CP, Thorp EB, Han S,
Jehle AW, Saraswathi V, Pridgen B, Kanter JE, Li R, Welch CL, Hasty
AH, Bornfeldt KE, Breslow JL, Tabas I and Tall AR: Defective
phagocytosis of apoptotic cells by macrophages in atherosclerotic
lesions of ob/ob mice and reversal by a fish oil diet. Circ Res.
105:1072–1082. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Yancey PG, Blakemore J, Ding L, Fan D,
Overton CD, Zhang Y, Linton MF and Fazio S: Macrophage LRP-1
controls plaque cellularity by regulating efferocytosis and Akt
activation. Arterioscler Thromb Vasc Biol. 30:787–795. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Jehle AW, Gardai SJ, Li S, Linsel-Nitschke
P, Morimoto K, Janssen WJ, Vandivier RW, Wang N, Greenberg S, Dale
BM, Qin C, Henson PM and Tall AR: ATP-binding cassette transporter
A7 enhances phagocytosis of apoptotic cells and associated ERK
signaling in macrophages. J Cell Biol. 174:547–556. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
He X, Andersson G, Lindgren U and Li Y:
Resveratrol prevents RANKL-induced osteoclast differentiation of
murine osteoclast progenitor RAW 264.7 cells through inhibition of
ROS production. Biochem Biophys Res Commun. 401:356–362. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Peled T, Shoham H, Aschengrau D, Yackoubov
D, Frei G, Rosenheimer GN, Lerrer B, Cohen HY, Nagler A, Fibach E
and Peled A: Nicotinamide, a SIRT1 inhibitor, inhibits
differentiation and facilitates expansion of hematopoietic
progenitor cells with enhanced bone marrow homing and engraftment.
Exp Hematol. 40:342–355. 2012. View Article : Google Scholar
|
|
53
|
Zhang JG, Zhao G, Qin Q, Wang B, Liu L,
Liu Y, Deng SC, Tian K and Wang CY: Nicotinamide prohibits
proliferation and enhances chemosensitivity of pancreatic cancer
cells through deregulating SIRT1 and Ras/Akt pathways.
Pancreatology. 13:140–146. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Stein S, Lohmann C, Schäfer N, Hofmann J,
Rohrer L, Besler C, Rothgiesser KM, Becher B, Hottiger MO, Borén J,
McBurney MW, Landmesser U, Lüscher TF and Matter CM: SIRT1
decreases Lox-1-mediated foam cell formation in atherogenesis. Eur
Heart J. 31:2301–2309. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Guarente L and Franklin H: Epstein
Lecture: Sirtuins, aging, and medicine. N Engl J Med.
364:2235–2244. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Chen Lf, Fischle W, Verdin E and Greene
WC: Duration of nuclear NF-kappaB action regulated by reversible
acetylation. Science. 293:1653–1657. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Albani D, Polito L, Batelli S, De Mauro S,
Fracasso C, Martelli G, Colombo L, Manzoni C, Salmona M, Caccia S,
Negro A and Forloni G: The SIRT1 activator resveratrol protects
SK-N-BE cells from oxidative stress and against toxicity caused by
alpha-synuclein or amyloid-beta 1–42 peptide. J Neurochem.
110:1445–1456. 2009.
|
|
58
|
Wu Y, Li X, Zhu JX, Xie W, Le W, Fan Z,
Jankovic J and Pan T: Resveratrol-activated AMPK/SIRT1/autophagy in
cellular models of Parkinson’s disease. Neurosignals. 19:163–174.
2011.PubMed/NCBI
|
|
59
|
Takeda-Watanabe A, Kitada M, Kanasaki K
and Koya D: SIRT1 inactivation induces inflammation through the
dysregulation of autophagy in human THP-1 cells. Biochem Biophys
Res Commun. 427:191–196. 2012. View Article : Google Scholar : PubMed/NCBI
|