Introduction
Osteoarthritis (OA), a highly prevalent joint
disease, exhibits a number of histological characteristics,
including a gradual degradation of the extracellular matrix (ECM)
and reduced cartilage cellularity, as well as a disruption of the
articular cartilage surface, belonging to the GuBi of Traditional
Chinese Medicine (TCM) (1–3).
Chondrocytes, the only cell population of the articular cartilage,
are capable of responding to structural changes in the surrounding
ECM by maintaining the dynamic equilibrium between production of
the ECM and its enzymatic degradation; however, the capacity of
chondrocytes to regenerate the normal ECM architecture is limited
and declines with aging due to abnormal responsiveness to anabolic
stimuli and cell death (4,5). A
number of factors may be involved in the development of OA;
however, one of the most important risk factors is cell death. Cell
death diminishes the ability of cells to proliferate, mainly due to
an increase in apoptosis, which is thought to be a major cause of
chondrocyte depletion during OA progression. Several studies have
demonstrated that another type of cell death, autophagy, is
involved in chondrocyte depletion during OA progression (6–8).
Autophagy, a cellular homeostatic mechanism, plays
an important role in nutrient and energy regulation, and in
targeting dysfunctional and altered cytosolic macromolecules,
membranes and organelles for delivery to lysosomes for recycling
and degradation (9–11). At the cellular level, failure of
autophagy leads to the increased production of abnormal gene
expression, reactive oxygen species, and may cause cell death
(12). The consequences of
autophagy failure at the tissue and organismal level are abnormal
skeletal development, cardiomyopathies, neurodegeneration and
premature death (7,13,14). The mammalian target of rapamycin
(mTOR), an important suppressor of autophagy, functions upstream of
the autophagy-related (Atg) proteins and is crucially regulated by
multiple upstream signaling pathways involving adenosine
monophosphate (AMP)-activated protein kinase and phosphoinositide 3
(PI3)-kinase/Akt (15,16). In articular cartilage, which is
characterized by a very low rate of cell turnover, autophagy
appears to be essential to maintain cellular integrity, survival
and function (7,8). Previous studies have verified that
autophagy is a constitutively active and apparently protective
process for the homeostatic state in normal cartilage (17). A reduced expression of Atg genes
has been observed in OA in humans and mice and is accompanied by an
increase in chondrocyte apoptosis, indicating a protective and
survival-promoting function of autophagy (7,8,17).
Tougu Xiaotong capsule (TXC; Medical License number:
MINZHIZI Z20100006) is composed of a combination of 4 natural
products, including Morindae officinalis, Radix Paeoniae
Alba, Rhizoma Chuanxiong and Sarcandra glabra.
According to the theories of TCM, these natural products mixed
together confer the TXC properties of nourishing Shen, filling in
Sui, supplementing Jing, dredging the meridians and collaterals to
limber the joints and strengthen bones and tendons. TXC has been
widely used for the therapy of OA in the Second People’s Hospital
affiliated to Fujian University of TCM for 2 decades and has been
shown to control pain and improve dysfunction in patients with OA
(18). We have previously
reported that TXC inhibits tidemark replication and cartilage
degradation by the regulation of chondrocyte autophagy (19). However, the precious molecular and
cellular mechanisms responsible for the effects of TXC on the
regulation of chondrocyte autophagy remain largely unknown. Thus,
the aim of this study was to establish a proof-of-principle that
the pharmacological enhancement of autophagy may be an effective
therapeutic approach for OA by regulating the
Atg12/microtubule-associated protein 1 light chain 3 (LC3)
conjugation systems. Our data demonstrate that TXC reduces the
severity of chondrocyte damage, at least in part by activating
autophagy, suggesting that TXC promotes chondrocyte autophagy,
contributing to the regulation of cartilage homeostasis.
Materials and methods
TXC extracts and fingerprint
analysis
TXC herbs were dried for 24 h in an air-circulating
oven at 50°C and then shredded and crushed to an appropriate
particle size in a high-speed rotary cutting mill (ZN-400A;
Zhongnan Pharmaceutical Machinery Factory, Changsha, China).
According to the proportion of TXC (Morinda
officinalis:Radix Paeoniae Alba:Rhizoma
Chuanxiong:Sarcandra glabra = 2:2:1:1), 108 g of herbal
powder were extracted with 1.5 l distilled water by reflux for 2
times, 2 h per times. The filtrate withdrawn from the TXC was
evaporated using a rotary evaporator (RE-2000; Shanghai Yarong
Biochemical Instrument Factory, Shanghai, China) and was then dried
to constant weight in a vacuum drying oven (DZF-300; Shanghai
Hengke Electronic Technology Co., Ltd., Shanghai, China). The TXC
extracts were dissolved in Dulbecco’s modified Eagle’s medium
(DMEM; Gibco, Grand Island, NY, USA) as a 20 mg/ml stock
solution.
The quality control of the TXC extracts was analyzed
by high performance liquid chromatography (HPLC)fingerprint on an
Agilent 1200 HPLC system (Agilent, Santa Clara, CA, USA) using an
Ultimate™ XB-C18 column (4.60×250.00 mm, 5 μm, Welch Materials,
Inc., Ellicott City MD, USA). The conditions for analysis were
methanol-0.1% phosphoric acid as a mobile phase and a detection
wavelength at 277 nm, a flow rate of 1 ml/min and a column
temperature of 30°C. The gradient procedure was as follows: 5% A at
0–5 min, 5–20% A at 5–10 min, 20–42% A at 15–25 min, 42–65% A at
25–40 min, 65–80% A at 40–55 min and 80–100% A at 55–70 min.
Paeoniflorin, ferulic acid, isofraxidin and rosmarinic acid
(National Institute for Pharmaceutical and Biological Products
Control, Beijing, China) were used as standard substances.
Chondrocyte isolation and culture
Animal use protocols were performed according to the
Guide for the Care and Use of Laboratory Animals approved by the
Animal Care and Use Committee of Fujian University of TCM.
Chondrocytes were collected from the knee articular cartilage of
4-week-old Sprague-Dawley (SD) rats (Shanghai Slack Laboratory
Animal Co., Shanghai, China). Chondrocytes were isolated using 0.2%
type II collagenase (Sigma, St. Louis, MO, USA) in pH 7.4
magnesium- and calcium-free phosphate-buffered saline (PBS; Sigma)
for 1 h at 37°C (20).
Chondrocytes were resuspended in low-glucose DMEM (Gibco)
supplemented with 10% fetal calf serum (Gibco), streptomycin (100
μg/ml) and penicillin (100 U/ml), and seeded in monolayer at a
density of 5×105 cells/cm2. The chondrocytes
used in this study were subjected to the second passage cells,
identified by type II collagen immunohistochemistry and scanning
electron microscopy (SEM). Cobalt chloride (CoCl2)
powder (Sigma) was dissolved in DMSO as a 1 mM stock solution. The
second-passage chondrocytes cultured till approximately 80%
confluency were treated with 100 μM of CoCl2 with or
without TXC for 48 h.
Assessment of cell viability by MTT
assay
The cells were seeded in a 96-well plate at a
density of 1.2×104 cells/cm2 and allowed to
adhere overnight, then treated with either CoCl2 (0, 50,
100, 200, 300, 400 μM) for 24 h or with 100 μM of CoCl2
for 6, 12, 24, 48, 72 h and either TXC (0, 50, 100, 200, 300, 400
μg/ml) + 100 μM CoCl2 for 48 h or with 200 μg/ml of TXC
+ 100 μM CoCl2 for 6, 12, 24, 48 and 72 h. Following
treatment, 20 μl MTT stock solution (5 mg/ml) were added to each
well, and the cells were incubated at 37°C for 4 h. Thereafter, the
medium was aspirated followed by the addition of 200 μl DMSO, and
the cells were shaken for 10 min. The color formed was determined
by an ELISA plate reader (EXL 800; BioTek, Winooski, VT, USA).
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from the chondrocytes using
TRIzol reagent (Invitrogen, Carlsbad, CA, USA). Reverse
transcription was performed using random primers and Superscript™
III (Invitrogen). PCR reactions were conducted on an ABI Prism
7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City,
CA, USA). Primers for the amplifications were designed as follows:
hypoxia-inducible factor-1α (HIF-1α) forward, 5′-GCA TCT CCA CCT
TCT ACC C-3′ and reverse, 5′-TTC TGC TCC ATT CCA TCC T-3′, 386 bp;
beclin 1 forward, 5′-GCT CAG TAC CAG CGA GAA TA-3′ and reverse,
5′-GTC AGG GAC TCC AGA TAC GA-3′, 350 bp; mTOR forward, 5′-GGA CGG
TGT AGA ACT TGG A-3′ and reverse, 5′-GAG ATG TCG CTT GCT TGA T-3′,
230 bp; Atg3 forward, 5′-GGA GGC TAT CAT TGA AGA AG-3′ and reverse,
5′-TGG GAG GTG AGG ATG GTT T-3′, 481 bp; Atg5 forward, 5′-ACG CTG
GTA ACT GAC AAA G-3′ and reverse, 5′-CAC ATG ACA TAA AGT GAG CC-3′,
250 bp; Atg7 forward, 5′-TGG GAG AAG AAC CAG AAA GG-3′ and reverse,
5′-TCA CGG GAT TGG AGT AGG AG-3′, 280 bp; Atg10 forward, 5′-GTG CCC
GTT CTG TAC TTT AGG-3′ and reverse, 5′-TCA TTT GTC TTG CAG GGA
TGT-3′, 188 bp; Atg12 forward, 5′-GAG ACA CTC CCA TAA TGA A-3′ and
reverse, 5′-GTA GGA CCA GTT TAC CAT C-3′, 207 bp; LC3 I forward,
5′-CTT CGC CGA CCG CTG TAA-3′ and reverse, 5′-ATC CGT CTT CAT CCT
TCT CCT G-3′, 287 bp; LC3 II forward, 5′-CTA ACC AAG CCT TCT TCC
TCC-3′ and reverse, 5′-GGT GCC TAC GTT CTG ATC TGT G-3′, 261 bp;
β-actin forward, 5′-CAC CCG CGA GTA CAA CCT TC-3′ and reverse,
5′-CCC ATA CCC ACC ATC ACA CC-3′, 207 bp.
Western blot analysis
Total protein was extracted from the chondrocytes
using RIPA buffer, and protein concentrations were determined using
a Bio-Rad protein assay (Bio-Rad, Hercules, CA, USA). Total protein
(50 μg) was fractionated by SDS-PAGE, and transferred onto a PVDF
membrane (Invitrogen). The PVDF membrane was blocked with 5%
non-fat milk and incubated with antibodies to HIF-1α, beclin 1,
mTOR, Atg3, Atg7, Atg12, LC3 I/II and β-actin (Santa Cruz
Biotechnology, Santa Cruz, CA, USA). The blots were developed using
a horseradish peroxidase-conjugated secondary antibody (Beyotime
Institute of Biotechnology, Shanghai, China). Immunoreactive
proteins were visualized by western blotting chemiluminescence
luminol reagent (Santa Cruz Biotechnology). Immunoblotting band
gray values were calculated using the Tocan 190 protein assay
system (Bio-Rad).
Transmission electron microscopy
(TEM)
The chondrocytes were fixed in 3% glutaraldheyde and
1.5% paraformaldehyde solution (pH 7.3) at 4°C for 24 h, post-fixed
with 1% osmic acid and 1.5% potassium hexacyanoferrate (II)
solution (pH 7.3) at 4°C for 2 h, rinsed with water, dehydrated in
a graded series of ethanol followed by propylene oxide, kept
overnight and embedded in Epon-Araldite resin. Ultrathin sections
were obtained using a Leica ultramicrotome and stained with 2%
aqueous uranyl acetate, counterstained with 0.3% lead citrate and
observed under a transmission electron microscope (H7650; Hitachi
High-Technologies Corp., Tokyo, Japan).
Statistical analysis
All data were collected from at least 3 independent
experiments. Statistical analysis was performed using SPSS 13.0
software. All data are presented as the means ± standard deviation
(SD) and analyzed by the Student’s t-test and ANOVA. Statistical
significance was set at P<0.05.
Results
Quality control of TXC
Compared to the spectrogram and chromatographic peak
of retention time with the reference substance (Fig. 1A), the composition of TXC was
identified (Fig. 1B), and
contained paeoniflorin, ferulic acid, isofraxidin and rosmarinic
acid.
Identification of chondrocytes
Type II collagen has been identified as the major
molecular form of collagen in articular cartilage and is
responsible for tensile strength, whereas proteoglycans provide the
compressive stiffness necessary for normal articulation and
function (5). The second
generation of chondrocytes was cultured for 3 days followed by type
II collagen immunohistochemical staining. The cytoplasm was stained
with brown, representing positive expression in chondrocytes
(Fig. 2A and B). The second
generation of chondrocytes showed a typical morphology of
chondrocytes with a polygonal or spherical shape (Fig. 2C and D). Therefore, we used the
second generation of chondrocytes in the subsequent
experiments.
TXC increases the cell viability of
CoCl2-exposed chondrocytes
CoCl2 is commonly used to activate
autophagic death by inducing HIF-1α. To establish the cell model of
autophagic death, the chondrocytes were treated with various
concentrations and for different periods of time with
CoCl2 to determine the effective concentration and
treatment duration time by MTT assay. As shown in Fig. 3A, in the cells that were treated
with CoCl2 concentrations of 50 μM (68.43±3.78%), 100 μM
(50.38±4.12%), 200 μM (42.84±1.95%), 300 μM (41.13±3.20%) and 400
μM (36.10±4.89%) for 24 h, a dose-dependent decrease in cell
viability was observed compared to the untreated cells (100±0.00%)
(P<0.01). Cell viability gradually decreased with the increase
in the duration of treatment in the chondrocytes treated with 100
μM CoCl2 for different periods of time (Fig. 3B), suggesting that
CoCl2 inhibited cell viability in a dose- and
time-dependent manner due to the CoCl2-induced
autophagic death.
To explore the effects of TXC on
CoCl2-treated chondrocytes, we examined the cell
viability of CoCl2-exposed chondrocytes treated with
various concentrations of TXC and for different periods of time by
MTT assay. As shown in Fig. 3C,
the cell viability of the CoCl2-exposed chondrocytes
treated with TXC concentrations of 50 μg/ml (105.65±3.38%), 100
μg/ml (111.38±3.04%), 200 μg/ml (121.43±5.67%), 300 μg/ml
(118.44±3.48%) and 400 μg/ml (119.81±2.77%) for 48 h was enhanced
in a dose-dependent manner compared to that of the cells treated
with CoCl2 only (100±0.00%) (P<0.01). Cell viability
gradually increased with the increase in the duration of treatment
in the CoCl2-exposed chondrocytes treated with 200 μg/ml
TXC for different periods of time (Fig. 3D), indicating that TXC promoted
the survival of CoCl2-exposed chondrocytes.
TXC enhances chondrocyte survival by
promoting cell autophagy
TXC has been reported to delay cartilage degradation
by activating chondrocyte autophagy in vivo; however, it
remains to be seen whether TXC enhances chondrocyte survival by
increasing the expression of Atg genes. To verify the effects of
TXC on chondrocyte autophagy, we observed the morphology of
CoCl2-exposed chondrocytes treated with or without TXC
under a phase-contrast microscope. As shown in Fig. 4, many of the chondrocytes treated
with CoCl2 for 24 and 48 h became rounded and shrunken,
and were detached from each other or floated in the medium,
compared to the untreated chondrocytes that showed densely
disorganized multilayers. However, TXC inhibited the changes in the
morphology of CoCl2-exposed chondrocytes, suggesting
that TXC enhanced chondrocyte survival by inhibiting the
CoCl2-induced autophagic death.
To explore the role of TXC in chondrocyte autophagy,
the mRNA and protein expression of Atg genes in
CoCl2-exposed chondrocytes treated with or without TXC
was examined by RT-PCR and western blot analysis, respectively. The
results revealed that the mRNA expression of beclin 1, Atg3, Atg5,
Atg7, Atg10, Atg12 and LC3 II/LC3 I in the chondrocytes treated
with TXC increased, compared to the untreated cells (P<0.05,
P<0.01) (Fig. 5). The protein
expression levels of these Atg genes were similar to their
respective mRNA expression levels (Fig. 6), suggesting that TXC promoted
chondrocyte autophagy by regulating the Atg12/LC3 conjugation
systems.
 | Figure 5Tougu Xiaotong capsule (TXC)
regulates the mRNA expression of autophagy-related (Atg) genes in
cobalt chloride (CoCl2)-exposed chondrocytes. (A) mRNA
expression of Atg genes in CoCl2-exposed chondrocytes
treated with or without TXC measured by RT-PCR. The mRNA expression
of (B) hypoxia-inducible factor-1α (HIF-1α), (C) beclin 1, (D)
mammalian target of rapamycin (mTOR), (E) Atg3, (F) Atg5, (G) Atg7,
(H) Atg10, (I) Atg12 and (J) microtubule-associated protein 1 light
chain 3 (LC3) II/LC3 I. β-actin was used as the internal control
for the quantification analysis. Data are the means ± standard
deviation (SD) and SD is shown as vertical bars,
*P<0.05, **P<0.01, compared to
untreated cells; ΔP<0.05, ΔΔP<0.01,
compared to CoCl2-exposed-chondrocytes. |
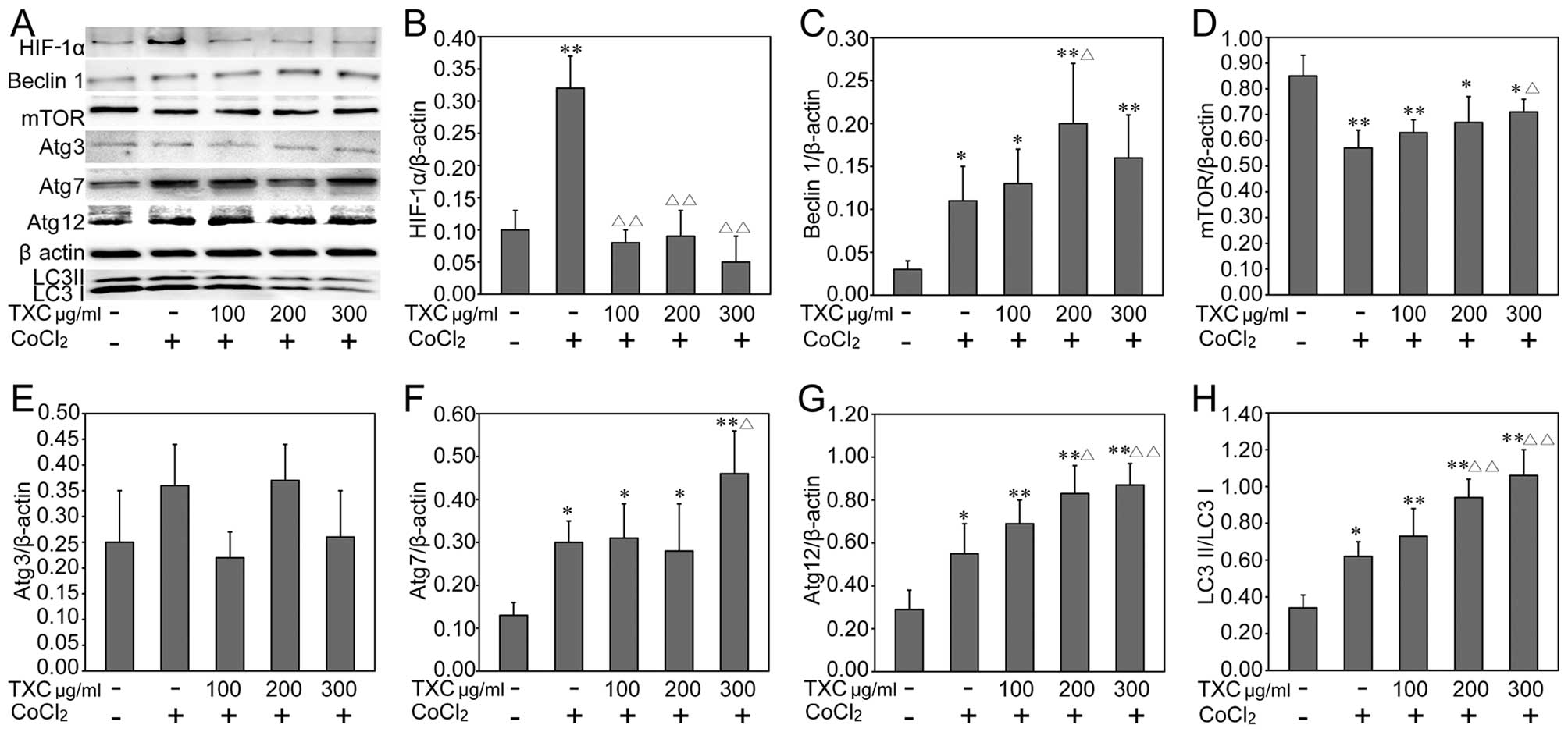 | Figure 6Tougu Xiaotong capsule (TXC)
regulates the protein expression of autophagy-related (Atg) genes
in cobalt chloride (CoCl2)-exposed chondrocytes. (A)
Protein expression of Atg genes in CoCl2-exposed
chondrocytes measured by western blot analysis. Protein levels of
(B) hypoxia-inducible factor-1α (HIF-1α), (C) beclin 1, (D)
mammalian target of rapamycin (mTOR), (E) Atg3, (F) Atg7, (G)
Atg12, and (H) microtubule-associated protein 1 light chain 3 (LC3)
II/LC3 I. β-actin was used as the internal control for the
quantification analysis. Data are the means ± standard deviation
(SD) and SD is shown as vertical bars, *P<0.05,
**P<0.01, compared to untreated cells;
ΔP<0.05, ΔΔP<0.01, compared to
CoCl2-exposed-chondrocytes. |
TXC promotes the formation of
autophagosomes in CoCl2-exposed-chondrocytes
We examined the ultrastructural changes of
CoCl2-exposed chondrocytes treated with or without TXC
by TEM. The results revealed that the nuclei of the untreated
chondrocytes had a normal appearance, a Golgi apparatus, rough
endoplasmic reticulum (ERr), vesicles and mitochondria (Fig. 7A). By contrast, the chondrocytes
treated with TXC displayed an increase in the number of
autophagosomes (Fig. 7B–H). These
autophagosomes were recognized as double-membrane structures with
contents ranging from degenerated organelles to granular cytoplasm,
protein aggregates and endoplasmic reticulum. The fusion of
autophagosomes with lysosomes was also observed (Fig. 7G and H), indicating that TXC
enhanced chondrocyte autophagy by promoting the formation of
autophagosomes in CoCl2-exposed chondrocytes.
Discussion
The present study systematically investigated the
effects of TXC on CoCl2-induced chondrocyte autophagic
death in vitro. Our results clearly demonstrated that TXC
enhanced the viability of chondrocytes exposed to CoCl2
and increased the formation of autophagosomes in the
CoCl2-exposed chondrocytes by upregulating the
expression of beclin 1, Atg3, Atg5, Atg7, Atg10, Atg12 and LC3
II/LC3 I. Taken together, these results indicate that TXC is a
potential therapeutic agent for the reduction of cartilage
degradation that occurs in OA.
Current treatments for the management of OA do not
reverse the degradation process of articular cartilage.
Non-steroidal anti-inflammatory drugs (NSAIDs) have been used in
the treatment of OA for the past several years; however, their
therapeutic effects remain unsatisfactory due to the serious
adverse side-effects, such as gastrointestinal and cardiovascular
diseases (21–23). Accordingly, the development of
novel drugs from natural herbs, which can provide cartilage
protection and be safely used in the prolonged treatment of OA, is
required. TXC has been used to control tidemark replication and
cartilage degradation by inhibiting chondrocyte apoptosis and
promoting chondrocyte autophagy (18,19). Therefore, in the present study, we
sought to determine the efficacy of TXC on chondrocyte autophagy,
as a chondroprotective agent.
Excessive mechanical loading of articular cartilage
causes damage to chondrocytes and the ECM, and initiates a
pathogenetic process that occurs due to abnormal chondrocyte
activation and results in the spreading of chondrocyte death and
damage to the ECM beyond the initial area that was exposed to the
highest mechanical load (24,25). The understanding of the mechanisms
involved in these cellular changes may provide the potential to
identify targets for pharmacological interventions in order to
attenuate or prevent the subsequent development of OA. In the
articular cartilage, autophagy activated by different types of
stress has been shown to be a constitutively active and protective
process for the survival of chondrocytes (26). A previous study reported that the
expression of 3 markers for different stages of autophagy,
including uncoordinated-51-like kinase (ULK), beclin 1 and LC3, was
decreased in the cartilage of patients with OA and in a mouse model
of OA (8).
In the present study, we used a
CoCl2-induced model of cell autophagic death to assess
the molecular mechanisms responsible for the promoting effects of
TXC on chondrocyte autophagy in vitro. In order to determine
the inducer concentration of CoCl2 in chondrocytes, cell
viability was examined. Following exposure to CoCl2 at
various concentrations and for different periods of time, cell
viability was inhibited due to the CoCl2-induced
autophagic death as shown by MTT assay. Our results revealed that
CoCl2 induced autophagic death in a dose- and
time-dependent manner. The changes observed in cell morphology
suggested that the cells underwent autophagic death 24 h following
incubation with the concentration of CoCl2 selected
based on the results of MTT assay. To determine the effective
concentration of TXC on the viability of CoCl2-exposed
chondrocytes, MTT assay was performed. These results indicated that
TXC enhanced chondrocyte survival, as shown by the increased cell
viability.
Autophagy, which is in part related to the reduced
expression of autophagic regulators, is compromised in
osteoarthritic cartilage. In articular cartilage, autophagy does
not only occur in response to mechanical injury, but is also
deficient with aging (8,27). Autophagy is characterized by the
formation of autophagosomes and their fusion with lysosomes. At the
late stage of autophagy, lysosomes fuse with and release lysosomal
enzymes into the autophagosome to degrade its contents (28). Atg genes control the autophagy
process leading to the induction and nucleation of autophagic
vesicles, their fusion and expansion with lysosomes, following
enzymatic degradation and recycling (29,30). Atg12 undergoes an ubiquitin-like
conjugation to Atg5 through an internal lysine residue and a
COOH-terminal glycine, respectively. This process is activated by
the Atg7 protein, which is homologous to the E1 family of
ubiquitin-activating enzymes, and Atg10, which functions as a
protein-conjugating enzyme (31).
The Atg12-Atg5 conjugates recruit Atg16 dimers. Atg16 is a bivalent
molecule, which leads to the formation of large multimeric
complexes, and these are thought to play a key role in the
nucleation of both cytoplasm-to-vacuole targeting vesicles and
autophagosomes (32). The number
of lysosomes increases in the process of autophagy, accompanied by
an increased expression of beclin 1 and LC3 (33). Beclin 1, forming a complex with
type III PI3 kinase and Vps34, participates in the nucleation of
the autophagic vesicle (19). The
involvement of LC3 in the protein conjugation system is required in
the expansion of the autophagosome. There are 2 forms of LC3,
including LC3 I, in the cytoplasm and LC3 II bound to the
autophagosome membrane. During autophagy, LC3 I is converted to LC3
II by the ubiquitin-like system. Thus, the level of LC3 II and the
ratio of LC3 II to LC3 I closely reflect the extent of autophagy
(34).
To gain insight into the mechanisms responsible for
the effects of TXC on CoCl2-induced autophagic death,
the expression of mTOR and HIF-1α, as modulators of autophagy, was
assessed in chondrocytes. mTOR plays an important role in multiple
cellular functions, such as cell metabolism, proliferation and
autophagy (35). All Atg genes
have been shown to act downstream of mTOR. HIF-1α, a heterodimeric
transcription factor that mediates adaptive responses to hypoxia,
serves to regulate both autophagy and apoptosis (36). The present results indicated that
the expression of beclin 1, Atg3, Atg5, Atg7, Atg10, Atg12 and LC3
II/LC3 I in the CoCl2-exposed chondrocytes treated with
or without TXC significantly increased, compared to the untreated
cells. In addition, ultrastructural analysis revealed that the
chondrocytes treated with TXC contained more autophagosomes than
the untreated cells, suggesting that TXC increases the formation of
autophagosomes in chondrocytes to clear the
CoCl2-induced autophagic death.
Based on these results, we hypothesized that TXC
promotes chondrocyte autophagy by regulating the Atg12/LC3
conjugation systems. Since autophagy serves to delay the onset of
apoptosis, experiments are curently in progress in order to explore
whether there is a direct association between the induction of
autophagy and apoptosis.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81102609), the Key Project
of Fujian Provincial Department of Science and Technology (grant
no. 2012Y0046), the Natural Science Foundation of Fujian Province
(grant no. 2011J05074) and the Young Talent Scientific Research
Project of Fujian Province Universities (grant no. JA12165).
References
|
1
|
Taniguchi N, Caramés B, Ronfani L, et al:
Aging-related loss of the chromatin protein HMGB2 in articular
cartilage is linked to reduced cellularity and osteoarthritis. Proc
Natl Acad Sci USA. 106:1181–1186. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ruiz-Romero C, Calamia V, Mateos J, et al:
Mitochondrial dysregulation of osteoarthritic human articular
chondrocytes analyzed by proteomics: a decrease in mitochondrial
superoxide dismutase points to a redox imbalance. Mol Cell
Proteomics. 8:172–189. 2009. View Article : Google Scholar
|
|
3
|
Li XH, Liang WN and Liu XX: Clinical
observation on curative effect of dissolving phlegm-stasis on 50
cases of knee osteoarthritis. J Tradit Chin Med. 30:108–112. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Loeser RF: Aging and osteoarthritis: the
role of chondrocyte senescence and aging changes in the cartilage
matrix. Osteoarthritis Cartilage. 17:971–979. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pennock AT, Robertson CM, Emmerson BC,
Harwood FL and Amiel D: Role of apoptotic and matrix-degrading
genes in articular cartilage and meniscus of mature and aged
rabbits during development of osteoarthritis. Arthritis Rheum.
56:1529–1536. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lotz MK and Caramés B: Autophagy and
cartilage homeostasis mechanisms in joint health, aging and OA. Nat
Rev Rheumatol. 7:579–587. 2011.PubMed/NCBI
|
|
7
|
Caramés B, Hasegawa A, Taniguchi N, Miyaki
S, Blanco FJ and Lotz M: Autophagy activation by rapamycin reduces
severity of experimental osteoarthritis. Ann Rheum Dis. 71:575–581.
2012.PubMed/NCBI
|
|
8
|
Caramés B, Taniguchi N, Otsuki S, Blanco
FJ and Lotz M: Autophagy is a protective mechanism in normal
cartilage, and its aging-related loss is linked with cell death and
osteoarthritis. Arthritis Rheum. 62:791–801. 2010.PubMed/NCBI
|
|
9
|
Levine B and Kroemer G: Autophagy in the
pathogenesis of disease. Cell. 132:27–42. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cecconi F and Levine B: The role of
autophagy in mammalian development: cell makeover rather than cell
death. Dev Cell. 15:344–357. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mathew R, Karp CM, Beaudoin B, et al:
Autophagy suppresses tumorigenesis through elimination of p62.
Cell. 137:1062–1075. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Komatsu M, Waguri S, Ueno T, et al:
Impairment of starvation-induced and constitutive autophagy in
Atg7-deficient mice. J Cell Biol. 169:425–434. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hara T, Nakamura K, Matsui M, et al:
Suppression of basal autophagy in neural cells causes
neurodegenerative disease in mice. Nature. 441:885–889. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang Q and Guan KL: Expanding mTOR
signaling. Cell Res. 17:666–681. 2007. View Article : Google Scholar
|
|
16
|
Wullschleger S, Loewith R and Hall MN: TOR
signaling in growth and metabolism. Cell. 124:471–484. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cuervo AM: Autophagy and aging: keeping
that old broom working. Trends Genet. 24:604–612. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li XH, Wu MX, Ye HZ, et al: Experimental
study on the suppression of sodium nitroprussiate-induced
chondrocyte apoptosis by Tougu Xiaotong capsule-containing serum.
Chin J Integr Med. 17:436–443. 2011. View Article : Google Scholar
|
|
19
|
Li X, Lang W, Ye H, et al: Tougu Xiaotong
capsule inhibits the tidemark replication and cartilage degradation
of papain-induced osteoarthritis by the regulation of chondrocyte
autophagy. Int J Mol Med. 31:1349–1356. 2013.PubMed/NCBI
|
|
20
|
Li X, Ye H, Yu F, et al: Millimeter wave
treatment promotes chondrocyte proliferation via G1/S cell cycle
transition. Int J Mol Med. 29:823–831. 2012.PubMed/NCBI
|
|
21
|
Schnitzer TJ, Burmester GR, Mysler E, et
al: Comparison of lumiracoxib with naproxen and ibuprofen in the
Therapeutic Arthritis Research and Gastrointestinal Event Trial
(TARGET), reduction in ulcer complications: randomised controlled
trial. Lancet. 364:665–674. 2004. View Article : Google Scholar
|
|
22
|
Lichtenberger LM, Zhou Y, Dial EJ and
Raphael RM: NSAID injury to the gastrointestinal tract: evidence
that NSAIDs interact with phospholipids to weaken the hydrophobic
surface barrier and induce the formation of unstable pores in
membranes. J Pharm Pharmacol. 58:1421–1428. 2006. View Article : Google Scholar
|
|
23
|
Farkouh ME, Greenberg JD, Jeger RV, et al:
Cardiovascular outcomes in high risk patients with osteoarthritis
treated with ibuprofen, naproxen or lumiracoxib. Ann Rheum Dis.
66:764–770. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Otsuki S, Brinson DC, Creighton L, et al:
The effect of glycosaminoglycan loss on chondrocyte viability: a
study on porcine cartilage explants. Arthritis Rheum. 58:1076–1085.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
D’Lima DD, Hashimoto S, Chen PC, Colwell
CW Jr and Lotz MK: Human chondrocyte apoptosis in response to
mechanical injury. Osteoarthritis Cartilage. 9:712–719.
2001.PubMed/NCBI
|
|
26
|
Goldring SR and Goldring MB: Clinical
aspects, pathology and pathophysiology of osteoarthritis. J
Musculoskelet Neuronal Interact. 6:376–378. 2006.PubMed/NCBI
|
|
27
|
Caramés B, Taniguchi N, Seino D, Blanco
FJ, D’Lima D and Lotz M: Mechanical injury suppresses autophagy
regulators and pharmacologic activation of autophagy results in
chondroprotection. Arthritis Rheum. 64:1182–1192. 2012.PubMed/NCBI
|
|
28
|
Kimura S, Noda T and Yoshimori T:
Dynein-dependent movement of autophagosomes mediates efficient
encounters with lysosomes. Cell Struct Funct. 33:109–122. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Srinivas V, Bohensky J, Zahm AM and
Shapiro IM: Autophagy in mineralizing tissues: microenvironmental
perspectives. Cell Cycle. 8:391–393. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cajee UF, Hull R and Ntwasa M:
Modification by ubiquitin-like proteins: significance in apoptosis
and autophagy pathways. Int J Mol Sci. 13:11804–11831. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Klionsky DJ and Emr SD: Autophagy as a
regulated pathway of cellular degradation. Science. 290:1717–1721.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Abeliovich H and Klionsky DJ: Autophagy in
yeast: mechanistic insights and physiological function. Microbiol
Mol Biol Rev. 65:463–479. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ohsumi Y and Mizushima N: Two
ubiquitin-like conjugation systems essential for autophagy. Semin
Cell Dev Biol. 15:231–236. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Caramés B, Kiosses WB, Akasaki Y, et al:
Glucosamine activates autophagy in vitro and in vivo. Arthritis
Rheum. 65:1843–1852. 2013.
|
|
35
|
Srinivas V, Bohensky J and Shapiro IM:
Autophagy: a new phase in the maturation of growth plate
chondrocytes is regulated by HIF, mTOR and AMP kinase. Cells
Tissues Organs. 189:88–92. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Bohensky J, Shapiro IM, Leshinsky S,
Terkhorn SP, Adams CS and Srinivas V: HIF-1 regulation of
chondrocyte apoptosis: induction of the autophagic pathway.
Autophagy. 3:207–214. 2007. View Article : Google Scholar : PubMed/NCBI
|



















