Introduction
Innate immunity offers the first defense in mammals,
depending on its capacity to rapidly detect invading pathogens and
then eliminating the ‘foreign danger’ (1). However, innate immunity is also
involved in the development of inflammatory responses that occur in
a sterile milieu (2).
Inflammasomes are intracellular multiprotein complexes that
regulate the activity of caspase-1 and can be activated by various
cellular dangers that trigger the processing and release of
pro-inflammatory cytokines to engage innate immunity (3). Nucleotide-binding oligomerization
domain (NOD)-like receptors (NLRs) have recently been identified as
innate immune receptors that play a pivotal role in initiating the
inflammatory response (4). The
NLR family, pyrin domain-containing 3 (NLRP3) inflammasome is
currently one of the most fully characterized inflammasomes. The
NLRP3 inflammasome consists of the NLRP3 scaffold, the
apoptosis-associated speck-like protein containing a caspase
recruitment domain (CARD) (ASC) adaptor and caspase-1 (5,6).
The NLRP3 inflammasome is activated in response to a variety of
signals that include pathogen-associated molecular patterns (PAMPs)
or danger-associated molecular patterns (DAMPs) (6). The NLRP3 inflammasome is a
multiprotein complex that activates cysteine protease caspase-1,
which leads to the maturation and secretion of pro-inflammatory
cytokines, such as interleukin-1β (IL)-1β and IL-18 (7).
The molecular mechanisms of NLRP3 inflammasome
assembly and activation have been gradually explored. Previous
studies have indicated that reactive oxygen species (ROS)
generation, potassium (K+) efflux, and the release of
cathensin B can activate the NLRP3 inflammasome (3). It is also well recognized that the
activation of the NLRP3 inflammasome requires double signals: one
is a priming signal from PAMPs that controls the expression of
NLRP3; the second stimulus is DAMPs that induce the activation of
caspase-1 (3,5,8,9).
Lipopolysaccharide (LPS), the major outer membrane component in
Gram-negative bacteria, is one of the most characterized PAMPs
(10). Adenosine triphosphate
(ATP), released from dying cells, has a high concentration within
the cell. Extracellular ATP, one of the DAMPs, serves as the second
signal for NLRP3 inflammasome activation (11). The IL-1 family has been recognized
to play important roles in inflammation with pro-inflammatory
properties, associated with acute and chronic inflammation, and
plays a significant role in the innate immune defense (12). There are several cytokines in the
IL-1 family, including IL-1α, IL-1β, IL-18 and IL-33. Among the
IL-1 family of cytokines, IL-1β is the most characterized cytokine;
studies have demonstrated that IL-1β plays an essential role in the
pathophysiology of autoimmune diseases (13,14). Importantly, IL-1β maturation
requires 2 steps: first, the upregulation of the precursor of IL-1β
(pro IL-1β) transcription through Toll-like receptor (TLR) ligands,
such as LPS; and second, the activation of the cysteine protease,
caspase-1 (15). Caspase-1 is a
proteolytic enzyme that processes the inactive precursor of IL-1β
into the mature form, and is termed IL-1β converting enzyme (ICE).
Caspase-1 itself acts as an inactive precursor in the cytoplasm and
can be activated by NLRP3 inflammasome assembly and subsequent
proteolytic self-processing (16).
It has been widely accepted that the NLRP3
inflammasome plays critical roles in inflammation and immune system
regulation. However, whether LPS and/or ATP affect the regulation
of NLRP3 inflammasome components, the induction of caspase-1, the
cleavage and release of IL-1β, IL-18 and IL-33 remains unclear.
Thus, in this study, we performed a series of experiments using
RAW264.7 murine macrophage cells. Our findings suggested that a
high concentration of potassium chloride or the silencing of NLRP3
with small interfering (siRNA) partially abrogated the LPS-induced
secretion of pro-inflammatory cytokines in vitro.
Materials and methods
Chemicals and reagents
Dimethyl sulfoxide (DMSO), ATP, ultrapure LPS and
potassium chloride were purchased from Sigma (St. Louis, MO, USA).
Phenylmethanesulfonyl fluoride (PMSF) and RIPA lysis buffer were
both from the Beyotime Institute of Biotechnology (Shanghai,
China). Rabbit anti-NLRP3 polyclonal antibody, rabbit anti-ASC
polyclonal antibody, rabbit anti-caspase-1 polyclonal antibody,
mouse anti-β-actin monoclonal antibody were all purchased from
Santa Cruz Biotechnology, Inc. (Santa Cruz CA, USA). Dulbecco’s
modified Eagle’s medium (DMEM) and fetal bovine serum (FBS) were
obtained from HyClone Laboratories, Inc. (Logan, UT, USA) and
Tianchang Heng Sheng Medical Devices Co., Ltd. (Huzhou, China),
respectively. The mouse IL-1β, IL-18 and IL-33 enzyme-linked
immunosorbent assay (ELISA) kits were purchased from R&D
Systems (Minneapolis, MN, USA). Small interfering RNA targeting
NLRP3 were purchased from GenePharma Corp. (Shanghai, China). All
other chemicals were of reagent grade.
Cell culture and treatment
The RAW264.7 murine macrophages cells were obtained
from the American Type Culture Collection (ATCC; Rockville, MD,
USA). The cells were cultured in high glucose DMEM medium (HyClone
Laboratories, Inc.) supplemented with 10% (v/v) heat-inactivated
FBS (Tianchang Heng Sheng Medical Devices Co., Ltd.), 100 U/ml
penicillin and 100 mg/ml streptomycin (both from Beyotime Institute
of Biotechnology) and kept in a 37°C incubator under a humidified
atmosphere containing 5% CO2. The cells were allowed to
adhere overnight (37°C, 5% CO2) and washed with fresh
medium to remove unattached cells prior to the experiment. Our
experiment on RAW264.7 cells included 2 groups. In the first group,
cells were treated with LPS, ATP and a high concentration of
K+, including 8 subgroups. RAW264.7 cells were
stimulated with LPS (200 ng/ml) for 6 h and 2 mM ATP for an
additional 30 min (LPS + ATP) or ATP alone. Potassium chloride (150
mM) was additionally added to the cell culture medium for 30 min
prior to LPS and/or ATP stimulation in the last 4 subgroups. In the
second group, the cells were treated with LPS, ATP and NLRP3 siRNA,
including 7 subgroups. RAW264.7 cells without treatment (untreated
group) were used as the controls. In the second group, RAW264.7
cells were stimulated with LPS and/or ATP as indicated in the first
group. We transfected the RAW264.7 cells with NLRP3 siRNA prior to
stimulation with LPS and/or ATP in the last 3 subgroups. After
these treatments, supernatants were collected for ELISA assay. The
cells were washed with PBS 3 times, and then the cell lysis (the
mixture of RIPA and PMSF, v/v=100:1) was added into the culture
plate without any culture medium. The whole progress of protein
extraction was carried out on ice. The mixture, including cell
lysis and the cells was sucked out and then place into a 1.5 ml EP
tube. The protein sample of RAW264.7 cells for western blot
analysis was obtained by vortexing the mixture evenly and
centrifugation at 12,000 rpm, at 4°C for 40 min, then removing the
supernatant and adding protein loading buffer.
ELISA
The RAW264.7 cells (2×106 cells/well)
were seeded in a 6-well-plate, and the cells are treated as
indicated. IL-1β, IL-18 and IL-33 levels in the cell supernatants
were measured using ELISA kits according to the manufacturer’s
instructions.
Quantitative reverse
transcription-polymerase chain reaction (qRT-PCR)
Total RNA was extracted from the RAW264.7 cells
using TRIzol reagent (Invitrogen, Carlsband, CA, USA). cDNA was
obtained using a ThermoScript RT-PCR synthesis kit (Fermentas, San
Diego, CA, USA) according to the manufacturer’s instructions. The
sequences of primers used fro qRT-PCR are listed in Table I. qRT-PCR analyses for the mRNA
expression of NLRP3, ASC, caspase-1 and β-actin were performed
using QuantiFast SYBR-Green RT-PCR kits (Qiagen, Hilden, Germany)
and analyzed using the PikoReal 96 Real-Time PCR system (Thermo
Scientific, Vantaa, Finland). The mRNA level of β-actin was
measured as an internal control. Data were computed using the
PikoReal 96 Real-Time PCR system software. Three experiments from 3
independent RNA samples were performed.
 | Table IPrimers used for qRT-PCR. |
Table I
Primers used for qRT-PCR.
| Gene | Forward primer | Reverse primer |
|---|
| NLRP3 |
5′-CCTGACCCAAACCCACCAGT-3′ |
5′-TTCTTTCGGATGAGGCTGCTTA-3′ |
| ASC |
5′-TGAGCAGCTGCAAACGACTA-3′ |
5′-ACACTGCCATGCAAAGCATC-3′ |
| Caspase-1 |
5′-ATGAATCACCAACACCAG-3′ |
5′-CTTGACGCATCCTAATCC-3′ |
| β-actin |
5′-CCCATCTATGAGGGTTACGC-3′ |
5′-TTTAATGTCACGCACGATTTC-3′ |
Western blot analysis
The RAW264.7 cells were collected and lysed in lysis
buffer (Beyotime Institute of Biotechnology), then the whole cell
lysate was separated by SDS-PAGE and further transfered onto PVDF
membranes (Millipore Corp., Billerica, MA, USA). The membranes were
then incubated at room temperature for 3 h with 5% non-fat milk in
Tris-buffered saline (TBS) solution with the detergent, Tween-20
(TBST), and were subsequently incubated with specific primary
antibody at 4°C for 12 h in blocking solution. Specific primary
antibodies targeting NLRP3, ASC, caspase-1 and β-actin were used
1:1,000, 1:500, 1:500, 1:500, respectively. Following 3 washes with
TBST, the membranes were incubated at room temperature for 1 h with
HRP-conjugated secondary antibody (anti-rabbit and anti-mouse,
respectively). The protein blots were detected using the
ECL-chemiluminescent kit (Thermo Scientific).
RNA interference
siRNAs targeting NLRP3 were obtained from GenePharma
Corp. and contained the following sequences: NLRP3 siRNA (sense,
5′-GGCGAGACCUCUGGGAAA ATT-3′ and antisense,
5′-UUUUCCCAGAGGUCUCGCCTT-3′); negative control (sense,
5′-UUCUCCGAACGUGUCACGUTT and antisense, 5′-ACGUGACACGUUCGGAG
AATT-3′). The RAW264.7 (2×106/ml cells) were cultured in
6-well plates with DMEM containing 10% FBS for 24 h and maintained
in a 37°C incubator under a humidified atmosphere containing 5%
CO2 (until the density of the transfected cells reached
30 to 80%). The transfection mixture was prepared using 5 μl of
Lipofectamine 2000 and 4.2 μl of NLRP3 siRNA, to 300 μl of
Opti-MEM. The siRNAs were then transfected into the RAW264.7 cells
using Lipofectamine 2000 according to the instructions of the
manufacturer. Knockdown efficiency was determined by qRT-PCR and
western blot analysis. Following transfection, the cells were
incubated for 6 h with antibiotics-free Opti-MEM, and were then
subjected to LPS/ATP stimulation with DMEM containing 10% FBS and
used for the assays. Three independent transfection experiments
were performed.
Statistical analysis
Statistic analysis was performed using SPSS 10.01
for Windows (SPSS, Inc., Chicago, IL, USA). P-values were assessed
using unpaired, two-tailed Student’s t-tests or one-way analysis of
variance (ANOVA) with the Tukey-Kramer method. Values are reported
as the means ± standard deviation (SD). Data shown are
representative of at least triplicate experiments. A P-value
<0.05 was considered to indicate a statistically significant
difference.
Results
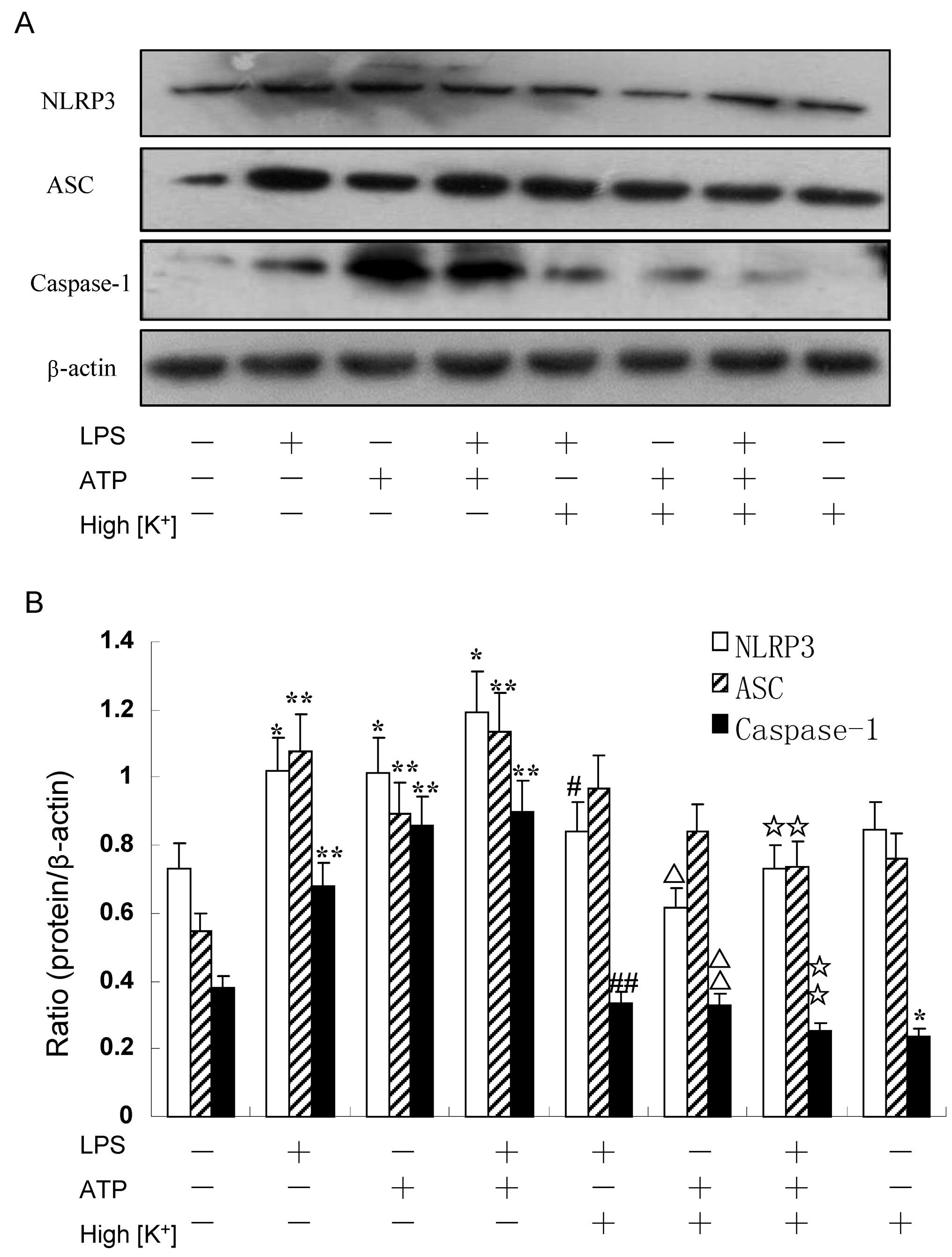
LPS and ATP induce NLRP3 inflammasome
activation, exerting a synergistic effect
Treatment with LPS (200 ng/ml) prior to treatment
with 5 mM ATP induced the upregulation of each component of the
NLRP3 inflammasome in the RAW264.7 murine macrophages. The
expression of NLRP3 and ASC in the RAW264.7 cells treated with LPS
alone was higher than that in those stimulated with ATP alone, as
shown by western blot analysis (Figs.
1 and 4). On the contrary,
the expression of caspase-1 was further induced by ATP alone
compared to stimulation with LPS alone (Figs. 1 and 4). Nevertheless, at the same time, dual
stimulation with LPS and ATP, had a synergistic effect on the
expression of the NLRP3 inflammasome (Figs. 1 and 4). Moreover, the mRNA levels of NLRP3,
ASC and caspase-1 in the RAW264.7 cells, were further detected by
qRT-PCR assay. The results revealed that the expression of NLRP3
and caspase-1 was significantly enhanced in the LPS + ATP group
(P=0.003 and P=0.002 for NLRP3 and caspase-1, respectively)
(Fig. 3), whereas the ASC levels
were modestly upregulated (P=0.040). However, treatment with ATP
had no effect on the mRNA expression of ASC compared with the
control group (P=0.140) (Fig. 3).
Collectively, our findings showed that the mRNA and protein
expression levels of the NLRP3 inflammasome were similar (Fig. 4).
High concentration of extracellular
K+/knockdown of the NLRP3 gene suppresses the activation
of NLRP3 and caspase-1 induced by LPS and/or ATP, but has no effect
on ASC
The RAW264.7 cells were cultivated in a 150 nM
potassium environment for 30 min prior to stimulation with LPS
and/or ATP. The protein expression of NLRP3 was moderately reduced
compared with the other corresponding groups (P<0.05) (Fig. 1B). However, the RAW264.7 cells
cultured in a high potassium milieu only demonstrated no difference
compared with the control group as regards the NLRP3 level
(P=0.064) (Fig. 1B). The ASC
levels slightly decreased following stimulation with LPS + ATP in
high K+ cell culture medium (P=0.033) (Fig. 1B). Nevertheless, the expression of
caspase-1 significantly decreased in the LPS group, ATP group and
the LPS + ATP group (P=0.004, P=0.003, P=0.001, respectively)
(Fig. 1B). The expression of
caspase-1 was low even without any stimuli when the RAW264.7 cells
were cultured in high K+ culture medium compared with
the control (P=0.023) (Fig. 1B).
Using specific siRNA to knockdown the NLRP3 gene, we found that the
expression of NLRP3 and caspase-1 significantly decreased, while
the ASC levels increased compared with the other corresponding
groups, as shown by qRT-PCR (Fig.
3). The results of western blot analysis for the protein
expression of the NLRP3 inflammasome were similar to those obtained
by qRT-PCR (Fig. 4). Of note, the
decrease in the ASC levels was not statistically significant with
the increasing extracellular K+ ion concentration in the
culture mediem (Fig. 1); NLRP3
siRNA had no effect on ASC at the mRNA and protein level. The
underlying mechanisms remain unclear, and require further
investigation.
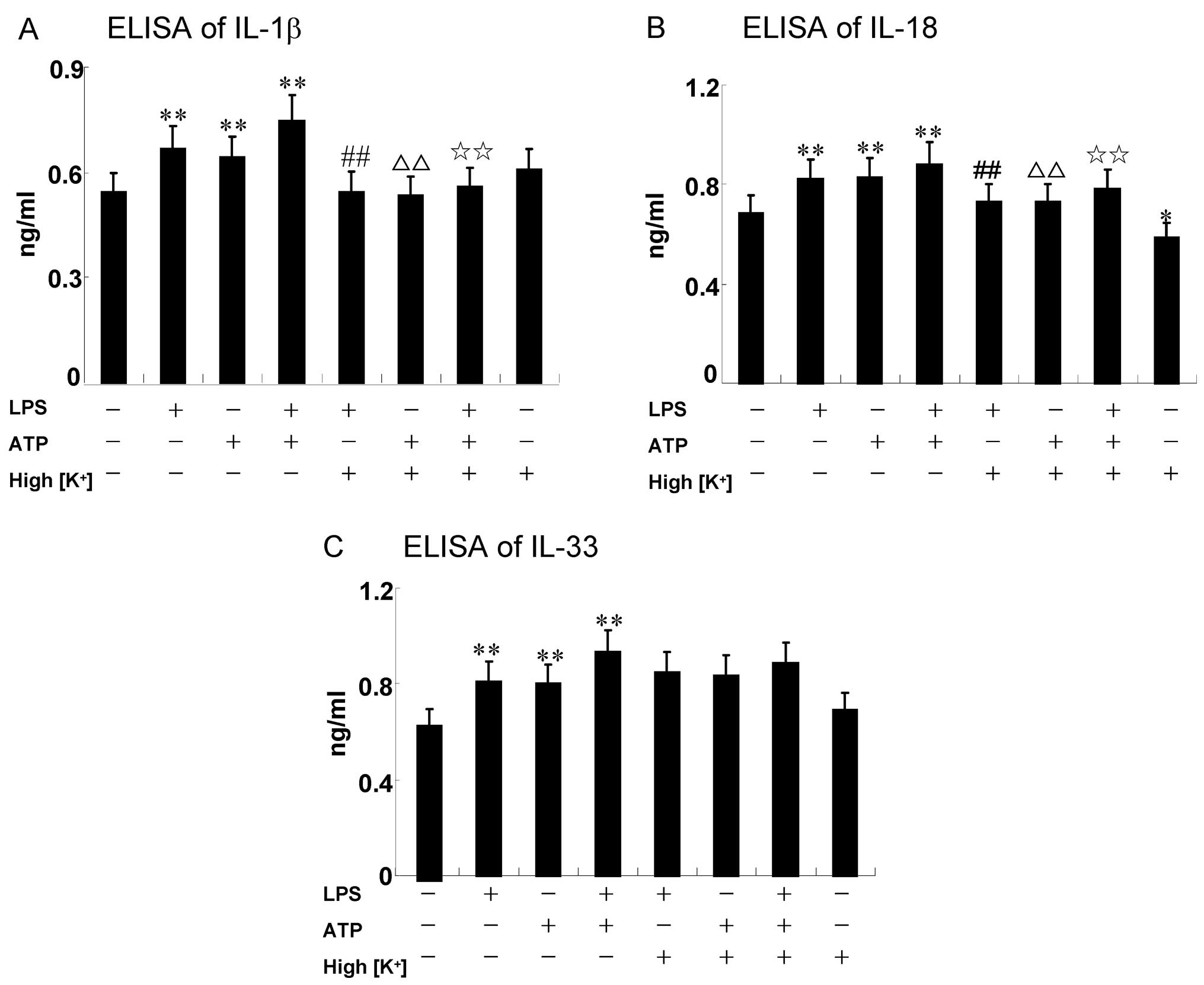
ATP enhances the LPS-induced expression
of IL-1β, IL-18 and IL-33 in RAW264.7 cells
The secretion of IL-1β and IL-18 requires the
induction of pro-IL-1β and pro-IL-18 expression, followed by its
proteolytic processing to mature IL-1β and IL-18 by the NLRP3
inflammasome (17). The
processing and generation of IL-33 are not yet clear. Caspase-1
plays a proteolytic role among the NLRP3 inflammasome complex. The
RAW264.7 cells were treated with 5 mM ATP for an additional 30 min
in the presence or absence of 200 ng/ml LPS for 6 h. The results
revealed that the expression of IL-1β, IL-18 and IL-33 increased
when the cells were treated with a combination of LPS and ATP
(0.75±0.08, 0.88±0.09 and 0.93±0.09, respectively; Fig. 2); (0.85±0.09, 0.91±0.11 and
0.87±0.08 ng/ml, respectively; Fig.
5).
High concengtration of extracellular
K+/NLRP3 siRNA attenuate the secretion of IL-1β and
IL-18 induced by LPS and/or ATP, but have no effect on IL-33
Potassium efflux is essential for the LPS and/or
ATP-induced NLRP3 inflammasome activation (18). Preventing K+ efflux
inhibits NLRP3 inflammasome assembly, mainly by elevating
extracellular the K+ concentration (19). In the present study, RAW264.7
cells were treated with a high concentration of K+ (150
nM) prior to any stimulation for 30 min. The expression of IL-1β
and IL-18 induced by LPS/ATP decreased significantly following
treatment with K+, as shown by ELISA (P<0.01)
(Fig. 2A and B). It is noteworthy
that the secretion of IL-18 still decreased without any external
stimuli when the RAW264.7 cells were cultured in high K+
culture medium compared with the control (P=0.034) (Fig. 2B). However, a high concentration
of K+ (150 mM) blocked the expression and release of
IL-1β and IL-18, but not that of IL-33 (P>0.05) (Fig. 2C). The low protein expression of
NLRP3 may abrogate NLRP3 inflammasome assembly. Thus, similar
results were observed on these 3 cytokines when the NLRP3 gene was
knocked down using NLRP3 siRNA (Fig.
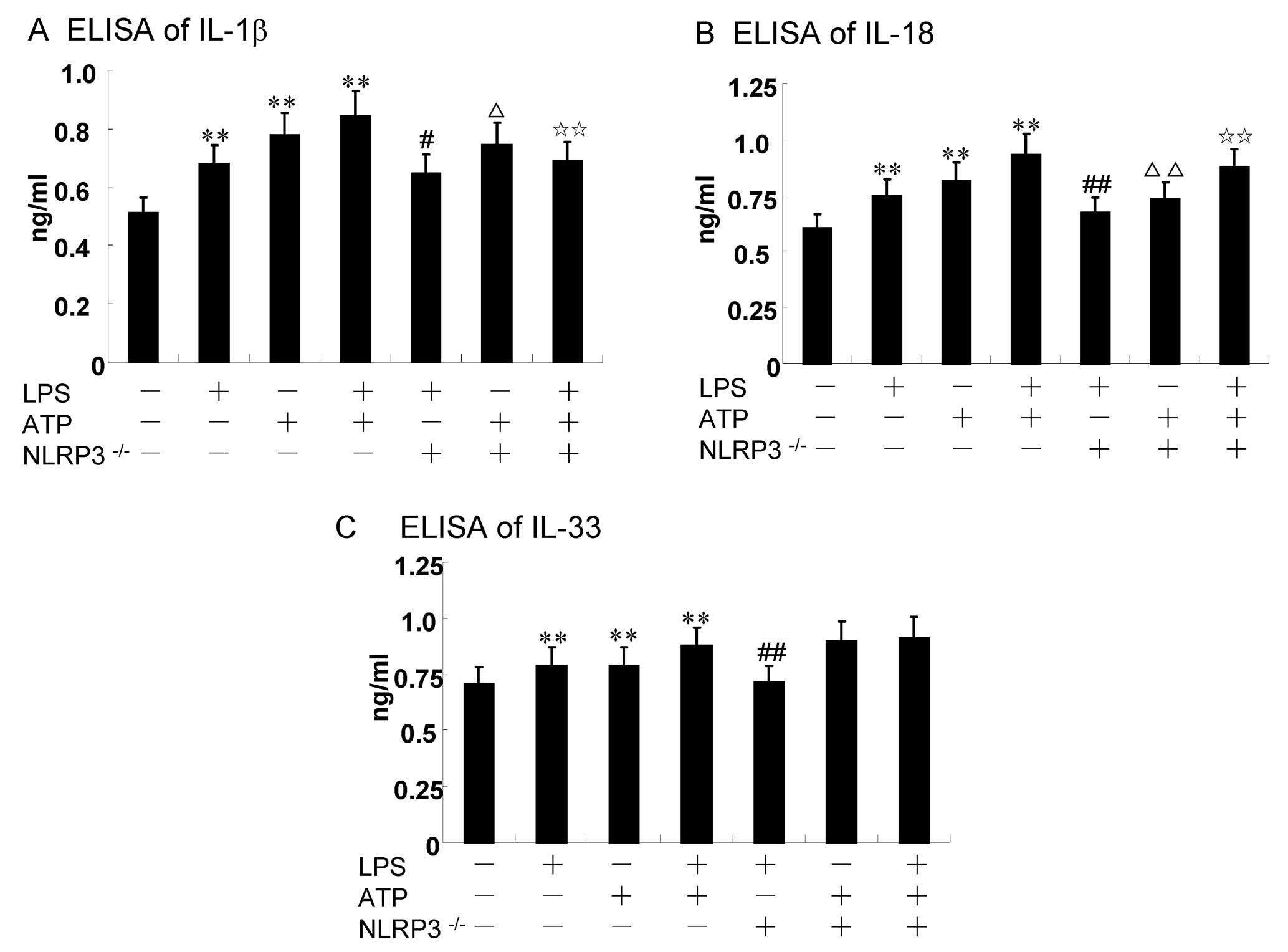
5). Specific siRNAs were used to knock down the NLRP3 gene in
order to investigate the effects on the expression and release of
cytokines in RAW264.7 cells (Fig.
5). The results revealed that the NLRP3 knockdown decreased
IL-18 expression (P<0.01) (Fig.
5B), and a certain decrease in IL-1β expression was also
observed (P<0.05) (Fig. 5A).
We also found a non-significant, but minor increase in IL-33
expression after the knockdown of NLRP3, compared to the ATP alone
and LPS + ATP group (P>0.05) (Fig.
5C). However, the IL-33 levels were decreased compared with the
LPS group (P=0.037) (Fig.
5C).
Discussion
The significance of innate immunity lies not only in
comprising the first line of defense against pathogenic and
non-pathogenic insults, but also in developing an efficient
adaptive immune response (20–24). The NLRP3 inflammasome provides
multi-protein molecular platforms for the activation of caspase-1
and subsequent processing and the secretion of IL-1 family members,
to engage innate immune defense (5,24).
Given the importance of IL-1 family members associated with acute
and chronic inflammation, understanding the role of the NLRP3
inflammasome in the initiation of innate immune response cannot be
ignored (24). Therefore, we
examined the effects of LPS/ATP on the expression of inflammatory
cytokines and the components of the NLRP3 inflammasome in RAW264.7
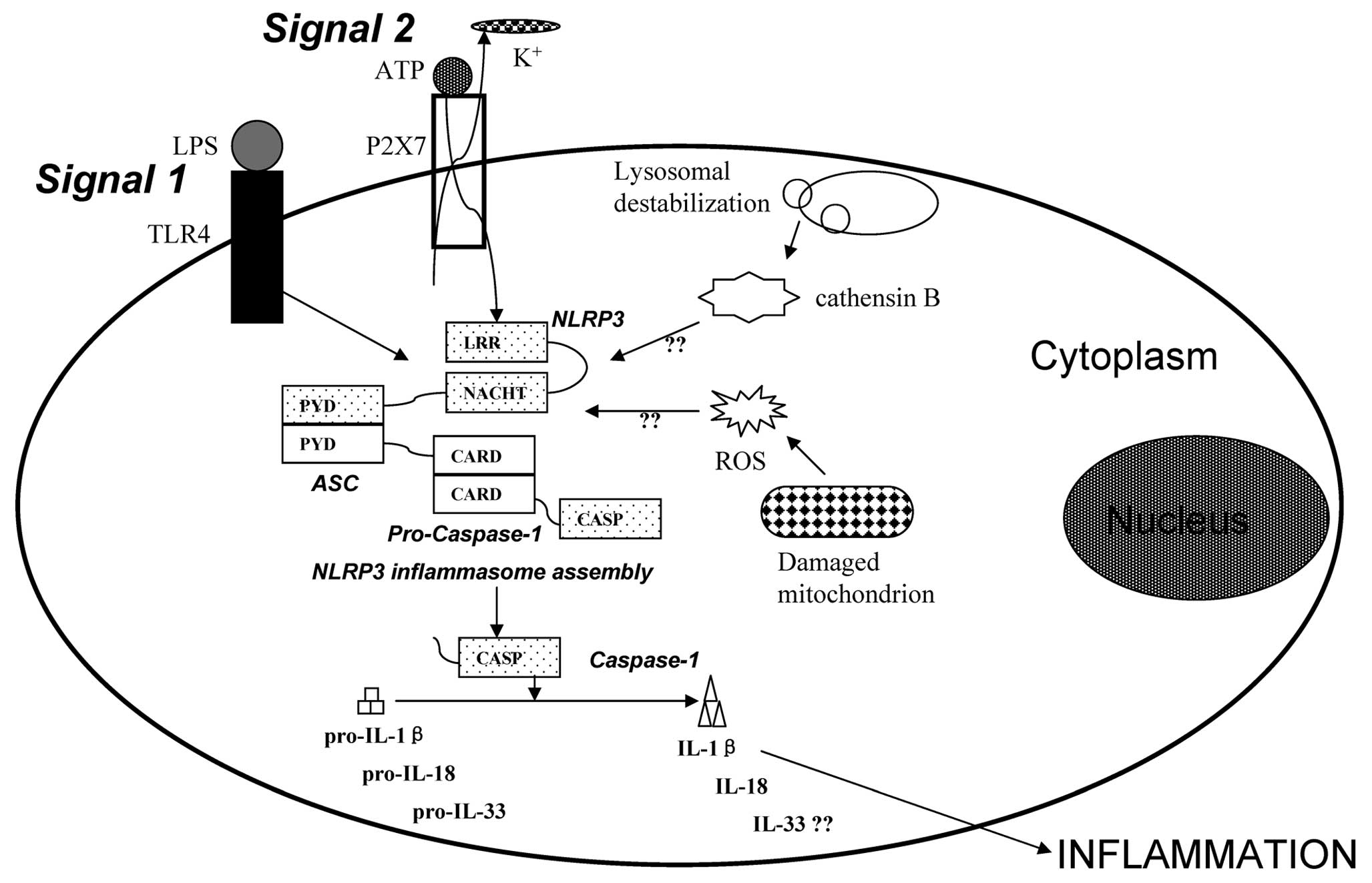
macrophage cells. There are two signals required for the activation
of the NLRP3 inflammasome in macrophages, the priming of the cells
triggered by LPS, and extracellular ATP, which enables the
subsequent orchestration of a robust processing and the release of
inflammatory cytokines, such as IL-1β and IL-18 (Fig. 6) (25). In our study, we found that LPS
and/or ATP stimulation induced the activation of the NLRP3
inflammasome and the sequential use of LPS and ATP exerted
synergistic effects on the protein expression of the NLRP3
inflammasome (Figs. 1 and
4).
Understanding the molecular mechanisms of NLRP3
inflammasome activation has become a focus or research. Mariathasan
et al (11) found that the
NLRP3 inflammasome can be activated by extracellular ATP
stimulation. In addition, hyaluronan and β-amyloid plaque, the
causative agents of Alzheimer’s disease, elevate plasma glucose, as
well as a diversity of environmental insults, including silica,
have been found to induce NLRP3 inflammasome activation (26–32). Similarly, exposure to UVB
irradiation can trigger NLRP3 inflammasome activation (33). TLR4 can bind to LPS directly (one
of the PAMPs), and ATP (one of the DAMPs) can bind to P2X7R (one of
the purine receptors). However, given the divergent structures of
DAMPs and PAMPs, it is likely that these agonists cannot bind to
NLRP3 directly. Thus, the mechanisms through which PAMPs and DAMPs
activate the NLRP3 inflammasome remain ambiguous. These mechanisms
have thus far proved to be elusive (34,35). However, current opinion suggests
that 3 models, including K+ efflux, lysosomal
destabilization and ROS production, can activate the signaling
pathway that results in NLRP3 inflammasome activation (Fig. 6) (36). Extracellular ATP connects with the
ATP-gated cation channel P2X7R when cells are under attack by
various insults, and then the connection of ligand and receptor can
cause membrane pore formation to trigger K+ efflux.
Thus, suppressing K+ efflux can inhibit the expression
of the NLRP3 inflammasome. In the present study, 150 mM
K+ (much more than the physiological concentration of
K+ inside the cell) was used to suppress K+
efflux. Our findings revealed that the expression of NLRP3
(P<0.05) and caspase-1 (P<0.01) were suppressed by the
increased concentration of extracellular K+ (Fig. 1). It has been known that
transfecting osteoblasts with siRNA can knock down NLRP3 expression
(37). Accordingly, the present
study found that the mRNA and protein levels of NLRP3 and caspase-1
decreased following the transfection of NLRP3 siRNA into RAW264.7
cells (Figs. 3 and 4). NLRP3 can undergo
self-oligomerization in the processing of PAMP or DAMP recognition,
and then recruits ASC, which is the crucial adaptor protein by
PYD-PYD domain interaction (Fig.
6) (38,39). ASC subsequently bridges to
caspase-1 through CARD-CARD interaction (Fig. 6) (40,41). Prior to NLRP3 inflammasome
activation, ASC is located in the nucleus of cells completely in
order to prevent caspase-1 activation and the subsequent processing
and generation of cytokines, such as IL-1β and IL-18 (42). Once NLRP3 molecular platforms are
activated by PAMPs and DAPMs, ASC is redistributed from the nucleus
to the cytosol, where it aggregates with NLRP3 and caspase-1 and
then transforms into perinuclear structures to achieve the assembly
of the NLRP3 inflammasome (43).
Available evidence suggests that the redistribution of ASC may
function as a checkpoint to prevent spontaneous and harmful
inflammasome activation (42).
Furthermore, the quantity of the protein expression of ASC and
NLRP3 is not in parallel (42).
In agreement with this viewpoint, our study indicated that a high
concontration of extracellular [K+]/NLRP3 siRNA
downregulated the expression of NLRP3 and caspase-1 induced by LPS
and/or ATP, but had little effect on ASC (Figs. 1, 3 and 4). Nevertheless, the precise mechanisms
that regulate inflammasomes at the level of ASC require further
clarification.
Caspase-1 is the first identified caspase, which is
generally presented in the cytosol of phagocytic cells as
pro-caspase-1 in the form of inactive zymogen (44). Following stimulation with various
microbial or various endogenous signals, pro-caspase-1 is
self-cleavaged by proteolytic cleavage from zymogen into the
enzymatically active heterodimer, composed of two 10 and 20 kDa
subunits (45). Active caspase-1
is essential for the cleavage of pro-IL-1β and pro-IL-18 into their
biologically active mature forms. Mature IL-1β is involved in many
immune reactions, including the recruitment of inflammatory cells
to the site of infection, whereas IL-18 plays an essential role in
the production of interferon-γ (IFN-γ) and enhancement of the
cytolytic activity of natural killer (NK) cells (46). When the expression of caspase-1 is
upregulated or downregulated, the secretion of IL-1β and IL-18 will
increase or decrease correspondingly. Our data indicated that LPS
and ATP stimulation induced the activation of caspase-1 (Figs. 1 and 4), and the robust release of IL-1β and
IL-18 (Figs. 2 and 5). However, the decreased levels of
caspase-1 (Figs. 1 and 4) accompanied with the reduction of
IL-1β and IL-18 (Figs. 2 and
5) were detected in both the
extracellular high K+ milieu- and NLRP3 siRNA-treated
groups. IL-33 belongs to the IL-1 subfamily (12), is closely related to IL-1β and
IL-18 as there is a caspase-1 site in the structure of the IL-33
precursor (47). The construction
of IL-33 is closer to IL-18 than IL-1β. However, evidence suggests
that the caspase-1 cleavage site is similar to the consensus
sequence for caspase-3 and that the intracellular IL-33 precursor
is also a substrate for caspase-3 (48). Neutrophil proteinase 3 (PR3) can
process the precursor IL-33 into an active mature form as IL-33,
but increasing PR3 incubation time downregulates the biological
activity of IL-33 (49). In
addition, neutrophil elastase and cathepsin G can cleave the IL-33
precursor, which results in the maturation of precursor IL-33
(50). Thus, extracellular IL-33
is secreted as a precursor and can be processed by a diversity of
enzymes apart from caspase-1. Given the complexity of IL-33
maturity processing, our data indicated that the expression of
IL-33 (P>0.05) did not decrease with the downregulation of
caspase-1 by the addition of a high concentration of K+
into the cell culture medium or by using siRNA to knock down the
NLRP3 gene (Figs. 2 and 5). This may elucidate the underlying
mechanisms of the processing of precursor IL-33 for a variety of
hydrolytic enzymes participating in this process.
Autoimmune disease pathogenesis is related to
cytokines, such as IL-1β and IL-18 and anakinra has been used to
block IL-1β activity to treat rheumatoid arthritis (RA) (51). However, the single use of anakinra
is less effective as other anti-cytokine therapies are more
effective (51). Thus, it is
possible that the inhibition of the NLRP3 inflammasome could
provide a therapeutic target than specific IL-1 blockade, whereby
blocking the NLRP3 inflammasome could block IL-1β and IL-18
activity simultaneously, both of which play critical roles in the
progression of RA. Nevertheless, further studies are required to
confirm this hypothesis.
Collectively, in the present study, LPS/ATP was
utilized to induce the activation of the NLRP3 inflammasome. A high
concentration of extracellular K+ and the knockdown of
NLRP3 using siRNA can block the secretion of IL-1β and IL-18 in
RAW264.7 cells.
Acknowledgements
Our study was supported by grants from the Anhui
Science and Technology Tackling Fund (no. 1206c0805026), and the
National Science Foundation of China (nos. 81072685 and
81273526).
References
|
1
|
Martinon F, Mayor A and Tschopp J: The
inflammasomes: guardians of the body. Annu Rev Immunol. 27:229–265.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Franchi L, Eigenbrod T and Núñez G:
Cutting edge: TNF-alpha mediates sensitization to ATP and silica
via the NLRP3 inflammasome in the absence of microbial stimulation.
J Immunol. 183:792–796. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jin C and Flavell RA: Molecular mechanism
of NLRP3 inflammasome activation. J Clin Immunol. 30:628–631. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Conforti-Andreoni C, Ricciardi-Castagnoli
P and Mortellaro A: The inflammasomes in health and disease: from
genetics to molecular mechanisms of autoinflammation and beyond.
Cell Mol Immunol. 8:135–145. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schroder K and Tschopp J: The
inflammasomes. Cell. 140:821–832. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Chen M, Wang H, Chen W and Meng G:
Regulation of adaptive immunity by the NLRP3 inflammasome. Int
Immunopharmacol. 11:549–554. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Leemans JC, Cassel SL and Sutterwala FS:
Sensing damage by the NLRP3 inflammasome. Immunol Rev. 243:152–162.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Davis BK, Wen H and Ting JP: The
inflammasome NLRs in immunity, inflammation, and associated
diseases. Annu Rev Immunol. 29:707–735. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cassel SL, Joly S and Sutterwala FS: The
NLRP3 inflammasome: a sensor of immune danger signals. Semin
Immunol. 21:194–198. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hsu HY and Wen MH:
Lipopolysaccharide-mediated reactive oxygen species and signal
transduction in the regulation of interleukin-1 gene expression. J
Biol Chem. 277:22131–22139. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mariathasan S, Weiss DS, Newton K, McBride
J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y, Monack DM and
Dixit VM: Cryopyrin activates the inflammasome in response to
toxins and ATP. Nature. 440:228–232. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van de Veerdonk FL and Netea MG: New
insights in the immunobiology of IL-1 family members. Front
Immunol. 4:1672013.PubMed/NCBI
|
|
13
|
Qi J, Ye X, Ren G, Kan F, Zhang Y, Guo M,
Zhang Z and Li D: Pharmacological efficacy of anti-IL-1β scFv, Fab
and full-length antibodies in treatment of rheumatoid arthritis.
Mol Immunol. 57:59–65. 2014.
|
|
14
|
Voronov E, Dayan M, Zinger H, Gayvoronsky
L, Lin JP, Iwakura Y, Apte RN and Mozes E: IL-1 beta-deficient mice
are resistant to induction of experimental SLE. Eur Cytokine Netw.
17:109–116. 2006.PubMed/NCBI
|
|
15
|
Girardin SE: Knocking in the NLRP3
inflammasome. Immunity. 30:761–763. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Latz E: The inflammasomes: mechanisms of
activation and function. Curr Opin Immunol. 22:28–33. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Budai MM, Varga A, Milesz S, Tozser J and
Benko S: Aloe vera downregulates LPS-induced inflammatory cytokine
production and expression of NLRP3 inflammasome in human
macrophages. Mol Immunol. 56:471–479. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hussen J, Düvel A, Koy M and Schuberth HJ:
Inflammasome activation in bovine monocytes by extracellular ATP
does not require the purinergic receptor P2X7. Dev Comp Immunol.
38:312–320. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Pétrilli V, Papin S, Dostert C, Mayor A,
Martinon F and Tschopp J: Activation of the NALP3 inflammasome is
triggered by low intracellular potassium concentration. Cell Death
Differ. 14:1583–1589. 2007.PubMed/NCBI
|
|
20
|
Kawai T and Akira S: Innate immune
recognition of viral infection. Nat Immunol. 7:131–137. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bose S and Banerjee AK: Innate immune
response against nonsegmented negative strand RNA viruses. J
Interferon Cytokine Res. 23:401–412. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
O’Neill LA and Bowie AG: Sensing and
signaling in antiviral innate immunity. Curr Biol. 20:R328–R333.
2010.PubMed/NCBI
|
|
23
|
Rathinam VA and Fitzgerald KA:
Inflammasomes and anti-viral immunity. J Clin Immunol. 30:632–637.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ciraci C, Janczy JR, Sutterwala FS and
Cassel SL: Control of innate and adaptive immunity by the
inflammasome. Microbes Infect. 14:1263–1270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Mankan AK, Dau T, Jenne D and Hornung V:
The NLRP3/ASC/Caspase-1 axis regulates IL-1beta processing in
neutrophils. Eur J Immunol. 42:710–715. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yamasaki K, Muto J, Taylor KR, Cogen AL,
Audish D, Bertin J, Grant EP, Coyle AJ, Misaghi A, Hoffman HM and
Gallo RL: NLRP3/cryopyrin is necessary for interleukin-1beta
(IL-1beta) release in response to hyaluronan, an endogenous trigger
of inflammation in response to injury. J Biol Chem.
284:12762–12771. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Halle A, Hornung V, Petzold GC, Stewart
CR, Monks BG, Reinheckel T, Fitzgerald KA, Latz E, Moore KJ and
Golenbock DT: The NALP3 inflammasome is involved in the innate
immune response to amyloid-beta. Nat Immunol. 9:857–865. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou R, Tardivel A, Thorens B, Choi I and
Tschopp J: Thioredoxin-interacting protein links oxidative stress
to inflammasome activation. Nat Immunol. 11:136–140. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dostert C, Pétrilli V, Van Bruggen R,
Steele C, Mossman BT and Tschopp J: Innate immune activation
through Nalp3 inflammasome sensing of asbestos and silica. Science.
320:674–677. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hornung V, Bauernfeind F, Halle A, Samstad
EO, Kono H, Rock KL, Fitzgerald KA and Latz E: Silica crystals and
aluminum salts activate the NALP3 inflammasome through phagosomal
destabilization. Nat Immunol. 9:847–856. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Cassel SL, Eisenbarth SC, Iyer SS, Sadler
JJ, Colegio OR, Tephly LA, Carter AB, Rothman PB, Flavell RA and
Sutterwala FS: The Nalp3 inflammasome is essential for the
development of silicosis. Proc Natl Acad Sci USA. 105:9035–9040.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Eisenbarth SC, Colegio OR, O’Connor W,
Sutterwala FS and Flavell RA: Crucial role for the Nalp3
inflammasome in the immunostimulatory properties of aluminium
adjuvants. Nature. 453:1122–1126. 2008. View Article : Google Scholar
|
|
33
|
Feldmeyer L, Keller M, Niklaus G, Hohl D,
Werner S and Beer HD: The inflammasome mediates UVB-induced
activation and secretion of interleukin-1beta by keratinocytes.
Curr Biol. 17:1140–1145. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Bryant C and Fitzgerald KA: Molecular
mechanisms involved in inflammasome activation. Trends Cell Biol.
19:455–464. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
McGettrick AF and O’Neill LA: NLRP3 and
IL-1beta in macrophages as critical regulators of metabolic
diseases. Diabetes Obes Metab. 15(Suppl 3): 19–25. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lamkanfi M and Dixit VM: Inflammasomes:
guardians of cytosolic sanctity. Immunol Rev. 227:95–105. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
McCall SH, Sahraei M, Young AB, Worley CS,
Duncan JA, Ting JP and Marriott I: Osteoblasts express NLRP3, a
nucleotide-binding domain and leucine-rich repeat region containing
receptor implicated in bacterially induced cell death. J Bone Miner
Res. 23:30–40. 2008. View Article : Google Scholar
|
|
38
|
Duncan JA, Bergstralh DT, Wang Y,
Willingham SB, Ye Z, Zimmermann AG and Ting JP: Cryopyrin/NALP3
binds ATP/dATP, is an ATPase, and requires ATP binding to mediate
inflammatory signaling. Proc Natl Acad Sci USA. 104:8041–8046.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Faustin B, Lartigue L, Bruey JM, Luciano
F, Sergienko E, Bailly-Maitre B, Volkmann N, Hanein D, Rouiller I
and Reed JC: Reconstituted NALP1 inflammasome reveals two-step
mechanism of caspase-1 activation. Mol Cell. 25:713–724. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Srinivasula SM, Poyet JL, Razmara M, Datta
P, Zhang Z and Alnemri ES: The PYRIN-CARD protein ASC is an
activating adaptor for caspase-1. J Biol Chem. 277:21119–21122.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Stehlik C, Lee SH, Dorfleutner A,
Stassinopoulos A, Sagara J and Reed JC: Apoptosis-associated
speck-like protein containing a caspase recruitment domain is a
regulator of procaspase-1 activation. J Immunol. 171:6154–6163.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bryan NB, Dorfleutner A, Kramer SJ, Yun C,
Rojanasakul Y and Stehlik C: Differential splicing of the
apoptosis-associated speck like protein containing a caspase
recruitment domain (ASC) regulates inflammasomes. J Inflamm (Lond).
7:232010. View Article : Google Scholar
|
|
43
|
Bryan NB, Dorfleutner A, Rojanasakul Y and
Stehlik C: Activation of inflammasomes requires intracellular
redistribution of the apoptotic speck-like protein containing a
caspase recruitment domain. J Immunol. 182:3173–3182. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Franchi L1, Eigenbrod T, Muñoz-Planillo R
and Nuñez G: The inflammasome: a caspase-1-activation platform that
regulates immune responses and disease pathogenesis. Nat Immunol.
10:241–247. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Martinon F and Tschopp J: Inflammatory
caspases: linking an intracellular innate immune system to
autoinflammatory diseases. Cell. 117:561–574. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Arend WP, Palmer G and Gabay C: IL-1,
IL-18, and IL-33 families of cytokines. Immunol Rev. 223:20–38.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Schmitz J, Owyang A, Oldham E, Song Y,
Murphy E, McClanahan TK, Zurawski G, Moshrefi M, Qin J, Li X,
Gorman DM, Bazan JF and Kastelein RA: IL-33, an interleukin-1-like
cytokine that signals via the IL-1 receptor-related protein ST2 and
induces T helper type 2-associated cytokines. Immunity. 23:479–490.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cayrol C and Girard JP: The IL-1-like
cytokine IL-33 is inactivated after maturation by caspase-1. Proc
Natl Acad Sci USA. 106:9021–9026. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Bae S, Kang T, Hong J, Lee S, Choi J, Jhun
H, Kwak A, Hong K, Kim E, Jo S and Kim S: Contradictory functions
(activation/termination) of neutrophil proteinase 3 enzyme (PR3) in
interleukin-33 biological activity. J Biol Chem. 287:8205–8213.
2012. View Article : Google Scholar
|
|
50
|
Lefrancais E, Roga S, Gautier V,
Gonzalez-de-Peredo A, Monsarrat B, Girard JP and Cayrol C: IL-33 is
processed into mature bioactive forms by neutrophil elastase and
cathepsin G. Proc Natl Acad Sci USA. 109:1673–1678. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Shaw PJ, McDermott MF and Kanneganti TD:
Inflammasomes and autoimmunity. Trends Mol Med. 17:57–64. 2011.
View Article : Google Scholar : PubMed/NCBI
|