Introduction
Microglia are the resident innate immune cells of
the central nervous system (CNS) that regulate neuroinflammation
(1). Activated microglia execute
host defense and repair injured tissue in the CNS, but they also
contribute to the expansion of inflammation in the CNS by means of
antigen presentation and release of inflammation-related mediators
such as cytokines, chemokines, and nitric oxide (2–4).
For example, Toll-like receptor 4 (TLR4), which is expressed in
microglia through its ligation with lipopolysaccharide (LPS), has
been reported to be involved in microglial activation and cause
neuronal injury due to the production and release of
inflammation-related mediators (5). However, much remains elusive with
regard to how the immunoregulatory molecules controlling microglial
function are involved in neuroinflammation.
The roles that semaphorins and their receptors,
plexins, play in the CNS have been previously investigated.
Semaphorins and plexins comprise two large families of molecules
that transmit signals crucial for the control of neuronal axon
guidance, cell migration, cell morphology, and cell-to-cell
interactions (6). The various
functions of semaphorins and plexins have also been exemplified in
the development of blood vessels and heart (7,8)
and in tumorigenesis (9).
Previous investigations have demonstrated a role for these
molecules in the immune system (10). For example, Plexin-C1 expressed in
dendritic cells moderately affect T-cell activation(11). Plexin-D1, which is highly
expressed in double-positive thymocytes, regulates the intrathymic
migration of immature thymocytes from the thymic cortex to medulla
(12), and Plexin-A4 is a
negative regulator in T-cell activation (13). Other studies have revealed the
diverse functions of plexins in the immune system. Plexin-A1
expressed in dendritic cells regulates the interaction between
dendritic cells and T cells to control adaptive immunity (14,15). Plexin-B1 expressed in microglia
and Plexin-A4 in macrophages exhibit a crucial role in promoting
the activation of these innate immune cells through the enhancement
of intracellular signals initiated by inflammatory stimuli. For
example, macrophages in Plexin-A4-deficient mice show the
significant attenuation of LPS-induced cytokine production
(16). Administration of a ligand
of Plexin-A4, Semaphorin 3A (Sema3A), to cultured peritoneal
macrophages enhances LPS-induced cytokine production in a
Plexin-A4-dependent manner (16).
Sema4D enhances the CD40-mediated activation of microglia through
Plexin-B1 (17).
In a previous study, we showed that
Plexin-A1-deficient microglial cells are significantly resistant to
LPS-induced inflammation and that Plexin-A1-mediated signaling
increased microglial NO production through crosstalk with the
LPS-stimulated TLR4 pathway (18). However, the role that
Plexin-A1-mediated signaling plays in the expression of other
LPS-induced inflammatory mediators in microglia has not been
clarified. Similarly, the intracellular signal transduction pathway
of Sema3A-Plexin-A1 signaling, which is involved in the increase of
LPS-induced microglial NO production remains to be clarified.
Against this background, using a BV-2 microglial
cell line, the aim of this study was to examine the intracellular
signal transduction pathway of Sema3A-Plexin-A1-mediated signaling,
which is involved in LPS-stimulated microglial activation.
Additionally, we aimed to demonstrate the necessity of Plexin-A1
for the optimal production of inflammatory mediators following
stimulation of the TLR4 pathway in BV-2 cells. In this study, the
targeted silencing of Plexin-A1 in BV2 cells resulted in the
significantly decreased activation of nuclear factor-κB (NF-κB) and
of extracellular signal regulated kinase 1/2 (ERK1/2) in the LPS
response. Sema3A serves as a ligand for Plexin-A1 in BV-2 cells and
enhances LPS-induced NO production in the cell line through ERK
activation in a Plexin-A1-dependent manner. Taking these findings
together, we propose the functional role of Plexin-A1 as a
regulator of the TLR4 pathway in microglia.
Materials and methods
Cell cultures and transfection
BV-2 murine microglial cells immortalized by
infection with v-raf/c-myc recombinant retrovirus (19) were kindly provided by Dr N. Kaneda
(Faculty of Pharmacy, Meijo University) with permission to use the
cell line from Dr E. Blasi (Department of Diagnostics, Clinical and
Public Health Medicine, University of Modena and Reggio Emilia,
Modena, Italy). In brief, BV2 cells were maintained in Dulbecco’s
modified Eagle’s medium (DMEM) (Wako, Osaka, Japan) supplemented
with 10% fetal bovine serum (FBS; Equitech Bio, Inc., Kerrville,
TX, USA), penicillin (20 U/ml), and streptomycin (20 μg/ml,
Sigma-Aldrich, St. Louis, MO, USA). For transfection with small
interfering RNA (siRNA), BV-2 cells were seeded in 6-well plates
for western blotting, 24-well plates for reverse
transcription-polymerase chain reaction (RT-PCR) and quantitative
RT-PCR, and 96-well plates for the Griess reaction and
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl-tetrazolium bromide (MTT)
assay at cell densities of 1×105, 2×104, and
0.3×104, respectively. Plated cells were grown in DMEM
with 10% FBS overnight, and were then transfected with
siRNA-Plexin-A1 (MS0178728; Takara Bio Inc., Shiga, Japan) and
siRNA-control (SNC01; Takara Bio Inc.) using the Lipofectamine
RNAiMAX transfection reagent (Life Technologies, Carlsbad, CA, USA)
48 h prior to treatment with reagents such as LPS and Sema3A,
according to the manufacturer’s protocol.
Mice
Plexin-A1-deficient mice were produced by the
gene-targeting method with E14.1 embryonic stem (ES) cells
(15). The gene-targeting vector
was designed to replace the exon covering the initiation codon with
a neomycin-resistance gene and then transfected into ES cells by
electroporation. G418 and ganciclovir-resistant clones were
screened by PCR and confirmed by Southern blotting. Mutant ES cells
were introduced into mouse blastocysts, then transferred into
pseudopregnant mice to generate chimeras. Chimeras were bred with
Balb/c mice to transmit the mutant allele to the germline. Pairs of
resultant heterozygous mice were bred to gain homozygous knockout
mice, which were backcrossed with Balb/c mice. This study used F10
generation knockout mice, with their wild-type littermates as
controls. Mice were housed in the animal facilities of Wakayama
Medical University and the animal center of the Faculty of
Pharmacy, Meijo University. The care and use of mice and other
experimental protocols were conducted in accordance with the
guidelines of the Physiological Society of Japan as well as the
guidelines on animal experimentation of both Wakayama Medical
University and Meijo University. The Animal Ethics Review Committee
of both institutions approved the experimental protocol.
Genotype analysis
Genotyping was performed by PCR with mouse tail DNA
as the template and a Plexin-A1 gene-specific primer set as
previously reported (15).
RT-PCR analysis for Plexin-A1 and
Neuropilin-1 gene transcripts
RNA was extracted from primary microglia, the BV-2
microglial cell line, or murine hippocampus by the SV Total RNA
Isolation System (Promega, Madison, WI, USA). Reverse transcription
of RNA was performed with SuperScript III reverse transcriptase and
random primers (Life Technologies). All the samples were normalized
with β-actin amplification for semiquantification. The following
oligonucleotides were used for PCR amplification: Plexin-A1,
forward: 5′-GTGTGTGGATAGC CATCA-3′ and reverse:
5′-CCAGCCTCTCGAACACT-3′; Neuropilin-1, forward:
5′-GGCCTCCTGCGATTCGTTACTGC T-3′ and reverse:
5′-CTTAGCCTTGCGCTTGCTGTCATC-3′; and Sema3A, forward:
5′-ATTGAATTCAACTATGCAAACGG AAAGAA-3′ and reverse:
5′-TAAGCGGCCGCGACACTTCTG GGTGCCCGCT-3′. Control primers used were:
β-actin, forward: 5′-GGGACGACATGGAGAAGATC-3′ and reverse: 5′-AGG
TCTTTACGGATGTCAACG-3′. All the primers were annealed at 63°C, and
35 cycles of amplification were performed.
Western blotting
For western blotting, tissue extracts were prepared
by homogenizing murine hippocampal tissue in T-PER Tissue Protein
Extraction Reagent (Thermo Scientific Inc., Waltham, MA, USA)
containing the protease inhibitor, α-complete (Roche Applied
Science, Penzberg, Germany). Fifteen micrograms of each sample were
adjusted to give a final solution of 60 mM Tris-HCl (pH 6.8), 2%
SDS, 10% glycerol, 0.1% bromophenol blue, and 5% β-mercaptoethanol.
The solution was heated at 95°C for 5 min, electrophoresed through
a 10% SDS-polyacrylamide gel, and transferred to polyvinylidine
difluoride membranes (Amersham Pharmacia Biotech, Buckinghamshire,
UK). Plexin-A1, Neuropilin-1, TLR4, iNOS, p-NF-κB, NF-κB, p-IκB-α,
IκB-α, p-ERK1/2, ERK1/2, and β-actin were detected with their
respective antibodies using an enhanced chemiluminescence western
blot detection system (Amersham Pharmacia Biotech) in accordance
with the manufacturer’s instructions. Antibodies used were
anti-Plexin-A1 antibody (Abcam, Cambridge, MA, USA),
anti-Neuropilin1 antibody (Abcam), anti-TLR4 antibody (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), anti-iNOS antibody (Merck
Millipore, Darmstadt, Germany), anti-p-NF-κB antibody (Cell
Signaling Technology, Beverly, MA, USA), anti-NF-κB antibody (Cell
Signaling Technology), anti-p-IκB-α antibody (Cell Signaling
Technology), anti-IκB-α antibody (Cell Signaling Technology),
anti-p-ERK1/2 antibody (Cell Signaling Technology), anti-ERK1/2
antibody (Cell Signaling Technology), anti-Sema3A antibody (Abcam),
and anti-β-actin antibody (Cell Signaling Technology).
NO assay and cell viability assay
To investigate the effect of Sema3A on NO production
in the BV-2 microglial cell line, the nitrite content was measured
with Griess reagent [1% sulfanilamide/0.1%
N-(1-naphthyl)-ethylenediamine dihydrochloride in 5% phosphoric
acid] according to the manufacturer’s instructions. BV-2 microglial
cells were plated at 0.3×104 cells/well in a 96-well
plate and cultured overnight in a CO2 incubator at 37°C,
combined with control siRNA or Plexin-A1-specific siRNA, and
incubated for two additional days. Eighteen hours after stimulation
of BV-2 cells with LPS and Sema3A or control IgG, 50 μl of the
culture supernatant were mixed with 50 μl of Griess reagent and
incubated for 15 min. Absorbance values were measured at 540 nm in
a plate reader (Bio-Rad, Gladesville, NSW, Australia), with fresh
DMEM as a blank in all the experiments. NO concentration was
calculated with reference to the nitrite standard curve. To analyze
cell viability of the BV-2 cells subjected to NO assay, 5 μl of MTT
(5 mg/ml; Sigma, Tokyo, Japan) were added to the BV-2 cells and
incubated at 37°C for 4 h. After the medium was removed, formazan,
a product of the MTT tetrazolium ring that was precipitated by the
action of mitochondrial dehydrogenases, was dissolved with 0.1 N
HCl in anhydrous isopropanol containing 10% Triton X-100 and
quantified spectrophotometrically at 595 nm for measurement of cell
viability.
Quantitative RT-PCR for analysis of
inflammatory gene transcripts
RNA was isolated from BV-2 microglial cells cultured
on a 24-well plate using the SV Total RNA Isolation System
(Promega). Quantitative RT-PCR with SYBR-Green (Qiagen, Tokyo,
Japan) was performed on a 7500 Fast Real-Time PCR System (Applied
Biosystems, Tokyo, Japan). Amplification of Rn18s was performed for
sample normalization. For the detection and quantification of
Rn18s, TNF-α, IL-1β, and iNOS transcripts, quantitative RT-PCR was
performed using QuantiTect Primer Assays following the
manufacturer’s protocol (Mm_Rn18s_2_SG QT01036875Mm_Tnf_1_SG
QT00104006, Mm_Il1b_2_SG QT01048355, and Mm_iNOS_1_SG QT00100275,
respectively; Qiagen).
Reagents for cell cultures
LPS (Escherichia coli serotype 055:B5),
Griess reagent, and MTT were obtained from Sigma-Aldrich.
Recombinant Sema3A, which consists of the extracellular region of
Sema3A and the Fc portion of murine IgG2A (Sema3A-Fc) and murine
IgG2A, was purchased from R&D Systems (Minneapolis, MN, USA).
ERK1/2 inhibitor PD98059 was purchased from Cell Signaling
Technology.
Statistical analysis
Data were expressed as the means ± standard error of
mean (SEM). Statistical analyses were performed with the Student’s
t-test or one-way analysis of variance followed by post-hoc
analysis. P<0.05 was considered statistically significant.
Results
Plexin-A1 is expressed by BV-2 cells
In a previous study, we found that murine primary
microglia expressed Plexin-A1 (18). In the present study, the
expression of Plexin-A1 in the BV-2 mouse microglial cell line was
confirmed by RT-PCR analysis and western blotting (Fig. 1).
Plexin-A1 is required for iNOS, IL-1β and
TNF-α expression in BV-2 cells
As in primary microglia, LPS stimulation induces an
increased expression of inflammatory mediators such as iNOS, IL-1β,
and TNF-α in BV-2 cells (20–22). Although we previously demonstrated
that Plexin-A1 is involved in LPS-induced NO production in primary
microglia (unpublished data), the involvement of Plexin-A1 in the
expression of other TLR4-induced inflammatory mediators has yet to
be adequately examined. In the present study, we examined the role
of Plexin-A1 in inflammatory factor production in response to TLR4
activation in BV-2 cells. After BV-2 cells were treated with
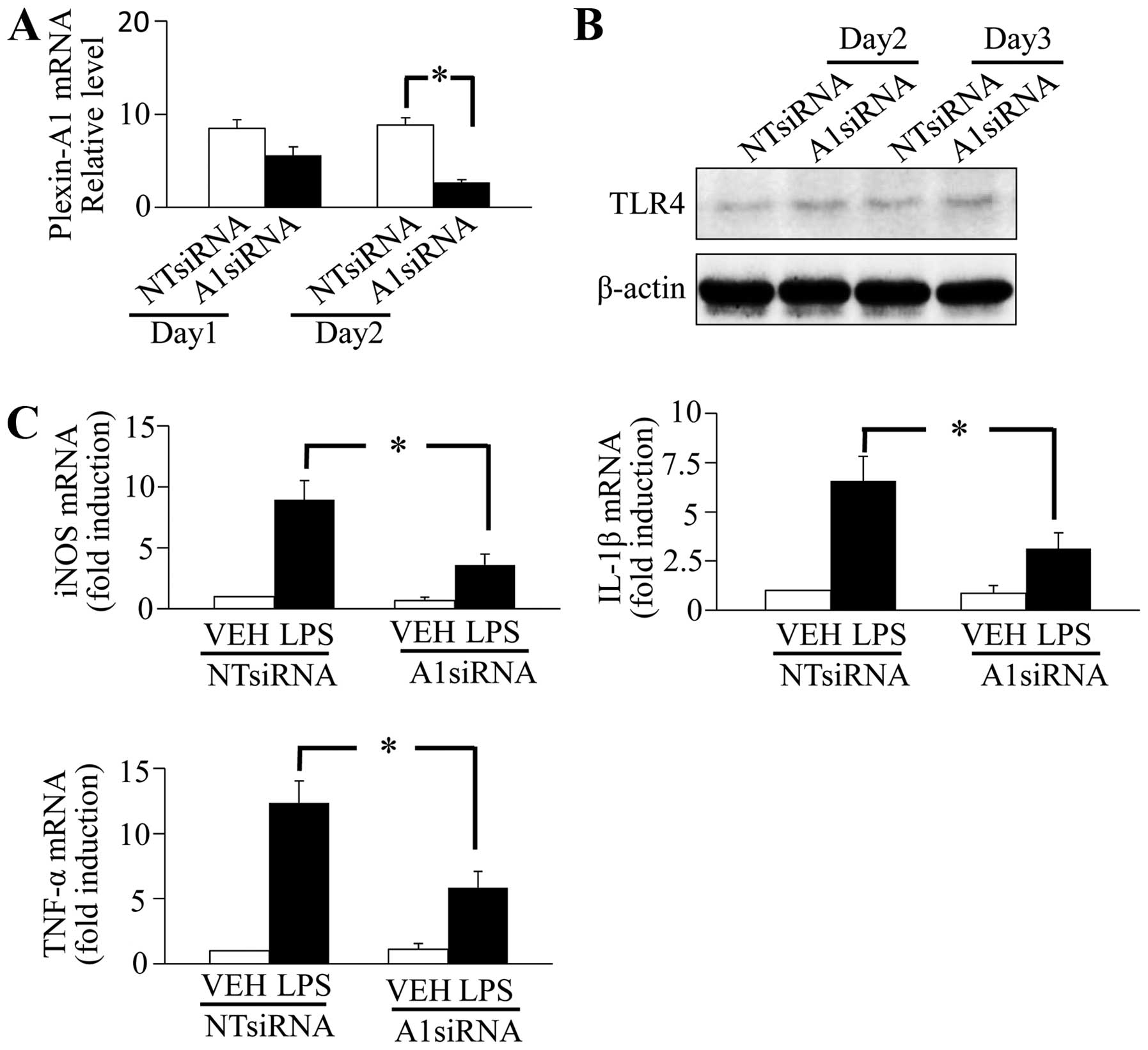
Plexin-A1-specific siRNA and cultivated for 1 or 2 days, the
knockdown efficiency of Plexin-A1 was confirmed using quantitative
RT-PCR, and a significant decrease of Plexin-A1 mRNA was observed
in the BV-2 cells that were cultured for 2 days after siRNA
treatment (Fig. 2A). To examine
the effect of Plexin-A1 on TLR4 expression, TLR4 expression level
was assessed using western blotting in Plexin-A1 knockdown cells;
however, no significant change was observed (Fig. 2B). To examine the role of
Plexin-A1 in the LPS-TLR4 signaling pathway in BV-2 cells, the
expression levels of iNOS, IL-1β and TNF-α were assessed with
quantitative RT-PCR in Plexin-A1 knockdown cells treated with
vehicle or LPS. During TLR4 stimulation with LPS, a significantly
lower amount of iNOS, IL-1β and TNF-α mRNA was generated in BV-2
cells with Plexin-A1 knockdown than BV-2 cells treated with control
non-target (NT) siRNA (Fig. 2C).
These findings showed that Plexin-A1 is required for the
TLR4-mediated generation of proinflammatory factors in BV-2 cells.
Assessment of LPS-induced iNOS protein expression using western
blotting showed a significantly lower level of iNOS protein
expression in BV-2 cells with targeted silencing of Plexin-A1 than
in control siRNA-treated BV-2 cells (Fig. 3A and B). Assessment of LPS-induced
NO production by the Griess reaction revealed a significant
decrease in NO in Plexin-A1-specific siRNA-treated BV-2 cells
compared with control siRNA-treated cells (Fig. 3C). MTT assay showed no significant
differences in cell viability among any BV-2 cell treatments
(Fig. 3D).
Plexin-A1 mediates the activation of
NF-κB and ERK
During the microglial response to TLR agonists and
microbial pathogens, NF-κB and mitogen-activated protein kinase
(MAPK) signaling pathways are known to regulate the production of
inflammatory mediators (23–26). Since the generation of iNOS,
IL-1β, and TNF-α was decreased by Plexin-A1 knockdown (Figs. 2C and 3), we investigated the role of Plexin-A1
in these signaling pathways in BV-2 microglial cells. In a previous
study, we reported that Sema3A-Plexin-A1 signaling increased NO
production through crosstalk with the TLR4 pathway in primary
microglia (18). Sema4D-Plexin-B1
signaling also increased NO production through ERK1/2 activation,
which is dependent on inflammatory stimulation in primary microglia
(17). Activation of ERK1/2 also
plays a crucial role in the induction of iNOS expression in
TLR4-stimulated microglia (27).
In neuronal cells, Plexin-A1 is involved in the determination of
the direction of axonal extension and the induction of growth cone
collapse in an ERK1/2 activation-dependent manner through crosstalk
with Plexin-B1 (28–32). Plexin-A1-mediated signaling in
microglia may therefore be involved in the production of
inflammatory factors through ERK1/2 activation by cooperating with
the TLR4 pathway. Accordingly, to examine whether Plexin-A1 is
involved in the activation of NF-κB and ERK1/2 in LPS-stimulated
microglia, the phosphorylation state of these proteins was assessed
in vehicle- or LPS-treated BV-2 cells with either control siRNA or
Plexin-A1-specific siRNA. Control NT siRNA-treated BV-2 cells
exhibited enhanced phosphorylation of NF-κB, IκB-α, and ERK1/2
after LPS stimulation. In contrast, BV-2 cells with targeted
knockdown of Plexin-A1 exhibited a significant decrease in NF-κB
and IκB-α phosphorylation compared with cells treated with control
siRNA (Fig. 4). LPS stimulation
induced a significant increase of ERK1/2 phosphorylation in control
siRNA-treated BV-2 cells, but not in Plexin-A1-specific
siRNA-treated BV-2 cells (Fig.
4). These findings indicate that Plexin-A1 deficiency leads to
a significant defect in NF-κB and ERK1/2 activation in
TLR4-mediated signaling in BV-2 microglial cells.
Sema3A enhances LPS-induced nitrite
production through ERK activation in a Plexin-A1-dependent
manner
Since Plexin-A1 knockdown suppressed the expression
of LPS-induced inflammatory mediators, the BV-2 cell itself may
produce the Plexin-A1 ligand Sema3A. A possible mechanism may be
that the production and/or secretion of Sema3A in BV-2 cells is
facilitated by LPS, after which Sema3A binds with Plexin-A1 to
activate BV-2 cells in an autocrine manner. To test the
possibility, BV-2 cells were treated with LPS and examined for
evidence of Sema3A mRNA production and Sema3A protein secretion in
the culture medium. Two-hour LPS treatment of BV-2 cells
significantly increased Sema3A mRNA levels (Fig. 5A). Furthermore, 4-h LPS treatment
of BV-2 cells induced the appearance of proteolytically processed
Sema3A (65 kDa; Fig. 5B) in the
supernatant, as shown in other experimental systems (33–35). The data suggest that the
production and secretion of Sema3A induced by the TLR4 activation
of BV-2 cells may cause Plexin-A1 to function in a signaling loop,
thereby enhancing inflammatory signals and cytokine production. The
above results suggested that Sema3A-Plexin-A1 signaling is involved
in the increase of LPS-induced NO production through ERK1/2 in BV-2
cells (Figs. 3 and 4). Thus, BV-2 cells transduced with
control NT or Plexin-A1 siRNA were treated with Sema3A in the
presence (100 ng/ml) or absence of LPS to functionally examine
whether Sema3A functions as a ligand for Plexin-A1 in BV-2 cells.
The Sema3A ligand was composed of Sema3A fused with the murine
IgG2A Fc fragment, and the IgG2A Fc fragment alone was utilized as
a negative control. In the presence of LPS, nitrite levels were
increased (Fig. 5C); in its
absence, Sema3A did not induce nitrite production in BV-2 cells.
Following the addition of soluble Sema3A (Fig. 5C), LPS-induced NO production was
intensified in BV-2 cells transduced with NT siRNA. By contrast,
the addition of the IgG2A Fc control made no effect. Enhancement of
LPS responsiveness was not observed in BV-2 cells with Plexin-A1
knockdown (Fig. 5C). Accordingly
it is indicated that Sema3A ligation to Plexin-A1 intensifies NO
generation of BV-2 cells only in the presence of LPS, which
suggests that in BV-2 cells, both LPS and Sema3A are necessary for
the establishment of optimal intracellular signaling to produce
inflammatory mediators. To clarify whether the increase in NO
production from Sema3A-Plexin-A1 signaling is dependent on the
activation of ERK1/2, we examined whether treatment with ERK1/2
inhibitor suppressed the increase in LPS-induced NO production
following the addition of Sema3A in BV-2 cells. BV-2 cells treated
with LPS, Sema3A, and ERK1/2 inhibitor showed significantly
decreased NO production, as compared with BV-2 cells treated with
only LPS and Sema3A (Fig. 5C).
These findings indicated that Sema3A-Plexin-A1 signaling was
involved in the increased production of NO through ERK1/2
activation in the microglial response to LPS. These in vitro
results suggested that Plexin-A1 binding by Sema3A is synergized
with LPS engagement of TLR4 on microglia to expand innate
inflammatory responses in the brain.
Discussion
Findings of this study have shown three novel
findings concerning the roles of Plexin-A1 expressed in BV2
microglial cells. First, Plexin-A1 enhances the LPS-induced
increase in inflammatory mediators in BV-2 cells, suggesting that
Plexin-A1 plays a crucial role in the activation process of
microglia. Second, the results clarified that Plexin-A1 in BV-2
cells is involved in the activation of NF-κB and MAPK pathway in
the LPS response. Third, the results indicate that Sema3A-Plexin-A1
signaling increases LPS-induced NO production through ERK
activation in BV-2 cells, thereby demonstrating that part of the
intracellular Sema3A-Plexin-A1 signaling pathway operates in the
activation of microglia. Results of a previous study (18) have shown that Sema3A-Plexin-A1
signaling increases NO production through crosstalk with TLR4
signaling in primary microglia. However, the mechanisms involved in
the intracellular signaling pathway were not identified. Therefore,
to the best of our knowledge, this is the first study showing the
crucial involvement of Sema3A-Plexin-A1 signaling in the
LPS-induced production of inflammatory mediators through NF-κB and
ERK activation in microglia.
We confirmed the expression of Plexin-A1 and
Neuropilin-1 with RT-PCR and western blotting in BV-2 cells
(Fig. 1). The binding of Sema3A
to the receptor complex of Neuropilin-1 and Plexin-A1 on the cell
membrane transduces intracellular signaling (36). Our earlier study has confirmed the
involvement of Sema3A-Plexin-A1 signaling in increased LPS-induced
NO production in primary microglia (18). Similar to primary microglia,
LPS-stimulated BV-2 cells increase the expression of inflammatory
mediators such as iNOS, IL-1β and TNF-α (20–22), although previous studies have not
directly demonstrated that Plexin-A1 is involved in the induction
of inflammatory mediators by the stimulation of TLR4. To
investigate the role of Plexin-A1 in BV-2 cells stimulated by LPS,
Plexin-A1 was knocked down using Plexin-A1-specific siRNA and the
expression level of inflammatory mediators was assessed in
microglia with TLR4 activation. As a result, Plexin-A1-specific
siRNA-treated BV-2 cells showed a significant decrease in iNOS,
IL-1β and TNF-α mRNA expression in LPS-treated cells as compared
with control siRNA-treated cells (Fig. 2C). Quantification of the iNOS
protein with western blotting and NO in the culture supernatant
using the Griess reaction showed significant decreases of iNOS and
NO levels in Plexin-A1-specific siRNA treated-BV-2 cells compared
with control siRNA-treated cells (Fig. 3A–C). Since the assessment of cell
viability by MTT assay did not show any significant differences
between Plexin-A1-specific siRNA-treated and control NT
siRNA-treated BV-2 cells, the significant decrease in NO production
may not reflect group-based differences of cell viability.
Accordingly Plexin-A1 is required to increase the expression of
inflammatory mediators in LPS-stimulated BV-2 cells.
During the microglial response to microbial
pathogens and TLR agonists, MAPK and NF-κB signaling pathways are
known to regulate the production of inflammatory mediators
(23–26). The decreased generation of iNOS,
IL-1β and TNF-α after targeted silencing of Plexin-A1 led to the
investigation of the potential role of Plexin-A1 in these signaling
pathways (Figs. 2C, 3A and B). Previously, we clarified that
Sema3A-Plexin-A1 signaling is involved in NO production in primary
microglia by crosstalk with TLR4-mediated signaling (18). Activation of ERK1/2 plays a
crucial role in the induction of iNOS expression in TLR4-stimulated
microglia (27). Sema4D-Plexin-B1
signaling increases NO production through ERK1/2 activation
dependent on inflammatory stimulation in primary microglia
(17). In neuronal cells,
Plexin-A1 is involved in determining the direction of axonal
extension and growth cone collapse in an ERK1/2
activation-dependent manner through crosstalk with Plexin-B1
(28–32). In microglial cells, therefore,
Plexin-A1 signaling may be involved in the production of
inflammatory factors through ERK1/2 activation and cooperative
action with the TLR4 pathway. To clarify the intracellular
signaling molecules located downstream of Plexin-A1 signaling that
have crosstalk with the TLR4 pathway in microglial activation, we
assessed the activation level of NF-κB, IκB-α and ERK1/2 by western
blotting. Phosphorylated NF-κB and IκB-α significantly increased in
control siRNA-treated BV-2 cells 1 h after LPS stimulation as
compared with Plexin-A1-specific siRNA-treated BV2 cells. These
findings suggest that Plexin-A1 is involved in the phosphorylation
of NF-κB and IκB-α in the LPS response of BV-2 cells. In
Plexin-A1-specific siRNA-treated BV-2 cells, LPS treatment induced
a significant increase of phosphorylated NF-κB and IκB-α compared
with vehicle-treated cells (Fig.
4). This result may be explained by the imperfect removal of
Plexin-A1 or the significant phosphorylation of NF-κB and IκB-α
through a pathway other than that of Plexin-A1 signaling. Sema3A
secreted from macrophages enhances the LPS response by acting in an
autocrine manner to activate Plexin-A4-mediated signaling (16). Therefore, the involvement of
Plexin-A4 signaling in LPS-dependent activation in microglia may be
a useful explanation for the fact that even in cells with targeted
silencing of Plexin-A1, phosphorylated NF-κB and IκB-α were
significantly increased with LPS treatment compared with vehicle
treatment. Since Plexin-A1 signaling may strengthen microglial
activation through ERK activation in response to LPS, the level of
phosphorylation of ERK was quantified by western blotting.
Phosphorylated ERK was found to increase significantly in control
siRNA-treated BV-2 cells compared with Plexin-A1-specific
siRNA-treated cells. This result suggests that Plexin-A1 may be
involved in LPS-induced microglial activation through ERK
phosphorylation. Since the phosphorylation of ERK1/2 in LPS
treatment of Plexin-A1-specific siRNA-treated BV-2 cells was not
found to be significantly greater than in the vehicle-treated BV-2
cells with Plexin-A1 knockdown, phosphorylation of ERK in the LPS
response may be dependent on Plexin-A1. In the LPS response of
macrophages, Plexin-A4-mediated signaling is involved in c-jun
N-terminal kinase phosphorylation (16), suggesting that plexin receptors
may increase the LPS response through differential pathways,
depending on various cell types or receptor families. Thus,
Plexin-A1 is crucial for the amplification of the LPS response in
microglia.
In BV-2 microglial cells, Sema3A was found to be
secreted by the cell itself in an autocrine manner, as in
macrophages (Fig. 4B). Whereas
injured neurons are suggested to produce Sema3A and Sema3A induces
microglial apoptosis (37),
microglia are also suggested to produce Sema3A which appears to
increase LPS-induced microglial activation through
Plexin-A1-mediated signaling. In control siRNA-treated BV-2 cells,
Sema3A generated a significant increase of LPS-induced NO
production as compared with control IgG, and this increase was
significantly suppressed by pretreatment with ERK inhibitor. By
contrast, NO levels did not increase significantly in BV-2 cells
with targeted knockdown of Plexin-A1 even after treatment with
Sema3A, when compared with the IgG treatment. These results suggest
that Sema3A-Plexin-A1 signaling is involved in the increase of
LPS-induced NO production in activated microglia through ERK
activation. Therefore, both Plexin-A1 and Sema3A are potential
targets for the treatment of encephalopathy triggered by LPS and
neuroinflammation-related mental disorders.
Acknowledgements
We would like to thank the members of the Department
of Physiology of Meijo University for their helpful discussion and
technical assistance. The study was primarily supported by a
Grant-in-Aid for Scientific Research from the Ministry of
Education, Science, Sports and Culture, Japan (No. 22590195), and
research grants from Meijo University.
References
|
1
|
Ransohoff RM and Perry VH: Microglial
physiology: unique stimuli, specialized responses. Annu Rev
Immunol. 27:119–145. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heppner FL, Greter M, Marino D, et al:
Experimental autoimmune encephalomyelitis repressed by microglial
paralysis. Nat Med. 11:146–152. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jack C, Ruffini F, Bar-Or A and Antel JP:
Microglia and multiple sclerosis. J Neurosci Res. 81:363–373. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ponomarev ED, Shriver LP and Dittel BN:
CD40 expression by microglial cells is required for their
completion of a two-step activation process during central nervous
system autoimmune inflammation. J Immunol. 176:1402–1410. 2006.
View Article : Google Scholar
|
|
5
|
Lee SJ and Lee S: Toll-like receptors and
inflammation in the CNS. Curr Drug Targets Inflamm Allergy.
1:181–191. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Tran TS, Kolodkin AL and Bharadwaj R:
Semaphorin regulation of cellular morphology. Annu Rev Cell Dev
Biol. 23:263–292. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Gu Y, Filippi MD, Cancelas JA, et al:
Hematopoietic cell regulation by Rac1 and Rac2 guanosine
triphosphatases. Science. 302:445–449. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Toyofuku T, Zhang H, Kumanogoh A, et al:
Guidance of myocardial patterning in cardiac development by Sema6D
reverse signalling. Nat Cell Biol. 6:1204–1211. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Neufeld G and Kessler O: The semaphorins:
versatile regulators of tumour progression and tumour angiogenesis.
Nat Rev Cancer. 8:632–645. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Suzuki K, Kumanogoh A and Kikutani H:
Semaphorins and their receptors in immune cell interactions. Nat
Immunol. 9:17–23. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Walzer T, Galibert L and De Smedt T:
Dendritic cell function in mice lacking Plexin C1. Int Immunol.
17:943–950. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Choi YI, Duke-Cohan JS, Ahmed WB, et al:
PlexinD1 glycoprotein controls migration of positively selected
thymocytes into the medulla. Immunity. 29:888–898. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Yamamoto M, Suzuki K, Okuno T, et al:
Plexin-A4 negatively regulates T lymphocyte responses. Int Immunol.
20:413–420. 2008. View Article : Google Scholar
|
|
14
|
Wong AW, Brickey WJ, Taxman DJ, et al:
CIITA-regulated plexin-A1 affects T-cell-dendritic cell
interactions. Nat Immunol. 4:891–898. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Takegahara N, Takamatsu H, Toyofuku T, et
al: Plexin-A1 and its interaction with DAP12 in immune responses
and bone homeostasis. Nat Cell Biol. 8:615–622. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wen H, Lei Y, Eun SY and Ting JP:
Plexin-A4-semaphorin 3A signaling is required for Toll-like
receptor- and sepsis-induced cytokine storm. J Exp Med.
207:2943–2957. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Okuno T, Nakatsuji Y, Moriya M, et al:
Roles of Sema4D-plexin-B1 interactions in the central nervous
system for pathogenesis of experimental autoimmune
encephalomyelitis. J Immunol. 184:1499–1506. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ito T, Yoshida K, Negishi T, et al:
Plexin-A1is required for Toll-like receptor-mediated microglial
activation in the development of lipopolysaccharide-induced
encephalopathy. Int J Mol Med. 33:1122–1130. 2014.PubMed/NCBI
|
|
19
|
Blasi E, Barluzzi R, Bocchini V, Mazzolla
R and Bistoni F: Immortalization of murine microglial cells by a
v-raf/v-myc carrying retrovirus. J Neuroimmunol. 27:229–237. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zeng KW, Wang S, Dong X, Jiang Y and Tu
PF: Sesquiterpene dimer (DSF-52) from Artemisia argyi
inhibits microglia-mediated neuroinflammation via suppression of
NF-κB, JNK/p38 MAPKs and Jak2/Stat3 signaling pathways.
Phytomedicine. 21:298–306. 2013.PubMed/NCBI
|
|
21
|
Manivannan J, Tay SS, Ling EA and Dheen
ST: Dihydropyrimidinase-like 3 regulates the inflammatory response
of activated microglia. Neuroscience. 253:40–54. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Oh WJ, Jung U, Eom HS, Shin HJ and Park
HR: Inhibition of lipopolysaccharide-induced proinflammatory
responses by Buddleja officinalis extract in BV-2 microglial cells
via negative regulation of NF-κB and ERK1/2 signaling. Molecules.
18:9195–9206. 2013.PubMed/NCBI
|
|
23
|
Kaminska B: MAPK signalling pathways as
molecular targets for anti-inflammatory therapy - from molecular
mechanisms to therapeutic benefits. Biochim Biophys Acta.
1754:253–262. 2005. View Article : Google Scholar
|
|
24
|
Kaminska B, Gozdz A, Zawadzka M,
Ellert-Miklaszewska A and Lipko M: MAPK signal transduction
underlying brain inflammation and gliosis as therapeutic target.
Anat Rec (Hoboken). 292:1902–1913. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Koistinaho M and Koistinaho J: Role of p38
and p44/42 mitogen-activated protein kinases in microglia. Glia.
40:175–183. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Johnson GL and Lapadat R:
Mitogen-activated protein kinase pathways mediated by ERK, JNK, and
p38 protein kinases. Science. 298:1911–1912. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Bhat NR, Zhang P, Lee JC and Hogan EL:
Extracellular signal regulated kinase and p38 subgroups of
mitogen-activated protein kinases regulate inducible nitric oxide
synthase and tumor necrosis factor-α gene expression in
endotoxin-stimulated primary glial cultures. J Neurosci.
18:1633–1641. 1998.PubMed/NCBI
|
|
28
|
Bechara A, Nawabi H, Moret F, et al:
FAK-MAPK-dependent adhesion disassembly downstream of L1
contributes to semaphorin3A-induced collapse. EMBO J. 27:1549–1562.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lerman O, Ben-Zvi A, Yagil Z and Behar O:
Semaphorin3A accelerates neuronal polarity in vitro and in its
absence the orientation of DRG neuronal polarity in vivo is
distorted. Mol Cell Neurosci. 36:222–234. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Bagnard D, Sainturet N, Meyronet D, et al:
Differential MAP kinases activation during semaphorin3A-induced
repulsion or apoptosis of neural progenitor cells. Mol Cell
Neurosci. 25:722–731. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Campbell DS and Holt CE: Apoptotic pathway
and MAPKs differentially regulate chemotropic responses of retinal
growth cones. Neuron. 37:939–952. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kruger RP, Aurandt J and Guan KL:
Semaphorins command cells to move. Nat Rev Mol Cell Biol.
6:789–800. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
33
|
Adams RH, Lohrum M, Klostermann A, Betz H
and Püschel AW: The chemorepulsive activity of secreted semaphorins
is regulated by furin-dependent proteolytic processing. EMBO J.
16:6077–6086. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Catalano A, Caprari P, Moretti S, Faronato
M, Tamagnone L and Procopio A: Semaphorin-3A is expressed by tumor
cells and alters T-cell signal transduction and function. Blood.
107:3321–3329. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Lepelletier Y, Moura IC, Hadj-Slimane R,
et al: Immunosuppressive role of semaphorin-3A on T cell
proliferation is mediated by inhibition of actin cytoskeleton
reorganization. Eur J Immunol. 36:1782–1793. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Banks WA and Erickson MA: The blood-brain
barrier and immune function and dysfunction. Neurobiol Dis.
37:26–32. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Majed HH, Chandran S, Niclou SP, et al: A
novel role for Sema3A in neuroprotection from injury mediated by
activated microglia. J Neurosci. 26:1730–1738. 2006. View Article : Google Scholar : PubMed/NCBI
|