Introduction
Histone deacetylase (HDAC) activity has been shown
to be upregulated in cancer cells. It is considered that this
upregulation results in the repression of tumor suppressor gene
products, such as p53, rendering HDACs an attractive drug target.
In cell culture models, HDAC inhibitors (HDACIs) have been shown to
decrease proliferation rates, induce apoptosis and induce
autophagy-related cell death in several cancer cell lines. Due to
their relative specificity toward cancer cells, HDACIs represent a
new class of cancer treatment agents that are generally well
tolerated (1).
Short-chain fatty acids, such as butyric and
valproic acid (VPA) were the first HDACIs to be identified as tumor
growth inhibitors and inducers of apoptosis both in vitro
and in vivo. However, they were found to have low potency
(2). Despite such weak in
vitro activity, the anticancer effects of VPA have been
investigated in preclinical models of skin, breast, colon, prostate
and small cell lung cancer, and the drug is currently being used in
phase I-III clinical trials (3).
However, the therapeutic doses of VPA are very high and cause
side-effects severe enough to limit its usage.
Despite their promising activity in preclinical
models, HDACIs have demonstrated modest antitumor activity as
single-agent therapy in solid malignancies (4). Thus, their use in combination
treatments with other agents may be the key to optimizing their
efficacy (4). HDACIs have shown
synergistic or additive antitumor effects when combined with
conventional anticancer drugs, targeted agents and radiation
(4,5).
The rationale to targeting the proteasome in cancer
arises from evidence that indicates that malignant cells accumulate
misfolded/damaged proteins, which are disposed of by the
proteasome, suggesting that tumor cells have a stronger dependency
on proteasome activity compared to that of normal cells (6). The 26S proteasome is the primary
subcellular component of the protein degradation pathway that
regulates the turnover of proteins involved in cell cycle
progression and apoptosis, such as the p21 cyclin-dependent kinase
(CDK) inhibitor (CDKI), cyclins and IκB, a regulator of nuclear
factor-κB (NFκB) transcriptional activity. Validation of the
proteasome as a therapeutic target has come with the FDA approval
of bortezomib, an inhibitor of the 20S proteasome (7).
In vitro studies have demonstrated that the
simultaneous targeting of HDACs and the 26S proteasome produces a
synergistic inhibition of cell proliferation and induction of
apoptosis in pancreatic cancer cell lines (8), multiple myeloma (9), lung cancer and hepatoma cell lines
(10). These effects are greater
than those observed with treatment with either agent alone. In the
present study, we examined the effects of combining HDAC inhibition
via VPA with proteasome inhibition via MG132, PI-1 or PR-39 on
colorectal cancer cell lines in vitro, with respect to their
potential synergistic effects on cell proliferation. Our hypothesis
was that these two agents act synergistically to inhibit cell
growth and induce apoptosis, as well as potentiate the sensitivity
of cancer cells to conventional chemotherapeutic drugs with
different modes of action in human colorectal cancer cells. We also
sought to investigate the molecular mechanisms of such a synergy by
assessing the effects of the combination therapy on the cell cycle,
apoptosis and downstream effector pathways. The aim of our study
was to provide evidence to support a mechanism-based regimen in the
treatment of colorectal cancer.
Materials and methods
Cells and cell culture
Human colorectal cancer cell lines (SW1116 and
SW837) and normal human fibroblasts (CRL1554) were obtained from
the American Type Culture Collection (ATCC; Manassas, VA, USA). The
SW1116 and SW837 cells were cultured in 90% Leibovitz’s L15 medium
supplemented with 10% heat-inactivated fetal bovine serum (FBS) and
grown at 37°C in a non-CO2 incubator. The CRL1554 cells
were cultured in Eagle’s minimum essential medium (EMEM, 90%)
supplemented with 10% heat-inactivated FBS and grown at 37°C in the
presence of 5% CO2 and 95% ambient air.
Chemicals and reagents
VPA was purchased from Sigma-Aldrich (St. Louis, MO,
USA). It was dissolved in sterile ddH2O to a final
concentration of 100 mM and stored at −20°C. This stock solution
was diluted to the desired concentrations with L15 medium and used
in the various assays. The proteasome inhibitors, MG132, proteasome
inhibitor-1 (PI-1) and PR-39, were obtained from Biomol
International (Plymouth Meeting, PA, USA). Trypsin, Leibovitz’s
L-15 medium and EMEM, FBS and penicillin/streptomycin solution
(200X) were obtained from Mediatech, Inc. (Herndon, VA, USA). An
Annexin V-FITC apoptosis detection kit was obtained from BD
Hoffmann-Laroche Inc. (Nutley, NJ, USA). A DNA-prep kit was
obtained from Beckman Coulter (Miami, FL, USA). All reagents for
RT-PCR and real-time (quantitative) PCR (qPCR) were obtained from
Applied Biosystems (Foster City, CA, USA). Nuclear/cytosol and
mitochondria/cytosol fractionation kits were obtained from
BioVision, Inc. (Mountain View, CA, USA). A 20S Proteasome assay
kit for Drug Discovery was purchased from Biomol International. The
cDNA reverse transcription kit, primers and target gene expression
probes were obtained from Applied Biosystems. Antibodies against
phospho-c-Jun N-terminal kinase (p-JNK), phospho-extracellular
signal-regulated kinase 1/2 (p-ERK1/2) and phospho-Akt (p-Akt) were
purchased from Cell Signaling Technology (Beverly, MA, USA).
Anti-p65 and anti-β-actin antibodies were purchased from Santa Cruz
Biotechnology, Inc. (Santa Cruz, CA, USA and Cambridge, UK). All
other reagents were purchased from Sigma-Aldrich. Plasticware was
purchased from Falcon Laboratories (Franklin Lakes, NJ, USA).
Cell proliferation assay
Cell proliferation assay was performed using a
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay, as previously described (11). The cells were treated with various
concentrations of VPA (0.5–8 mM) for 24 h and then exposed to MG132
(0.15 and 0.25 μM), PI-1 (7.8 and 15.6 nM) or PR-39 (106 and 112
pM) for an additional 48 h. For single-agent treatment, the cells
were exposed to VPA for 72 h. Upon the completion of the
treatments, the media were discarded and 100 μl/well of MTT (5
mg/ml in culture medium filter-sterilized) was added, and the
plates were incubated for 4 h at 37°C. The resulting formazan
crystals were dissolved in 200 μl/well of DMSO:ethanol (1:1 v/v)
for 20 min at ambient temperature and the absorbance was recorded
at λ540 and 650 nm using an ELISA plate reader. The control cells
were incubated in medium supplemented with 0.2% DMSO alone. At this
concentration, DMSO has no effect on the growth and survival of
cells. The effects of the combined treatment with VPA (1 mM)/MG132
(0.2 μM), VPA (1 mM)/ PI-1 (10 nM) and VPA (1 mM)/PR-39 (212 pM) on
the normal human fibroblasts, CRL1554, were also monitored by MTT
assay and an inverted microscope.
Colony formation assay
A colony formation assay was carried out as
previously described (11) to
confirm the effects of VPA, PIs (MG132, PI-1 or PR-39) and their
combinations on the colorectal cancer cells. Briefly, the SW1116
and SW837 cells (2.5×105 cells/ml) were seeded into
24-well plates and incubated for 18 h. The cells were then treated
with VPA (2.5 mM) or the PIs MG132 (1.5 μM), PI-1 (36 nM), or PR-39
(2 nM), as well as combinations of VPA with each of the PIs. After
24 h, the cells were trypsinized, counted and plated at 500
cells/ml into a 6-well plate and incubated for 10–14 days. The
cells were finally fixed in 100% methanol for 30 min at room
temperature and stained with 0.1% crystal violet for 1 h. The
stained colonies were counted and compared with an untreated
control sample.
Determination of HDAC activity
A colorimetric HDAC activity assay kit (BioVision,
Inc.) was used to monitor HDAC activity in the cancer cell nuclear
extracts, according to the manufacturer’s instructions. The SW1116
and SW837 colorectal cancer cells (2.5×105 cells/ml)
were seeded into 24-well plates and incubated for 18 h, followed by
treatment with VPA (2.5 mM) for an additional 24 h. Nuclear
extracts of the untreated and VPA-treated cells (50 μg) were
diluted with ddH2O to a final volume of 85 μl in each
well of a 96-well plate. Only 85 μl ddH2O was added for
background reading. For a positive control, 10 μl of HeLa nuclear
extract were diluted with 75 μl ddH2O. For a negative
control, the tested nuclear extract was diluted to 83 μl, and then
2 μl of TSA (HDACI, 1 mM) were added. Alternatively, a known sample
containing no HDAC activity was used as a negative control. Ten
microliters of the 10× HDAC assay buffer and 5 μl of the HDAC
colorimetric substrate [Boc-Lys(Ac)-pNA, 10 mM] were added to each
well and mixed thoroughly, and the plates were then incubated at
37°C. After 1 h, the reaction was terminated by the addition of 10
μl of lysine developer, mixed well and then incubated at 37°C for a
further 30 min. Absorbance was recorded at 400 or 405 nm using an
ELISA plate reader.
Determination of proteasome activity
Proteasome activity, in the cancer cell extracts,
was monitored by using a 20S proteasome assay kit for Drug
Discovery (Biomol International) according to the manufacturer’s
instructions. Briefly, the SW1116 and SW837 cells
(2.5×105 cells/ml) were seeded into 24-well plates and
incubated for 18 h, and then treated with the PIs, MG132 (1.5 μM),
PI-1 (36 nM), or PR-39 (2 nM) for 24 h. A nuclear/cytosolic
fractionation kit (BioVision, Inc.) was used to prepare the cell
extracts of the untreated and treated cells. Finally, the cytosolic
extracts (0.5 μg), positive (epoxomicin) and negative (no 20S
proteosome) controls were incubated with 75 μM proteasome substrate
(Suc-LLVY-AMC) in 100 μl assay buffer (20 mM Tris-HCl, pH 8.0) for
90 min at 37°C. Fluorescence from 7-amido-4-methyl-coumarin (AMC)
was measured using a VersaFluor™ fluorometer with excitation at
λ360 nm and emission λ460 nm (Bio-Rad Laboratories, Inc., Hercules,
CA, USA).
Measurement of reactive oxygen species
(ROS) production
The extent of ROS generation was analyzed by
quantifying the fluorescence intensity of 2′,7′-dichlorofluorescein
diacetate (DCFH-DA) (Sigma-Aldrich), as previously described
(12). DCFH-DA is a stable,
non-fluorescent cell-permeable compound that fluoresces
(dichlorofluorescein) in the presence of active radicals and emits
green fluorescence upon excitation at 485 nm. The SW1116 and SW837
cells (2.5×105 cells/ml) were seeded into 24-well plates
and incubated for 18 h. The cells were then treated with VPA (2.5
mM) or the PIs: MG132 (1.5 μM), PI-1 (36 nM) or PR-39 (2 nM), or a
combination of VPA with each of the PIs, or the solvent alone as
described above. After 48 h, the cells were washed and subsequently
incubated with 10 μM DCFH-DA in phosphate-buffered saline (PBS) for
30 min at 37°C. Fluorescence was analyzed on a microtiter plate
reader using excitation at 485 nm and emission at 535 nm. The
production of ROS was determined by comparing the intensity of
fluorescence in the treated vs. untreated cells. The free radical
scavenger, L-N-acetylcysteine (L-NAC) (Sigma-Aldrich), was used to
assess the functional role of ROS generation in cell death. The
cells were pre-incubated with 15 mM L-NAC for 3 h, followed by
treatment with VPA, PIs, or combinations of both for 48 h. The
assessment of cell death was carried out as described above.
Cell cycle analysis
Flow cytometry was used to determine the
distribution of cells in the various cell cycle phases
(G0/G1, S and G2/M) by measuring
the DNA content of the nuclei labeled with propidium iodide as
previously described (12).
Briefly, the SW837 cells (2.5×105 cells/ml) were seeded
into 24-well plates and incubated for 18 h. The cells were then
treated with either VPA (2.5 mM), PIs MG132 (1.5 μM), PI-1 (36 nM)
or PR-39 (2 nM), or a combination of VPA and a PI for 24 h. The
untreated and drug-treated cancer cells were collected by
trypsinization, washed with cold PBS and counted. The cells were
then processed by using a DNA-prep kit (Beckman Coulter) and a
DNA-Prep EPICS workstation (Beckman Coulter) as follows: the cells
were treated with a cell-membrane permeabilizing agent followed by
propidium iodide and RNase. The samples were then incubated at room
temperature for 15 min before being analyzed using a FC500 flow
cytometer (Beckman Coulter, Nyon, Switzerland). The Phoenix
statistical software package with advanced DNA cell cycle software
(Phoenix Flow Systems, San Diego, CA, USA) was used to calculate
the percentage of cells in the various cell cycle phases.
Assessment of apoptosis
An Annexin V-FITC apoptosis detection kit (BD
Hoffmann-La Roche Inc.) was used to determine the level of
apoptotic cell death in the colorectal cancer cells treated with
VPA, PIs, or a combination of both according to the manufacturer’s
instructions. Briefly, the SW837 cells (2.5×105
cells/ml) were seeded into 24-well plates and incubated for 18 h.
The cells were then treated with VPA (2.5 mM), the PIs, MG132 (1.5
μM), PI-1 (36 nM) or PR-39 (2 nM), and a combination of VPA and one
of the inhibitors for 24 h. The control and treated cells were
resuspended in 100 μl of staining solution containing Annexin V
fluorescein and propidium iodide in HEPES buffer, incubated at room
temperature for 15 min, and finally analyzed by flow cytometry.
Annexin V binds to cells that express phosphatidylserine on the
outer layer of the cell membrane, while propidium iodide stains the
cellular DNA of cells with a compromised cell membrane. This allows
live cells (unstained with either of the fluorochromes) to be
discriminated from apoptotic cells (stained only with Annexin V)
and necrotic cells (stained with both Annexin V and propidium
iodide).
Western blot analysis
The colorectal cancer cells, SW1116 and SW837
(2.5×105 cells/ml), were seeded into 24-well plates and
allowed to grow for 18 h. The cells were then treated with VPA (2.5
mM), PI-1 (36 nM) or a combination of both for 24 h. Following
treatment, the cells were rinsed with ice-cold PBS and cytosol, and
then nuclear and mitochondrial fractions were prepared using
cytosol/nuclear and cytosol/mitochondria fractionation kits
(BioVision, Inc.) according to the manufacturer’s instructions.
Protein concentration was determined by the Bradford (13) method using bovine serum albumin
(BSA) as a standard. Western blot analysis was carried out as
previously described (14).
Briefly, the cell lysates (50 μg) were resolved by SDS-PAGE on
7.5–15% slab gels, and proteins were transferred onto
polyvinylidene difluoride membranes blocked in a PBS solution
containing 0.1% Tween-20 and 5% (w/v) dry skim milk for 30 min and
incubated at 4°C overnight with primary antibodies against Bax,
cytochrome c, NF-κB (p65), pAkt, pJNK, pERK1/2 and β-actin.
After washing with PBS containing 0.1% Tween-20, the blots were
incubated with an alkaline phosphatase-conjugated species specific
IgG secondary antibody for 2 h at room temperature. Bound
antibodies were detected using nitroblue tetrazolium and
5-bromo-4-chloro-3-indolyl phosphate. The specificities of the
antibodies used in this study were examined by testing their
reactivity with unrelated antigens, such as BSA. The signal
intensities of the respective bands were quantified using a GS-800
calibrated imaging densitometer (Bio-Rad Laboratories, Inc.).
Analysis of mRNA expression of genes
involved in the regulation of apoptosis and the cell cycle
The expression of genes involved in the regulation
of the cell cycle and apoptosis was determined in the cells from
the control and treated groups by reverse transcription-PCR and
real-time PCR (qPCR) using an ABI 7000 SDS system (Applied
Biosystems) and the comparative ΔΔCt method, as previously
described (11). Ready-made
assays-on-demand that target gene expression probes and primers
were obtained from Applied Biosystems. The targets and Applied
Biosystems assay numbers for the cell cycle-related genes were as
follows: CDK1 (Hs00364293_m1), CDK4 (Hs00364847_m1), CDC25A
(Hs00153168_m1), p15 (Hs00394703_m1), p19 (Hs00176481_m1), p21
(Hs00355782_m1) and p27 (Hs00197366_m1).
The targets and Applied Biosystems assay numbers for
the pro-apoptotic and anti-apoptotic genes and caspases that were
used in this study are as follows: Bax (Hs00180269_m1), Bad
(Hs00188930_m1), Bim (Hs00375807_m1), apoptotic protease activating
factor 1 (Apaf1; Hs00559441_m1), Bcl-2 (Hs00608023_m1), the
X-linked inhibitor of apoptosis (XIAP; Hs00236913_m1), caspase-3
(Hs00234387_m1), caspase-8 (Hs01018151_m1), caspase-9
(Hs00154260_m1), the apoptosis-inducing factor (AIF; Hs00269879_ml)
and FLIP (Hs00354474_ml). GAPDH was used as an endogenous control
to normalize the expression values for each sample. For the
comparative Ct method, we performed a two-step RT-PCR process using
cDNA and carried out qPCR using the target gene expression assays
and TaqMan Universal master mix (Applied Biosystems). The SW837
colorectal cancer cells were plated (2.5×105 cells/ml)
into 24-well plates and incubated in a non-CO2 incubator
for 18 h. The cells were then treated with either VPA (2.5 mM), the
PIs MG132 (1.5 μM), PI-1 (36 nM) or PR-39 (2 nM), or a combination
of VPA with each PI for 24 h. mRNA was extracted using a nucleospin
RNAII ready-to-use system (MACHEREY-NAGEL GmbH & Co., Düren,
Germany), and 200 ng/μl of mRNA was used in each RT reaction.
First, DNA was eliminated by DNase-I treatment for 20 min at 25°C,
followed by heat inactivation for 10 min at 65°C. cDNA synthesis
was performed using a high capacity cDNA Reverse Transcription kit
(Applied Biosystems) according to the manufacturer’s instructions.
For each sample, 2.5 μl of cDNA and 12.5 μl of TaqMan Universal
Master Mix (2X) were used, and the final volume was adjusted to 25
μl with nuclease-free water in an optical 96-well reaction plate
(Applied Biosystems). qPCR was performed on an ABI 7000 SDS system
using ABI Prism SDS collection software version 1.1 (Applied
Biosystems). The conditions for qPCR followed the instructions
provided with the TaqMan Universal Master Mix: step 1, 95°C for 10
min; step 2, 94°C for 15 sec; and step 3, 60°C for 1 min. The
samples were analyzed using ABI Prism SDS collection software
version 1.1 by setting the base line between 3 and 15 and the
threshold at 0.2. The amount of target normalized to an endogenous
reference and relative to a calibrator (untreated) was given by
2−ΔΔCt and the log comparative Ct was presented
graphically.
Analysis of the chemosensitizing activity
of VPA and PIs
The potential of the combined treatment with VPA and
PIs (MG132, PI-1 or PR-39) to sensitize human colorectal cancer
cells to standard chemotherapeutic drugs was investigated as
previously described (12), with
some modifications. Briefly, the SW1116 and SW837 colorectal cells
(27×103 cells/well) were seeded into 96-well plates and
incubated for 18 h. The cells were then treated for 24 h with
various concentrations of camptothecin (CPT;
64×10−10-1×10−4 M), 5-fluorouracil (5FU;
41.6×10−9-0.65×10−3 M), oxaliplatin (OXP;
4×10−10-0.06×10−4 M), doxorubicin (DOX;
55×10−11-0.85×10−5 M), carboplatin (CAP;
43.5×10−10-0.86×10−4 M), cisplatin (CIP;
26.88×10−9-0.42×10−3 M), taxol (TAX;
93.44×10−10-1.4×10−4 M), cyclophosphamide
(CPA; 14×10−9-0.22×10−3 M), vincristine (VCR;
16×10−11-2.5×10−5 M), etoposide (ETP;
25.6×10−10-0.4×10−4 M), ellipticine (ELP;
12.8×10−10-0.2×10−4 M), amsacrine (AMS;
80×10−11-1.25×10−5 M), homoharringtonine
(HHG; 12.8×10−11-0.2×10−5 M) and aphidicolin
(APD; 17.28×10−11-0.27×10−5 M). The drugs
were then removed from the samples. At the end of treatment, the
cells were washed with Hank’s balanced salt solution (HBSS) and
incubated with a combination of VPA (1 mM)/MG132 (0.25 μM), VPA (1
mM)/PI-1 (10 nM) and VPA (1 mM)/PR-39 (212 nM) for 72 h. An MTT
assay was used to monitor the growth of the treated cells.
Statistical analyses
The results are expressed as the means ± standard
error of the mean (SEM) of at least 3 independent experiments.
Statistical analyses were performed using SPSS-21 software. The
statistical significance of the differences between the control and
treated groups was determined by one-way ANOVA. P-values <0.05
were considered to indicate statistically significant
differences.
Results
PIs potentiate the antimitogenic effect
of VPA against human colorectal cancer cells
To determine whether the PIs (MG132, PI-1 and PR-39)
enhance the cytotoxicity of VPA toward colorectal cancer cells, the
cancer cells were treated with various concentrations of VPA
(0.5–8.0 mM) alone, as well as in the presence of the tested PIs:
MG132 (0.15 and 0.25 μM), PI-1 (7.8 and 15.6 nM) and PR-39 (106 and
212 pM). The concentrations used in this experiment were determined
by an independent dose response experiment (data not shown).
When treated with various concentrations of VPA, the
SW1116 and SW837 colorectal cancer cells displayed a dose- and
time-dependent growth inhibition. The IC50 values for
the SW1116 and SW837 cells were 2.75 and 7.5 mM, respectively,
indicating that the SW1116 cells are more sensitive than the SW837
cells, which demonstrated a resistance to VPA treatment. The
combined treatment of VPA (0.5–8 mM) and MG132 (0.15 μM) exerted a
greater growth inhibitory effect for both the SW1116
(IC50 2.25 mM, P≤0.803) (Fig. 1A-a) and SW837 cells
(IC50 4.75 mM, P≤0.036) (Fig. 1A-d), compared to the resistance
produced by single treatment with VPA. MG132 (0.15 μM) slightly
improved the sensitivity of the SW1116 [sensitization ratio (SR)
1.22] and SW837 (SR 1.60) cells to VPA. By contrast, the combined
treatment with VPA and MG132 (0.25 μM) led to a significantly
greater growth inhibitory effect on the SW1116 cells
(IC80 1.0 mM, P≤0.002) compared to treatment with VPA
alone (IC80 6.75 mM for SW1116 cells; Fig. 1A-a). The addition of MG132 (0.25
μM) markedly increased the sensitivity of the SW1116 cells (SR
6.75) to VPA. The combined treatment with VPA and MG132 (0.25 μM)
led to a greater growth inhibitory effect on the SW837 cells
(IC80 3.0 mM) compared to treatment with VPA alone
(IC50 7.5 mM; Fig.
1A-d).
Combined treatment with VPA (0.5–8.0 mM) and PI-1
(7.8 nM) exerted a greater, although not significant, growth
inhibitory effect on both the SW1116 (IC50 2.0 mM,
P≤0.607) (Fig. 1A-b) and SW837
cells (IC50 5.25 mM, P≤0.154) (Fig. 1A-e) compared to that produced by
treatment with VPA alone (IC50 3.0 mM for the SW1116
celks and IC50 6.75 mM for the SW837 cells) (Fig. 1A-b and e). PI-1 (7.8 nM)
moderately increased the sensitivity of both the SW1116 (SR 1.5)
and SW837 cells (SR 1.30) to VPA. However, the combined treatment
with VPA and PI-1 (15.6 nM) led to a much greater, statistically
significant growth inhibitory effect on the SW1116 (IC70
1.75 mM, P≤0.013; Fig. 1A-b) and
SW837 cells (IC50 3.25 mM, P≤0.002; Fig. 1A-e) compared to treatment with VPA
alone. PI-1 (15.6 nM) markedly increased the sensitivity of both
the SW1116 (SR 3.3) and SW837 cells (SR 2.1) to VPA.
Combined treatment with VPA (0.5–8.0 mM) and PR-39
(106 pM) exerted a slightly greater growth inhibitory effect on the
SW1116 (IC50 2.25 mM, P≤0.759) and SW837 cells
(IC50 6.75 mM, P≤0.388) than that produced by single
treatment with VPA (IC50 values equal to 3.0 mM for
SW1116 cells and 7.75 mM for SW837 cells; Fig. 1A-c and f). PR-39 slightly improved
the sensitivity of the SW1116 (SR 1.33) and SW837 cells (SR 1.15)
to VPA. Combined treatment with VPA and PR-39 (212 pM) exerted a
significantly greater growth inhibitory effect on the SW1116 cells
(IC50 1.25 mM, P≤0.013; Fig. 1A-c), and an even more significant
growth inhibitory effect on the SW837 cells (IC50 5.5
mM, P≤0.0001; Fig. 1A-f) compared
to the results obtained with treatment with VPA alone. Finally,
PR-39 (212 pM) increased the sensitivity of both the SW1116 (SR
2.4) and SW837 cells (SR 1.41) to VPA.
The results of the growth inhibitory effects were
confirmed by a colony formation assay. Treatment of the SW1116
cells with a combination of VPA and MG132 significantly inhibited
colony formation (mean number of colonies, 82, P≤0.0001) compared
to the untreated SW1116 cells (mean number of colonies, 245).
Colony formation by the SW1116 cells was also significantly
inhibited by the combined treatment with VPA and MG132 (P≤0.0001)
compared to treatment with either VPA (mean number of colonies,
169) or MG132 (mean number of colonies, 108) alone (Fig. 1B-a).
Treatment of the SW1116 cells with a combination of
VPA and PI-1 significantly inhibited colony formation (mean number
of colonies, 121, P≤0.0001) compared to the untreated SW1116 cells
(mean number of colonies, 245). The same treatment also
significantly inhibited the colony formation of the SW1116 cells
(P≤0.001) compared to treatment with VPA alone (mean number of
colonies, 169). This combined treatment decreased the number of
colonies formed compared to the cells treated with PI-1 alone (mean
number of colonies, 130). However, the difference in the number of
colony formations was statistically insignificant (P≤0.407)
(Fig. 1B-b).
Combined treatment with VPA and PR-39 significantly
inhibited the colony formation of the SW1116 cells (mean number of
colonies, 98, P≤0.0001) compared to the untreated SW1116 cells
(mean number of colonies, 245). The combined treatment also
significantly inhibited the colony formation of the SW1116 cells
compared to treatment with either VPA alone (mean number of
colonies, 169, P≤0.0001) or PR-39 alone (mean number of colonies,
126, P≤0.003) (Fig. 1B-c).
The morphological changes of the human colorectal
cancer cells following treatment with VPA, PIs, or a combination of
both (Fig. 1C-a-f) clearly
demonstrated the cytolytic effects of these treatments, and
confirmed the results of the experiments analyzing the inhibitory
effects.
Inhibition of HDAC and 26S proteasome
activities and induction of ROS generation in cancer cells
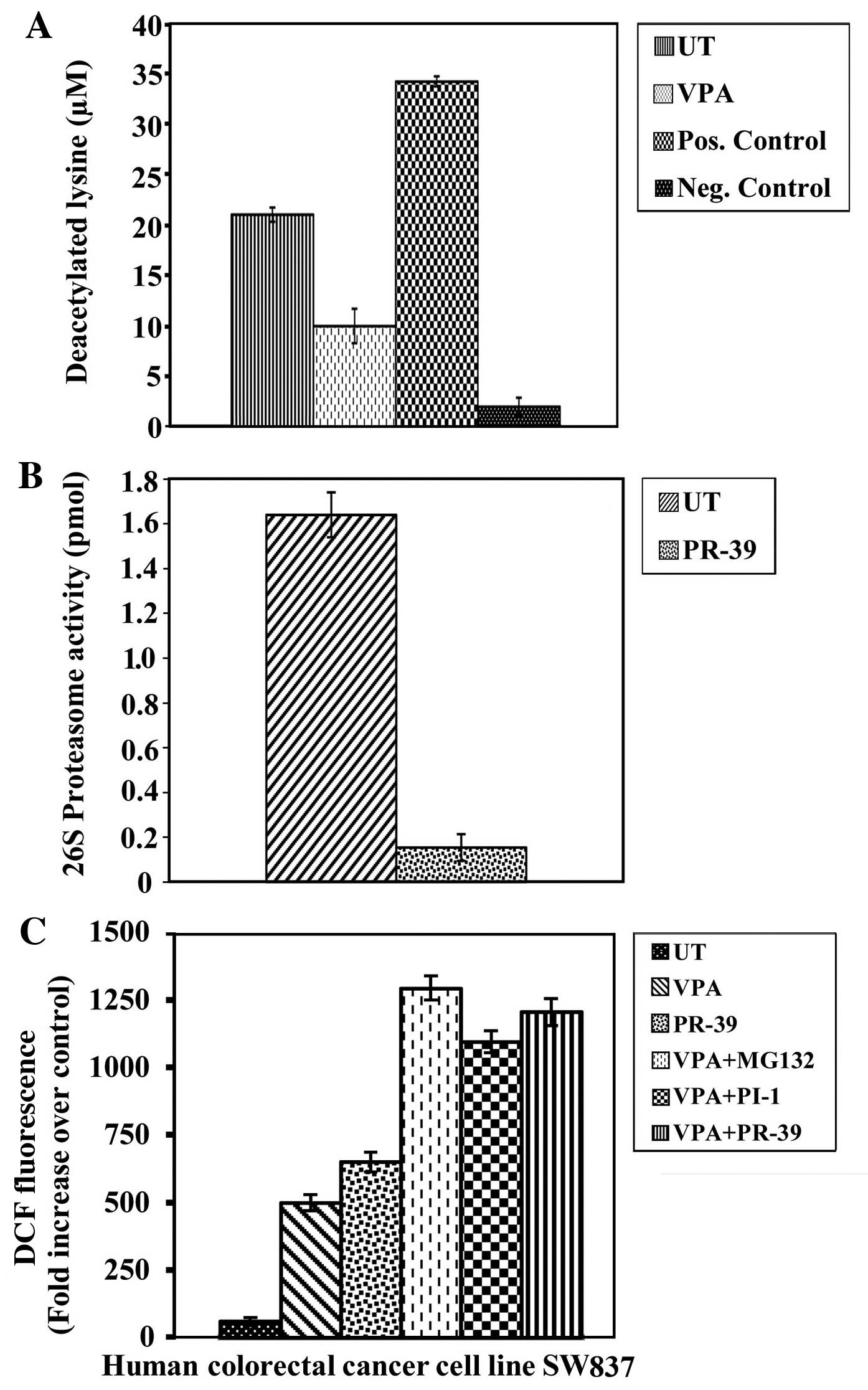
To determine whether the antimitogenic activity of
VPA is associated with the inhibition of intra-nuclear HDAC
activity, HDAC activity was measured in nuclear fractions of
untreated and VPA-treated SW837 colorectal cancer cells. The cancer
cells treated with VPA showed a significant inhibition of HDAC
activity (P≤0.0001) compared to the untreated cancer cells
(Fig. 2A).
To determine whether the antimitogenic activities of
the PIs included in this study are linked to the inhibition of
intracellular proteasome activity, 26S proteasome activity was
measured in both the untreated and PI-treated cancer cell extracts.
The PI, PR-39, exerted a significant inhibitory effect on 26S
proteasome activity (P≤0.0001) in the colorectal cancer cells
compared to the untreated cancer cells (Fig. 2B). A significant inhibition of 26S
proteasome activity was also observed following treatment with the
PIs, MG132 (P≤0.001) and PI-1 (P≤0.001), (data not shown).
ROS have been implicated in HDAC and PI-induced
cytotoxicity in a number of different types of malignancies
(15). Thus, the effects of VPA,
PIs, or a combination of VPA and PIs on ROS generation in the human
SW837 colorectal cancer cells were examined. The levels of ROS were
markedly increased (P≤0.0001) following the combined treatment with
VPA/MG132, VPA/PI-1 and VPA/PR-39 compared to the untreated cells
and cells treated with VPA or PIs alone (Fig. 2C). Additionally, solo treatments
with MG132 (P≤0.0001) or PI-1 (P≤0.004) significantly induced ROS
generation, compared to the untreated cancer cells. The induction
of ROS was abrogated by pre-treatment with L-NAC (data not
shown).
Alterations in the cell cycle and
induction of apoptosis in colorectal cancer cells treated with VPA,
PIs or a combination of both
To elucidate the effects of VPA, the tested PIs, and
the combinations of VPA with each PI on the cell cycle, the
distribution of cancer cells in the various cell cycle phases
(G0/G1, S and G2/M) was determined
by flow cytometry. Treatment of the SW837 cancer cells with VPA
resulted in the accumulation of cancer cells in the S phase [63.6
vs. 44.3% for the untreated cells (UT)] at the expense of a strong
decrease in the number of cells in the G0/G1
phase (31.9 vs. 41.5% for UT) and the G2/M phase (4.4
vs. 14.1% for UT) (Fig. 3A).
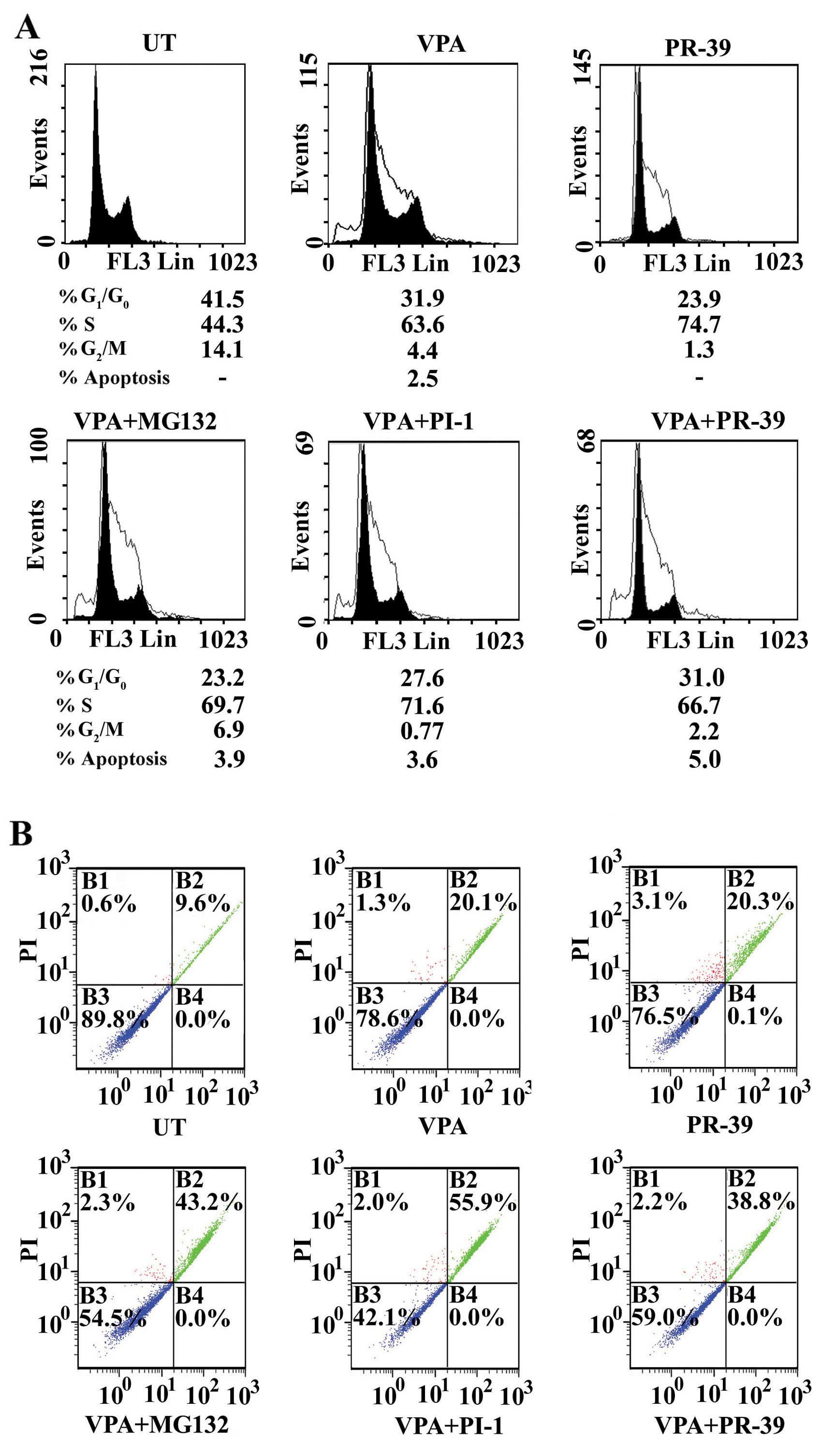 | Figure 3Modulation of cell cycle and
induction of apoptosis in colorectal cancer cells treated with
valproic acid (VPA), proteasome inhibitors (PIs) and their
combinations. (A) Cell cycle analysis: SW837 cells
(2.5×105 cell/ml) were treated with VPA (2.5 mM) and the
PIs, MG132 (1.5 μM), PI-1 (36 nM) or PR-39 (2 nM), and a
combination of both for 24 h: at least 3 samples were analyzed and
20,000 events were scored for each sample. The vertical axis
represents the relative number of events and the horizontal axis
represents the fluorescence intensity. (B) Monitoring of apoptosis.
SW837 cells were treated as described above. Cells were stained
with Annexin V-fluorescein and propidium iodide, and then analyzed
by flow cytometry. Quadrant B1, percentage of necrotic cells;
quadrant B2, percentage of late apoptotic cells; quadrant B3,
percentage of living cells; and quadrant B4, percentage of early
apoptotic cells. |
Treatment of the SW837 cells with PR-39 resulted in
a marked accumulation of cancer cells in the S phase (74.7 vs.
44.3% for UT) at the expense of a decrease in the number of cells
in the G0/G1 phase (23.9 vs. 41.5% for UT)
and the G2/M phase (1.3 vs. 14.1% for UT) (Fig. 3A). Treatment of the SW837 with
MG132 or PI-1 resulted in the growth arrest of cancer cells in the
S phase or the G0/G1 phase, respectively
(data not shown).
Combined treatment with VPA and MG132 arrested the
growth of the cancer cells in the S phase (69.7 vs. 44.3% for UT)
at the expense of a decrease in the number of cells in the
G0/G1 phase (23.2 vs. 41.5% for UT) and
G2/M phase (6.9 vs. 14.1% for UT) (Fig. 3A).
Treatment with a combination of VPA and PI-1
resulted in the accumulation of SW837 cells in the S phase (71.6
vs. 44.3% for UT) at the expense of a decrease in cells in the
G0/G1 phase (27.6 vs. 41.5% for UT) and the
G2/M phase (0.77 vs. 14.1% for UT) (Fig. 3A).
Combined treatment with VPA and PR-39 resulted in
the accumulation of cancer cells in the S phase (66.7 vs. 44.3% for
UT) at the expense of a decrease in the G0/G1
phase (31 vs. 41.5% for UT) and the G2/M phase (2.2 vs.
14.1% for UT) (Fig. 3A). In a
parallel experiment of the distribution of the cell cycle phase
using the SW1116 colorectal cancer cells treated with VPA, PIs, or
a combination of VPA with PIs, the results obtained were similar to
those obtained with the SW837 cells (data not shown).
To determine what effect, if any, the PIs (MG132,
PI-1 and PR-39) exert on the response of colorectal cancer cells to
VPA and to distinguish between the different types of cell death,
untreated and treated SW837 cells were double-stained with Annexin
V and propidium iodide and analyzed by flow cytometry. Annexin V
binding combined with PI labeling was performed for the distinction
between apoptosis (Annexin V+/propidium
iodide−) and necrosis (Annexin V+/propidium
iodide+).
Treatment of the SW837 cancer cells with the
combination of VPA and MG132 markedly induced apoptosis (0.0% early
apoptosis, 43.2% late apoptosis, and 2.3% necrosis) compared to the
untreated cells (0.0% early apoptosis, 9.6% late apoptosis and 0.6%
necrosis), VPA alone-treated SW837 cells (0.0% early apoptosis,
20.1% late apoptosis, and 1.3% necrosis) (Fig. 3B) and the MG132 alone-treated
SW837 cells (0.0% early apoptosis, 26.4% late apoptosis and 0.8%
late apoptosis) (data not shown).
In addition, the combination of VPA and PI-1
markedly induced the apoptosis of SW837 cells (0.0% early
apoptosis, 55.9% late apoptosis, and 2.0% necrosis) compared to the
untreated SW837 cells (0.0% early apoptosis, 9.6% late apoptosis,
and 0.6% necrosis) and the SW837 cells treated with VPA alone (0.0%
early apoptosis, 20.1% late apoptosis, and 1.3% necrosis) or the
PI-1 alone-treated SW837 cells (0.0% early apoptosis, 11.9% late
apoptosis, and 0.8% necrosis) (data not shown). Moreover, the
combined treatment of the SW837 cancer cells with VPA and PR-39
markedly enhanced apoptosis (0.0% early apoptosis, 38.8% late
apoptosis and 2.2% necrosis) compared to the untreated, VPA-treated
and PR-39 alone-treated SW837 cells (0.1% early apoptosis, 20.3%
late apoptosis and 3.1% necrosis) (Fig. 3B).
These results suggested that the tested PIs, MG132,
PI-1 and PR-9, markedly increased VPA-mediated lethality in human
colorectal cancer cells. The induction of apoptosis in the SW1116
colorectal cancer cells treated with VPA, PIs or a combination of
both provided similar results to those obtained with SW837 (data
not shown).
Expression of Bax, cytochrome c, NF-κB,
p-Akt, p-ERK1/2 and p-JNK in colorectal cancer cells treated with
VPA, PI-1 or their combination
A previous study, using several models of apoptosis
demonstrated that Bax translocates from the cytosol to the
mitochondria when overexpressed or in response to certain cell
death stimuli. Moreover, the translocation of Bax to the
mitochondria in a number of systems has been suggested to be
responsible for the release of cytochrome c from the
mitochondria to the cytosol and the activation of apoptosis
(16). In this study, to examine
whether this pathway was activated by combined treatment with VPA
and PI-1, the effects of single and combined treatment with VPA and
PI-1 on Bax translocation and cytochrome c release were
examined. The SW837 cells treated with VPA, PI-1 or the combination
of both were collected and separated into cytosolic and
mitochondrial fractions. The distributions of Bax and cytochrome
c in these fractions were examined by western blot analysis.
Elevated levels of Bax were detected in the mitochondria following
combined treatment with VPA and PI-1, concomitant with the decrease
in Bax expression in the cytosolic fraction (Fig. 4A and E). A marked increase in
cytochrome c release in the cytosolic fraction corresponded
with a decrease in cytochrome c release in the mitochondrial
fraction (Fig. 4B and F). These
results suggested that the combined treatment with VPA and PI-1
induced the translocation of Bax to the mitochondria, subsequently
releasing cytochrome c, preferentially into the cytosol of
the SW837 colorectal cancer cells.
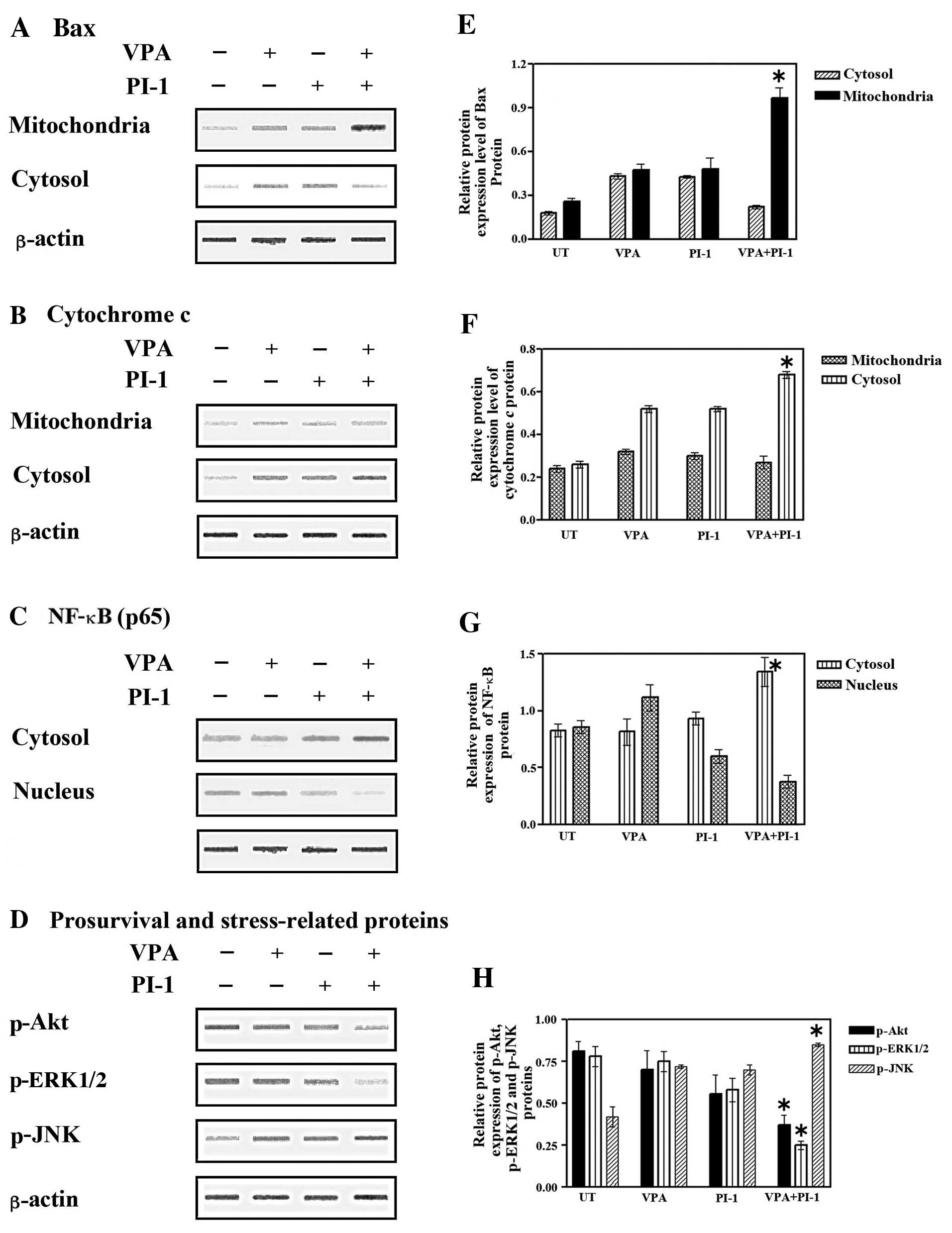 | Figure 4Effect of valproic acid (VPA),
proteasome inhibitor-1 (PI-1) or their combination on Bax
translocation, cytochrome c release and nuclear factor-κB (NF-κB),
phospho-Akt (p-Akt), phospho-extracellular signal-regulated kinase
1/2 (p-ERK1/2), as well as phospho-c-Jun N-terminal kinase (p-JNK)
signaling pathways in cancer cell extracts after solo or combined
treatment with VPA and PI-1. SW837 cells (2.5×105
cells/well) were plated into 24-well plate and incubated at 37°C
for 18 h. Cells were then treated with VPA (2.5 mM), PI-1 (36 nM)
or their combination for 24 h. Cytosolic, mitochondrial and nuclear
fractions were prepared as described in Materials and methods. (A)
Translocation of Bax from the cytosol to the mitochondria, (B)
cytochrome c release from the mitochondria to the cytosol,
(C) the levels of NF-κB in the nucleus and (D) cytoplasmic
fractions and the levels of p-Akt, p-ERK1/2 and p-JNK were analyzed
by western blot analysis. Relative protein expression of (E and F)
mitochondrial and cytosolic Bax and cytochrome c, of (G)
cytoplasmic and nuclear NF-κB, of (H) p-Akt, p-ERK and p-JNK after
normalization to β-actin, *P<0.05 vs. control
(untreated cells). Data were obtained from at least 3 replicate
experiments. |
The NF-κB pathway is a key pathway that promotes the
expression of proliferative and anti-apoptotic genes. The functions
of NF-κB depend on its cellular distribution (17). In this study, to determine whether
NFκB is involved in the synergistic apoptotic effects of VPA and
PI-1, the alterations in NF-κB distribution in the SW837 cells were
examined. Treatment with VPA or PI-1 alone had little effect on
cytoplasmic NF-κB expression compared to the untreated cells,
although nuclear NF-κB expression was slightly increased (Fig. 4C). Despite the elevated level of
NF-κB observed in the cytoplasm of the colorectal cancer cells,
following co-treatment with VPA and PI-1, there was a marked
decrease in the amount of nuclear NF-κB (Fig. 4C and G). These data clearly
indicate that the combination of VPA and PI-1 affects the nuclear
translocation of NF-κB in colorectal cancer cells, suggesting that
the dysfunction of NF-κB is involved in the synergistic apoptotic
effects of VPA and PIs.
To further elucidate the underlying molecular
mechanisms of the anticancer effects of VPA, PIs or the combination
of VPA with PIs on human colorectal cancer cells, the expression of
pro-survival-related (p-Akt and p-ERK1/2) and stress-related
(p-JNK) genes was examined by western blot analysis. As shown in
Fig. 4D, there was a slight
decrease in p-Akt and p-ERK1/2 expression in the SW837 cells
following treatment with VPA or PI-1 alone. More importantly,
co-treatment with VPA and PI-1 induced a marked reduction in the
levels of p-Akt and p-ERK1/2. On the other hand, p-JNK levels were
slightly increased in the cancer cells treated with either VPA or
PI-1 alone, while co-treatment with both VPA and PI-1 markedly
increased the levels of p-JNK (Fig.
4D and H). These data suggest that the Akt, ERK1/2 and JNK
pathways are associated with the enhanced apoptosis that is induced
by the combination of VPA and PIs.
mRNA expression of genes involved in the
regulation of the cell cycle and apoptosis in colorectal cancer
cells treated with VPA, PIs or a combination of both
To elucidate the molecular mechanisms responsible
for the increased lethality induced by VPA in the human colorectal
cancer cells following combination treatment with PIs, the effects
of co-treatment with VPA and PIs on the expression/activation of
various signaling molecules were examined. Treatment of the SW837
cells with a combination of VPA and MG132, PI-1 or PR-39 markedly
downregulated the mRNA expression of genes related to cell cycle
control, CDK1, CDK4 and CDC25A, compared to treatment with VPA,
PR-39 (Fig. 5A), MG132 or PI-1
alone (data not shown). By contrast, the same combined treatments
upregulated the mRNA expression of the p15, p19, p21 and p27 genes,
compared to treatment with only one agent (Fig. 5A).
Treatment of the SW837 cells with a combination of
VPA and PIs differentially upregulated the mRNA expression of the
pro-apoptotic genes, Bax, Bad, Bim, Apaf1, and caspase-3, -8 and
-9. However, the same combined treatment differentially
downregulated the expression of the anti-apoptotic genes, Bcl-2,
XIAP and FLIP, (Fig. 5B). The
cycle threshold values (Ct) of the target genes under investigation
in this study ranged from 20.826–30.981.
Chemosensitization of human colorectal
cancer cells by combined treatment with VPA and PIs
The ability of the combined treatments with VPA and
PIs, MG132, PI-1 or PR-39 to sensitize the cancer cells to standard
chemotherapeutic drugs was examined. The results are summarized in
Figs. 6–10 and Tables I and II.
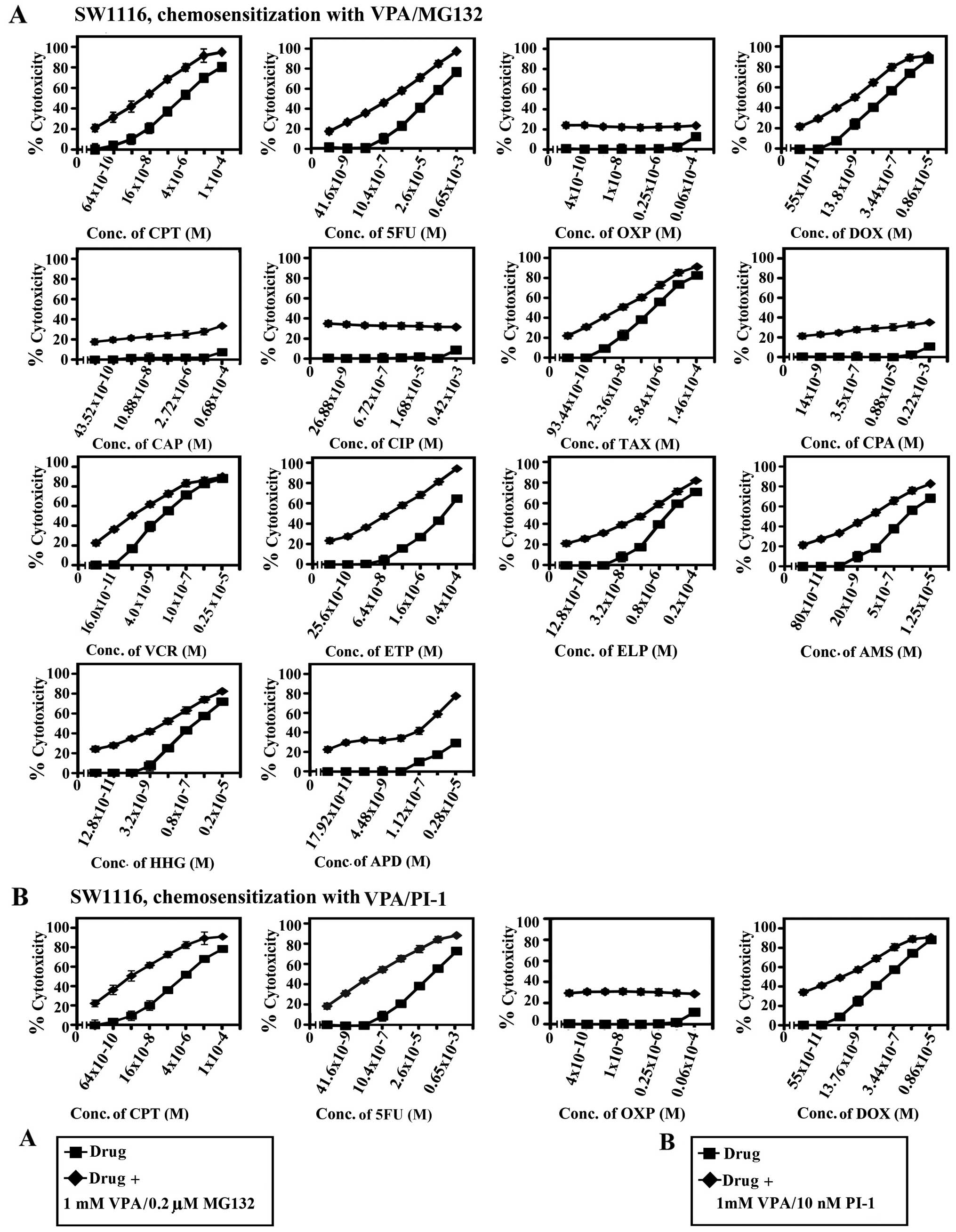 | Figure 6Chemosensitizing effect of combined
treatment with valproic acid (VPA) and MG132 or proteasome
inhibitor-1 (PI-1) on SW1116 colorectal cancer cells. Cancer cells
were inoculated (27×103 cells/well) into 96-well plates
and allowed to grow for 18 h. The cells were treated with various
concentrations of (A) camptothecin (CPT), 5FU, oxaliplatin (OXP),
doxorubicin (DOX), carboplatin (CAP), cisplatin (CIP), taxol (TAX),
cyclophosphamide (CPA), vincristine (VCR), etoposide (ETP),
ellipticine (ELP), amsacrine (AMS), homoharringtonine (HHG) and
aphidicolin (APD) or (B) CPT, 5-fluorouracil (5FU), OXP and DOX for
24 h. The drugs were then removed, cells washed with HBSS and
treated with a combination of (A) VPA (1.0 mM)/MG132 (0.2 μM) or
(B) VPA (1.0 mM)/PI-1 (10 nM) for 72 h. Cell proliferation was
monitored by MTT assay. conc., concentration. |
 | Figure 10Chemosensitizing effect of combined
treatment with valproic acid (VPA) and PR-39 on SW837 colorectal
cancer cells, and the effect of the combined treatment of VPA with
proteasome inhibitors (PIs) on normal human fibroblasts. (A) SW837
cells were plated (27×103 cells/well) into 96-well
plates and allowed to grow for 18 h. The cells were treated with
various concentrations of camptothecin (CPT), 5-fluorouracil (5FU),
oxaliplatin (OXP), doxorubicin (DOX), carboplatin (CAP), cisplatin
(CIP), taxol (TAX), cyclophosphamide (CPA), vincristine (VCR),
etoposide (ETP), ellipticine (ELP), amsacrine (AMS),
homoharringtonine (HHG) and aphidicolin (APD) for 24 h. The drugs
were then removed, cells washed with HBSS and treated with a
combination of VPA (1.0 mM)//PR-39 (212 pM) for 72 h. Cell
proliferation was monitored by MTT assay. (B) The effect of the
combined treatment with VPA (1 mM)/MG-132 (0.2 μM), VPA (1 mM)/PI-1
(10 nM) and VPA (1 mM)/PR-39 (212 pM) on normal human fibroblasts
(CRL1554). Control cells were treated with vehicle (DMSO at a final
concentration of 0.2%). Cell growth was monitored by MTT assay. UT,
untreated cells; conc., concentration. |
 | Table IIC50 and sensitization
ratios of cytotoxic drugs and their combinations with the HDACI,
VPA, and proteasome inhibitors in the human colorectal cancer cell
line, SW1116. |
Table I
IC50 and sensitization
ratios of cytotoxic drugs and their combinations with the HDACI,
VPA, and proteasome inhibitors in the human colorectal cancer cell
line, SW1116.
| SW1116 |
|---|
|
|
|---|
| Combined treatment
with the HDACI, VPA, and proteasome inhibitors | IC50
(M)a | Sensitization
ratiob | P-valuec |
|---|
| CPT
(64×10−10-1×10−4 M) + VPA (1 mM)/MG132 (2
μM) |
104.4×10−9 | 28.6 | 0.003 |
| 5FU
(41.6×10−9-0.65×10−3 M) + VPA (1 mM)/MG132 (2
μM) |
19.4×10−6 | 41.2 | 0.001 |
| DOX
(55×10−11-0.86×10−5 M) + VPA (1 mM)/MG132 (2
μM) |
14×10−9 | 14.1 | 0.013 |
| TAX
(93.44×10−10-1.46×10−4 M) + VPA (1 mM)/MG132
(2 μM) |
23.4×10−8 | 15.6 | 0.007 |
| VCR
(16.0×10−11-0.25×10−5 M) + VPA (1 mM)/MG132
(2 μM) |
0.8×10−9 | 18.7 | 0.028 |
| ETP
(25.6×10−10-0.4×10−4 M) + VPA (1 mM)/MG132 (2
μM) |
0.7×10−7 | 140 | 0.0001 |
| ELP
(12.8×10−10-0.2×10−4 M) + VPA (1 mM)/MG132 (2
μM) |
0.16×10−6 | 12.5 | 0.002 |
| AMS
(80×10−11-1.25×10−5 M) + VPA (1 mM)/MG132 (2
μM) |
0.625×10−7 | 31.2 | 0.0001 |
| HHG
(12.8×10−11-0.2×10−5 M) + VPA (1 mM)/MG132 (2
μM) |
11.25×10−9 | 17.8 | 0.001 |
| CPT
(64×10−10-1×10−4 M) + VPA (1 mM)/PI-1 (10
nM) |
29×10−9 | 138 | 0.001 |
| 5FU
(41.6×10−9-0.65×10−3 M) + VPA (1 mM)/PI-1 (10
nM) |
52.5×10−8 | 185 | 0.0001 |
| DOX
(55×10−11-0.86×10−5 M) + VPA (1 mM)/PI-1 (10
nM) |
51.7×10−10 | 41.6 | 0.05 |
| TAX
(93.44×10−10-1.46×10−4 M) + VPA (1 mM)/PI-1
(10 nM) |
29.25×10−8 | 14.9 | 0.004 |
| VCR
(16.0×10−11-0.25×10−5 M) + VPA (1 mM)/PI-1
(10 nM) |
5.7×10−10 | 20 | 0.002 |
| ETP
(25.6×10−10-0.4×10−4 M) + VPA (1 mM)/PI-1 (10
nM) |
0.84×10−7 | 170 | 0.0001 |
| ELP
(12.8×10−10-0.2×10−4 M) + VPA (1 mM)/PI-1 (10
nM) |
0.6×10−6 | 4.2 | 0.002 |
| AMS
(80×10−11-1.25×10−5 M) + VPA (1 mM)/PI-1 (10
nM) |
0.25×10−6 | 8.8 | 0.0001 |
| HHG
(12.8×10−11-0.2×10−5 M) + VPA (1 mM)/PI-1 (10
nM) |
3.75×10−9 | 53.3 | 0.0001 |
| CPT
(64×10−10-1×10−4 M) + VPA (1 mM)/PR-39 (212
pM) |
43.75×10−8 | 6.8 | 0.366 |
| 5FU
(41.6×10−9-0.65×10−3 M) + VPA (1 mM)/PR-39
(212 pM) |
22.7×10−6 | 3.58 | 0.224 |
| DOX
(55×10−11-0.86×10−5 M) + VPA (1 mM)/PR-39
(212 pM) |
42.84×10−9 | 4 | 0.288 |
| TAX
(93.44×10−10-1.46×10−4 M) + VPA (1 mM)/PR-39
(212 pM) |
73.12×10−8 | 5 | 0.327 |
| VCR
(16.0×10−11-0.25×10−5 M) + VPA (1 mM)/PR-39
(212 pM) |
0.4×10−8 | 3.75 | 0.393 |
| ETP
(25.6×10−10-0.4×10−4 M) + VPA (1 mM)/PR-39
(212 pM) |
1.48×10−6 | 6.7 | 0.12 |
| ELP
(12.8×10−10-0.2×10−4 M) + VPA (1 mM)/PR-39
(212 pM) |
0.5×10−6 | 3 | 0.122 |
| AMS
(80×10−11-1.25×10−5 M) + VPA (1 mM)/PR-39
(212 pM) |
0.05×10−5 | 3.9 | 0.228 |
| HHG
(12.8×10−11-0.2×10−5 M) + VPA (1 mM)/PR-39
(212 pM) |
6×10−8 | 2.5 | 0.18 |
| Table IIIC50 and sensitization
ratios of cytotoxic drugs and their combinations with the HDACI,
VPA, and proteasome inhibitors in the human colorectal cancer cell
line, SW837. |
Table II
IC50 and sensitization
ratios of cytotoxic drugs and their combinations with the HDACI,
VPA, and proteasome inhibitors in the human colorectal cancer cell
line, SW837.
| SW837 |
|---|
|
|
|---|
| Combined treatment
with the HDACI, VPA, and proteasome inhibitors | IC50
(M)a | Sensitization
ratiob | P-valuec |
|---|
| CPT
(64×10−10-1×10−4 M) + VPA (1 mM)/MG132 (2
μM) |
108×10−9 | 46.3 | 0.0001 |
| 5FU
(41.6×10−9-0.65×10−3 M) + VPA (1 mM)/MG132 (2
μM) |
39.48×10−8 | 3.3×1020 | 0.0001 |
| DOX
(55×10−11-0.86×10−5 M) + VPA (1 mM)/MG132 (2
μM) |
171.8×10−11 | 3.76×102 | 0.0001 |
| TAX
(93.4×10−10-1.46×10−4 M) + VPA (1 mM)/MG132
(2 μM) |
292.5×10−10 | 75.2 | 0.0001 |
| VCR
(16×10−11-0.25×10−5 M) + VPA (1 mM)/MG132 (2
μM) |
0.5×10−8 | 15 | 0.0001 |
| ETP
(25.6×10−10-0.4×10−4 M) + VPA (1 mM)/MG132 (2
μM) |
5.25×10−8 | 1.88×102 | 0.0001 |
| ELP
(12.8×10−10-0.2×10−4 M) + VPA (1 mM)/MG132 (2
μM) |
0.8×10−5 | 1.25 | 0.0001 |
| AMS
(80×10−11-1.25×10−5 M) + VPA (1 mM)/MG132 (2
μM) |
0.625×10−5 | 104 | 0.0001 |
| HHG
(12.8×10−11-0.2×10−5 M) + VPA (1 mM)/MG132 (2
μM) |
3.75×10−8 | 40 | 0.0001 |
| CPT
(64×10−10-1×10−4 M) + VPA (1 mM)/PI-1 (10
nM) |
104.4×10−9 | 95.7 | 0.0001 |
| 5FU
(41.6×10−9-0.65×10−3 M) + VPA (1 mM)/PI-1 (10
nM) |
21×10−8 | 6.2×102 | 0.0001 |
| DOX
(55×10−11-0.86×10−5 M) + VPA (1 mM)/PI-1 (10
nM) |
27.5×10−10 | 3.12×102 | 0.0001 |
| TAX
(93.44×10−10-1.46×10−4 M) + VPA (1 mM)/PI-1
(10 nM) |
46.8×10−9 | 1.09×102 | 0.0001 |
| VCR
(16.0×10−11-0.25×10−5 M) + VPA (1 mM)/PI-1
(10 nM) |
0.8×10−9 | 21.8 | 0.01 |
| ETP
(25.6×10−10-0.4×10−4 M) + VPA (1 mM)/PI-1 (10
nM) |
0.17×10−5 | 5.8 | 0.002 |
| ELP
(12.8×10−10-0.2×10−4 M) + VPA (1 mM)/PI-1 (10
nM) |
0.4×10−7 | 187 | 0.0001 |
| AMS
(80×10−11-1.25×10−5 M) + VPA (1 mM)/PI-1 (10
nM) |
0.2×10−7 | 405 | 0.0001 |
| HHG
(12.8×10−11-0.2×10−5 M) + VPA (1 mM)/PI-1 (10
nM) |
3.75×10−9 | 93.3 | 0.0001 |
| CPT
(64×10−10-1×10−4 M) + VPA (1 mM)/PR-39 (212
pM) |
14×10−8 | 53.7 | 0.001 |
| 5FU
(41.6×10−9-0.65×10−3 M) + VPA (1 mM)/PR-39
(212 pM) |
32.8×10−7 | 39.6 | 0.0001 |
| DOX
(55×10−11-0.86×10−5 M) + VPA (1 mM)/PR-39
(212 pM) |
17.1×10−9 | 37.8 | 0.001 |
| TAX
(93.44×10−10-1.46×10−4 M) + VPA (1 mM)/PR-39
(212 pM) |
23.4×10−8 | 9.4 | 0.0001 |
| VCR
(16.0×10−11-0.25×10−5 M) + VPA (1 mM)/PR-39
(212 pM) |
1.5×10−9 | 13.3 | 0.041 |
| ETP
(25.6×10−10-0.4×10−4 M) + VPA (1 mM)/PR-39
(212 pM) |
2.52×10−6 | 35.7 | 0.0001 |
| ELP
(12.8×10−10-0.2×10−4 M) + VPA (1 mM)/PR-39
(212 pM) |
0.16×10−6 | 46.8 | 0.0001 |
| AMS
(80×10−11-1.25×10−5 M) + VPA (1 mM)/PR-39
(212 pM) |
0.05×10−5 | 16 | 0.0001 |
| HHG
(12.8×10−11-0.2×10−5 M) + VPA (1 mM)/PR-39
(212 pM) |
0.8×10−7 | 5 | 0.013 |
The combination of VPA and MG132 exhibited a potent
chemosensitizing effect on the SW837 colorectal cancer cells to CPT
(46-fold), 5FU (330-fold), DOX (376-fold), TAX (75-fold), ETP
(188-fold), AMS (104-fold) and HHG (40-fold). The same combination
of VPA and MG132 enhanced the chemosensitivity of the SW1116
colorectal cancer cells to CPT (29-fold), 5FU (41-fold), ETP
(140-fold) and AMS (31-fold). Moreover, the combination of VPA and
PI-1 markedly increased the chemosensitivity of the SW1116
colorectal cancer cells to CPT (138-fold), 5FU (185), DOX
(42-fold), ETP (170-fold) and HHG (53-fold). This combination
exerted a more profound chemosensitizing effect on the SW837
colorectal cancer cells to CPT (96-fold), 5FU (620-fold), DOX
(312-fold), TAX (109-fold), ELP (187-fold), AMS (405-fold) and HHG
(93-fold) compared to that observed in the SW1116 colorectal cancer
cells. Furthermore, the combination of VPA and PR-39 exhibited a
marked enhancement in the chemosensitivity of the SW837 colorectal
cancer cells to CPT (54-fold), 5FU (40-fold), DOX (38-fold), ETP
(36-fold), ELP (47-fold) and AMS (16-fold), compared to the SW1116
colorectal cancer cells (Tables I
and II).
Collectively, these results clearly indicate the
potential of the combined treatment with VPA and the PIs, MG132,
PI-1 and PR-39, to significantly enhance the chemosensitivity of
colorectal cancer cells to standard chemotherapeutic drugs. This
combined effect is exerted through various mechanisms of action in
a combination- and cancer subtype-dependent manner. The effect of
the combined treatment with VPA/MG132, VPA/PI-1 and VPA/EPM on
normal human fibroblast cells CRL1545 was also examined
microscopically by an MTT assay. The results presented in Fig. 10B clearly demonstrate that these
combinations have little effect (≤10–15%) on CRL 1554 cells,
indicating their minimal cytotoxicity.
Discussion
VPA, a branched short-chain fatty acid, is widely
used as an anti-epileptic drug and a mood stabilizer. VPA was
classified as an HDACI, acting at the level of gene transcription
by inhibiting deacetylation and making transcription sites more
accessible (18). Due to the
increasing number of clinical trials involving VPA, and the
interesting results obtained from such studies, we hypothesize that
this molecule will be involved in an increasing number of therapies
in the near future. Nevertheless, numerous publications related to
the therapeutic effects of VPA have also indicated the possible
side-effects associated with its use in treatment (19,20). Favoring the advantageous effects
of VPA use by balancing the therapeutic potential with serious
side-effects will no doubt prove to be a challenging and complex
issue in clinical management (20). The enhanced effectiveness of VPA
against cancer may be achieved by combining VPA treatment with
agents that have different modes of action and diverse targets,
such as PIs. Few studies have focused on the therapeutic potential
of VPA in human colorectal cancer (21). In the present study, we examined
the effectiveness of combining HDAC inhibition via VPA with
proteasome inhibition via MG132, PI-1 or PR-39 on colorectal cancer
cell lines, with regard to their potential synergistic effects on
cell proliferation and apoptosis. We also investigated the
synergistic molecular mechanisms of the combined treatment by
assessing the effects on pro-survival- and stress-related gene
expression, and the potential of the combined treatments to
sensitize the human colorectal cancer cells to standard
chemotherapeutic drugs.
In the present study, combined treatment with VPA
and the PIs, MG132, PI-1 or PR-39, markedly decreased the survival
of human colorectal cancer cells when compared to treatment with
VPA or any of the tested PIs alone (Fig. 1). MG132 (0.25 μM) markedly
enhanced the sensitivity of the SW1116 cells (SR 6.75) (Fig. 1A-a and d) and the SW837 cells
(IC80 3 mM for combined treatment vs.
IC50=7.5 mM for VPA alone) to VPA. PI-1 (7.8 nM)
moderately increased the sensitivity of the SW1116 cells (SR 1.5)
and SW837 cells (SR 1.3), and at 15.6 nM, PI-1 further enhanced the
sensitivity of the SW1116 cells (SR 3.3) and the SW837 cells (SR
2.1) to VPA (Fig. 1A-b and e).
Furthermore, PR-39 (106 pM) slightly increased the sensitivity of
the SW1116 cells (SR 1.33) and SW837 cells (SR 1.15) to VPA. PR-39
at a concentration of 212 pM moderately enhanced the sensitivity of
the SW1116 cells (SR 2.4) and the SW837 cells (SR 1.4) to VPA
(Figs. 1A-c and f). The results
of the experiments on the inhibitory effects were confirmed by the
observation of inhibition of colony formation (Fig. 1B) and morphological changes
(Fig. 1C). These results are in
agreement with those of previous studies using diverse malignant
cell types and various combinations of HDACIs and PIs (15,22).
Normal cell function is dependent on the proper
maintenance of chromatin structure. The regulation of chromatin
structure is controlled by histone modifications that directly
influence chromatin architecture and genome function. Specifically,
the HDAC families of proteins modulate chromatin compaction and are
commonly deregulated in many tumors, including colorectal cancer
(23). HDACIs interfere with
tumorigenic HDAC activity; however, the precise mechanisms involved
in this process remain to be elucidated (23). The anti-proliferative effects of
VPA on human colorectal cancer cells may be based on the direct
inhibition of HDACs by a direct influence on enzymes that are
responsible for acetylation and deacetylation of nucleosomal
histones. In the present study, the intracellular deacetylase
activity was significantly reduced (P≤0.0001) following treatment
with VPA (Fig. 2A). These results
are consistent with those reported in other malignant cell types
(21,24). By contrast, other investigators
have found that intracellular deacetylase activity was inhibited in
non-small cell lung cancer cell lines, which were resistant to
HDACI-mediated cell death (25).
The proteasome is a fundamental non-lysosomal tool
that cells use to process or degrade a variety of short-lived
proteins. Proteolysis mediated by the ubiquitin-proteasome system
has been implicated in the regulation of apoptosis (26). The proteasome pathway functions
upstream of mitochondrial alterations and caspase activation, and
is involved in different systems, including NF-κB, Bax and Bcl-2
(27). PIs, employed alone or in
combination with other anticancer agents, have been suggested as a
new class of potential anticancer agents (28). This increasing interest stems not
only from their direct apoptosis-inducing activity, but also from
the possibility of the sensitization of neoplastic cells to other
antitumor agents. In vitro data support the idea that PIs
increase the sensitivity of tumor cells to the apoptotic action of
tumor necrosis factor, radiotherapy, or chemotherapy (29). In the present study, the
proteasomal activity of human colorectal cancer cells was
significantly decreased following exposure to the PIs, MG132
(P≤0.001), PI-1 (P≤0.001) or PR-39 (P≤0.0001) (Fig. B). Our results
are in line with those reported by other studies using a number of
different cell lines (30,31).
VPA and other class I/II HDACIs, i.e.,
suberoylanilide hydroxamic acid (SAHA), MS-275 and sodium butyrate,
have been reported to elevate the cellular levels of ROS (32,33). Although ROS induce toxicity to
cells at high levels, at physiological levels, they function as
part of normal cell signaling. Since the induction of cell death
resulting from proteasome inhibition is linked to increased levels
of ROS in leukemia cells (34),
in this study, experiments were carried out to measure the capacity
of proteasomal inhibition to modulate intracellular ROS content and
the lethality of combined treatments with VPA and the PIs, MG132,
PI-1 or PR-39, in human colorectal cancer cells. Combined treatment
with VPA/MG132, VPA/PI-1 and VPA/PR-39 induced a significant
(P≤0.0001) increase in ROS levels compared to the untreated cells
and cells treated with VPA or one of the PIs alone (Fig. 2C). Co-administration of the
antioxidant, L-NAC, (15 mM) substantially blocked the
VPA/PI-mediated increase in ROS levels (data not shown). Our
results are consistent with those of other studies using different
combinations of HDACs and PIs in various cell systems (15,35). HDACIs have been shown to raise
intracellular ROS levels (36),
and our results indicated that the combination of VPA and PIs
induced a greater increase in ROS levels than that observed with
either agent alone. Thus, it is conceivable that this greater
oxidative challenge may contribute to the synergistic effects.
Importantly, the blockade of ROS using the scavenger, L-NAC,
substantially reduced cell death, suggesting that ROS generation
plays a major role in the cytotoxicity of combined VPA and PI
treatment.
Cancer cells produce higher levels of ROS than
normal cells due to the increased metabolic stress and
proliferative capacity. Thus, cancer cells may be more sensitive to
oxidative stress as a result of high endogenous ROS levels, and
this may provide a selective mechanism to induce cell death
(37).
Flow cytometric analysis indicated that in human
colorectal cancer cells, combined treatment with VPA/MG132,
VPA/PI-1 and VPA/PR-39 induced an arrest of the cell cycle at the S
phase. Our results are in agreement with those reported in another
study using the melanoma cell line, SK-Mel-37: the combination of
VPA and hydroxyurea (HU) increased the number of cells in the S
phase (38). The results from
flow cytometry reported in that study are contradictory to those
reported by other groups using several different cell systems and
various combinations of HDACIs and PIs (35,39).
Cell proliferation and cell death induction are two
major targets in cancer therapy. There are two fundamental types of
cell death, apoptosis and necrosis, which can be defined by
morphological and biochemical criteria. Over the past several
years, the idea that cells can commit suicide by mechanisms other
than apoptosis has gained momentum (40). Apoptosis is marked by cellular
shrinking, condensation of chromatin and the loss of plasma
membrane integrity, resulting in the breaking up of the cell into
apoptotic bodies. Whereas apoptosis is an inherent, programmed
cellular death mechanism, its conceptual counterpart, necrosis, is
a more uncontrolled form of death and is characterized by cellular
swelling and disruption of the plasma membrane, leading to the
release of the cellular components and inflammatory tissue response
(41).
HDACIs are known to induce programmed cell death in
cancer cells, although the underlying mechanisms are far from being
understood and seem to be very heterogeneous. Many HDACI-mediated
death mechanisms have already been described, including the
induction of a senescence-like phenotype (42) and autophagic cell death (43). This diversity of cell death
mechanisms has also been observed for VPA (44). In the present study, the
co-administration of VPA with PI, MG132, PI-1 or PR-39 induced a
marked increase in apoptosis in human colorectal cancer cells. Our
results are consistent with those reported by other groups using
several different cell systems and various combinations of HDACIs
and PIs (1,15,22,35,39).
Mitochondria play a decisive role in apoptosis
(45), functioning as integrators
of different pro-apoptotic signaling pathways. In response to these
signals, mitochondria activate megachannels (also known as
permeability transition pores) that are present between the inner
and outer mitochondrial membrane. Bcl-2, a fundamental death
antagonist protein, inhibits megachannel opening, whereas, Bax a
death agonist protein, facilitates the opening of megachannels
(46). The increase in the
content of agonists and the concomitant decrease in the content of
antagonists stimulate the release of cytochrome c from the
mitochondria, with the consequent activation of caspases. In this
study, we examined whether VPA, PI-1 and their combination are
capable of inducing changes in the levels of Bax and cytochrome
c in both the mitochondria and cytosol. Our results
indicated that after 24 h of co-exposure to VPA and PI-1,
cytochrome c was released from the mitochondria. Such an
event may be responsible for the production of the apoptosome, with
the consequent activation of caspase-9, which in turn activates
caspase-3 (45). Our results
support the hypothesis that the release of cytochrome c is
induced in VPA/PI-1-treated colorectal cancer cells by the decrease
in the level of the anti-apoptotic factor, Bcl-2, and the
concomitant increase in the level of the pro-apoptotic factor,
Bax.
The NF-κB pathway is one of the most important
signaling cascades that suppresses apoptosis, enhances
chemoresistance and promotes tumorigenesis by increasing the
expression of cellular survival signals (47). Bortezomib prevents NF-κB nuclear
translocation and subsequent transactivation, which may lower the
threshold for HDACI lethality (48). Bortezomib has been shown to
sensitize B-lymphoma cells to HDACI-mediated apoptosis through the
inhibition of the NF-κB pathway (31). In the present study, we examined
the change in NF-κB distribution in the human SW837 colorectal
cancer cell line to determine whether NFκB is involved in the
synergistic apoptotic effects of combined treatment with VPA and
PI-1. Our results demonstrated an elevated level of NF-κB in the
cytoplasm of colorectal cancer cells, and a concomitant marked
decrease in the amount of nuclear NF-κB following co-treatment with
VPA and PI-1. These results are in line with those reported by
others (1,25,31). Our results demonstrated that
co-treatment with VPA/PI-1 affects the nuclear translocation of
NF-κB in colorectal cancer cells, suggesting that the dysfunction
of NF-κB may be involved in the synergistic apoptotic effects of
VPA and PIs.
It has been reported that HDACIs suppress NF-κB in
part by suppressing the cellular proteasome activity. In a study
using colon cancer cells, Place et al (49) found that HDACI reduced cellular
proteasome activity by reducing the expression of the catalytic
β-type subunits of the proteasome. HDAC inhibition was found to
specifically downregulate the expression of the three catalytic
β-subunits, β5, β1 and β2, which are responsible for all the
endopeptidase activities associated with the proteasome. The
reduced levels in these catalytic subunits results in the
intracellular suppression of proteasome activity (49). By downregulating proteasome
activity, HDACIs stabilize IκBα. Under normal conditions, IκBα is
phosphorylated, ubiquitinated and degraded by the proteasome,
releasing NF-κB.
Multiple potential molecular mechanisms may
contribute to the synergy between VPA and MG132, PI-1 or PR-39, and
add to the effects of proteasome and HDAC inhibition.
Cyto-protective pathways, such as NF-κB, ERK and AKT, confer a
survival advantage on colorectal cancer cells, not only by
protecting the cells from apoptosis, but also by rendering them
resistant to conventional cytotoxic drugs.
Interruption of the Raf-1/MEK/ERK pathway precedes
pro-apoptotic signaling (50).
Specific blockade of this pathway by bortezomib synergistically
potentiates the HDCI-induced apoptosis of myeloid leukemia cells
(51). In the present study,
combined treatment with PI, PI-1 and HDACI, VPA markedly
downregulated the levels of the phosphorylated form of ERK. These
findings further establish the functional complementation of both
agents in the inhibition of the ERK pathway. Our results are in
line with those reported by others (52). Our results also demonstrated that
the co-administration of VPA and PI-1 markedly attenuated the
levels of the phosphorylated form of AKT, indicating an additional
role in AKT signaling, which is to further down-modulate the
apoptotic threshold. Cross-talk between the ERK and AKT cascades
has been described (53) and
thus, in human colorectal cancer cells, the combined downregulation
of ERK and AKT may be considerably more lethal than the
interruption of either pathway alone. Our results are in agreement
with those reported by other groups using different cell lines and
various combinations of HDACIs and PIs (52,54).
Similar to ERK, JNK is also a member of the MAPK
pathways and is implicated in the control of cellular survival and
stress responses. However, differences do exist; ERK activation is
generally associated with enhanced cell survival, whereas JNK is
related to cell death. The bortezomib-mediated induction of
apoptosis in leukemia cells has been linked to JNK activation
(55). The upregulation of
stress-related cascades is also one of the important factors
involved in SAHA-mediated lethality (56). In the present study, when VPA was
combined with the PI, PI-1, a marked increase in JNK expression was
observed. Our results are in line with those reported in other
groups using different cell line systems and various combinations
of HDAC and PIs (52,55,56).
The mechanisms by which HDAC and PIs exert their
cytotoxic effects have not been fully delineated and are likely to
be multifactorial and cell line-specific. Numerous genes are
affected by PIs or VPA, and it is therefore likely that a
combination of these effects may lead to the synergy observed
between the two classes of agents. As cyclin-dependent kinases
(CDKs) and CDKIs are critical cell regulators, we investigated
whether their expression is affected by treatment with VPA/PIs. Our
results revealed that CDK1, CDK4 and CDC25A were downregulated. By
contrast, p15, p19, p21 and p27 were differentially upregulated
following co-treatment with VPA and PI-1. These results are in
agreement with those reported in previous studies (24,38).
Despite the relatively uniform synergistic
induction of apoptosis by HDACIs and PIs, the molecular pathways
involved in the response to the combination treatment are not yet
fully understood and may vary between different malignancies
(5,52). The synergistic induction of
apoptosis in our in vitro model of human colorectal cancer
was associated with the upregulation of the expression of
pro-apoptotic genes, including Bax, Bad, Bim, Apaf-1 and
AIF, and caspase-3, -8 and -9, as well with as the
downregulation of the anti-apoptotic genes, including Bcl-2, XIAP
and FLIP. These results are in agreement with those reported on
other malignancies using various HDACIs and PIs (24,57,58).
Cancer patients often develop chemoresistance, and
increasing the concentration of cytotoxic drugs fails to
significantly improve the therapeutic response. One hypothesis to
explain the development of chemoresistance is an acquired
resistance of the tumor cells to apoptosis (59), allowing tumors to withstand high
levels of chemotherapy. Tumor cells that are resistant to apoptosis
also exhibit an increased proliferative capacity.
Synergism has been observed when HDACIs are
combined with docetaxel, epothilone B and gemcitabine in breast
cancer (60), combined with 5FU
in colon cancer (61), combined
with etoposide, camptothecin, doxorubicin and cisplatin in
glioblastoma (62), combined with
topoisomerase II inhibitors, cytarabine and fludarabine in leukemia
(63) and combined with
cytarabine, etoposide and topotecan in non-Hodgkin’s lymphoma,
chronic lymphocytic leukemia (CCL) and multiple myeloma cells
(64).
In this study, combined treatment with VPA and the
PIs, MG132, PI-1 or PR-39, markedly decreased the apoptotic
threshold of colorectal cancer cells. These results led us to
examine the efficacy of the combined treatments of VPA and PIs to
enhance the sensitivity of human colorectal cancer cells to
conventional chemotherapeutic drugs of various modes of action. Our
results demonstrated that the various combinations of VPA and
MG132, PI-1 or PR-39 differentially increased the sensitivity of
colorectal cancer cells to conventional chemotherapeutic agents in
a VPA/PI combined- and colorectal cancer subtype-dependent manner
(Figs. 6–10 and Tables I and II). These results are consistent with
findings in other malignancies (59,65,66).
The net balance between cytoprotective pathways and
stress-related cascades is known to be a critical determinant of
cell death. VPA may interact with PIs by disrupting multiple
cytoprotective signaling pathways and by shifting the balance
toward their stress-related counterparts, which trigger
mitochondrial damage, caspase activation and eventually lead to
irreversible engagement of the apoptotic cascade in human
colorectal cancer cells.
Clinical trials have been conducted to determine
the safety, toxicity and maximum dose of VPA alone or in
combination with other drugs in solid tumor malignancies and to
define its clinical feasibility (67). Existing preclinical and
preliminary clinical data strongly suggest that VPA is a drug with
potential to eventually be used in combination therapies, either
with classical cytotoxic or other molecular-targeted drugs or
radiation (68). The issue of
drug costs cannot be underestimated. A contemporaneous and highly
discussed topic in cancer therapy is drug access for low and middle
income populations (69); thus,
it appears obvious that if the efficacy of VPA is confirmed in
cancer therapy, it will be widely available.
The findings reported in the present study have
important clinical implications. Molecular targeting of HDAC and
the proteasome in combination synergistically induces apoptosis and
may exert a therapeutic effect at lower concentrations of each
drug, thus reducing the toxicity of the therapeutic treatment. This
combination strategy provides a promising rationale for its use in
the treatment of colorectal cancer and other malignancies.
Acknowledgements
This study was supported by Kuwait University,
Research Grant no. SL05/11. Western blot scanning was carried out
by the Research Core Facility (RCF), AbdulMohsen AbdulRazzaq Health
Sciences Centre (HSC), Kuwait University (KU).
References
|
1
|
Pitts TM, Morrow M, Kaufman SA, et al:
Vorinostat and bortezomib exert synergistic antiproliferative and
proapoptotic effects in colon cancer cell models. Mol Cancer Ther.
8:342–349. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Göttlicher M, Minucci S, Zhu P, et al:
Valproic acid derivatives: A novel class of HDAC inhibitors
inducing differentiation of transformed cells. EMBO J.
20:6969–6978. 2001.
|
|
3
|
Tan J, Cang S, Ma Y, et al: Novel histone
deacetylase inhibitors in clinical trials as anti-cancer agents. J
Hematol Oncol. 3:52010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Nolan L, Johnson PWM, Ganesan A, et al:
Will histone deacetylase inhibitors require combination with other
agents to fulfil their therapeutic potential? Br J Cancer.
99:689–694. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Dai Y, Chen S, Wang L, et al: Bortezomib
interacts synergistically with belinostat in human acute myeloid
leukaemia and acute lymphoblastic leukaemia cells in association
with perturbations in NF-κB and Bim. Br J Haematol. 153:222–235.
2011.PubMed/NCBI
|
|
6
|
Adams J: The proteasome: a suitable
antineoplastic target. Nat Rev Cancer. 4:349–360. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Orlowski RZ and Kuhn DJ: Proteasome
inhibitors in cancer therapy: lessons from the first decade. Clin
Cancer Res. 14:1649–1657. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bai J, Demirjian A, Sui J, et al: Histone
deacetylase inhibitor trichostatin A and proteasome inhibitor
PS-341 synergistically induce apoptosis in pancreatic cells.
Biochem Biophys Res Commun. 4:1245–1253. 2006. View Article : Google Scholar
|
|
9
|
Pei XY, Dai Y and Grant S: Synergistic
induction of oxidative injury and apoptosis in human multiple
myeloma cells by the proteasome inhibitor bortezomib and histone
deacetylase inhibitors. Clin Cancer Res. 11:3839–3852. 2004.
View Article : Google Scholar
|
|
10
|
Emanuele S, Lauricella M, Carlisi D, et
al: SAHA induces apoptosis in hepatoma cells and synergistically
interact with proteasome inhibitor Bortezomib. Apoptosis.
12:1327–1338. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Abaza MS, Al-Safar A, Al-Sawan S, et al:
c-myc anti-sense oligonucleotides sensitize human colorectal cancer
cells to chemotherapeutic drugs. Tumour Biol. 29:287–303. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Abaza MSI: Augmentation of the anticancer
effects of proteasome inhibitors by combination with sodium
butyrate in human colorectal cancer cells. Exp Ther Med. 1:675–693.
2010.
|
|
13
|
Bradford MM: A rapid and sensitive method
for the quantitation of microgram quantities of protein utilizing
the principle of protein-dye binding. Anal Biochem. 72:248–254.
1976. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kwon SH, Ahn SH, Kim YK, et al: Apicidin,
a histone deacetylase inhibitor, induces apoptosis and Fas/Fas
ligand expression in human acute promyelocytic leukemia cells. J
Biol Chem. 277:2073–2080. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Premkumar DR, Jane EP, Agostino NR, et al:
Bortezomib-induced sensitization of malignant human glioma cells to
vorinostat-induced apoptosis depends on reactive oxygen species
production, mitochondrial dysfunction, noxa upregulation, Mcl-1
cleavage, and DNA damage. Mol Carcinog. 52:118–133. 2013.
View Article : Google Scholar
|
|
16
|
Murphy KM, Streips UN and Lock RB: Bcl-2
inhibits a Fas-induced conformational change in the Bax N terminus
and Bax mitochondrial translocation. J Biol Chem. 275:17225–17228.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Knupfer MM, Pulzer F, Schindler I, et al:
Different effects of valproic acid on proliferation and migration
of malignant glioma cells in vitro. Anticancer Res. 21:347–351.
2001.PubMed/NCBI
|
|
18
|
Barbetti V, Gozzini A, Cheloni G, et al:
Time- and residue-specific differences in histone acetylation
induced by VPA and SAHA in AML1/ETO-positive leukemia cells.
Epigenetics. 8:210–219. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sztanjnkrycer MD: Valproic acid toxicity:
overview and management. J Toxicol Clin Toxicol. 40:789–801. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chateauvieux S, Morceau F, Dicato M and
Diederich M: Molecular and therapeutic potential and toxicity of
valproic acid. J Biomed Biotechnol. 2010:4793642010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kostrouchová M, Kostrouch Z and
Kostrouchová M: Valproic acid, a molecular lead to multiple
regulatory pathways. Folia Biol (Praha). 53:37–49. 2007.PubMed/NCBI
|
|
22
|
Bastian L, Hof J, Pfau M, et al:
Synergistic activity of bortezomib and HDACi in preclinical models
of B-cell precursor acute lymphoblastic leukemia via modulation of
p53, PI3K/AKT, and NF-κB. Clin Cancer Res. 19:1445–1457.
2013.PubMed/NCBI
|
|
23
|
Stypula-Cyrus Y, Damania D, Kunte DP, et
al: HDAC up-regulation in early colon field carcinogenesis is
involved in cell tumorigenicity through regulation of chromatin
structure. PLoS One. 8:e646002013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sidana A, Wang M, Shabbeer S, et al:
Mechanism of growth inhibition of prostate cancer xenografts by
valproic acid. J Biomed Biotech. 2012:1803632012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Denlinger C, Keller M, Mayo M, et al:
Combined proteasome and histone deacetylase inhibition in non-small
cell lung cancer. J Thorac Cardiovasc Surg. 127:1078–1086. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Drexler HC, Risau W and Konerding MA:
Inhibition of proteasome function induces programmed cell death in
proliferating endothelial cells. FASEB J. 14:65–77. 2000.
|
|
27
|
Li B and Dou QP: Bax degradation by the
ubiquitin/proteasome-dependent pathway: involvement in tumor
survival and progression. Proc Natl Acad Sci. 97:3850–3855. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Adams J, Palombella VJ, Sausville EA, et
al: Proteasome inhibitors: a novel class of potent and effective
antitumor agents. Cancer Res. 59:2615–2622. 1999.PubMed/NCBI
|
|
29
|
Chandra J, Niemer I, Gilbreath J, et al:
Proteasome inhibitors induce apoptosis in glucocorticoid-resistant
chronic lymphocytic leukemic lymphocytes. Blood. 92:4220–4229.
1998.PubMed/NCBI
|
|
30
|
Miller CP, Ban K, Dujka ME, et al:
NPI-0052, a novel proteasome inhibitor, induces caspase-8 and
ROS-dependent apoptosis alone and in combination with HDAC
inhibitors in leukemia cells. Blood. 110:267–277. 2007. View Article : Google Scholar
|
|
31
|
Heider U, Metzler I, Kaiser M, et al:
Synergistic interaction of the histone deacetylase inhibitor SAHA
with the proteasome inhibitor bortezomib in mantle cell lymphoma.
Eur J Haematol. 80:133–142. 2008. View Article : Google Scholar
|
|
32
|
Defoort EN, Kim PM and Winn LM: Valproic
acid increases conservative homologous recombination frequency and
reactive oxygen species formation: a potential mechanism for
valproic acid-induced neural tube defects. Mol Pharmacol.
69:1304–1310. 2006. View Article : Google Scholar
|
|
33
|
Rahmani M, Reese E, Dai Y, et al:
Coadministration of histone deacetylase inhibitors and perifosine
synergistically induces apoptosis in human leukemia cells through
Akt and ERK1/2 inactivation and the generation of ceramide and
reactive oxygen species. Cancer Res. 65:2422–2432. 2005. View Article : Google Scholar
|
|
34
|
Dolado I, Swat A, Ajenjo N, et al:
p38alpha MAP kinase as a sensor of reactive oxygen species in
tumorigenesis. Cancer Cell. 11:191–205. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dasmahapatra G, Lembersky D, Son MP, et
al: Carfilzomib interact synergistically with histone deacetylase
inhibitors in mantle cell lymphoma cells in vitro and in
vivo. Mol Cancer Ther. 10:1686–1697. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ungerstedt JS, Sowa Y, Xu WS, et al: Role
of thioredoxin in the response of normal and transformed cells to
histone deacetylase inhibitors. Proc Natl Acad Sci USA.
102:673–678. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Chen Y, McMillan-Ward E, Kong J, et al:
Oxidative stress induces autophagic cell death independent of
apoptosis in transformed and cancer cells. Cell Death Differ.
15:171–182. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Krämer OH, Knauer SK, Zimmermann D, et al:
Histone deacetylase inhibitors and hydroxyurea modulate the cell
cycle and cooperatively induce apoptosis. Oncogene. 27:732–740.
2008.PubMed/NCBI
|
|
39
|
Nile D, Huang K, Yin S, et al:
Synergistic/additive interaction of valproic acid with bortezomib
on proliferation and apoptosis of acute myeloid leukemia cells.
Leuk Lymphoma. 53:2487–2495. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Proskuryakov SY, Konoplyannikov AG and
Gabai VL: Necrosis: a specific form of programmed cell death? Exp
Cell Res. 283:1–16. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Hirsch T, Marchetti P, Susin SA, et al:
The apoptosis-necrosis paradox. Apoptogenic proteases activated
after mitochondrial permeability transition determine the mode of
cell death. Oncogene. 15:1573–1581. 1997. View Article : Google Scholar
|
|
42
|
Place RF, Noonan EJ and Giardina C: HDACs
and the senescent phenotype of WI-38 cells. BMC Cell Biol.
6:372005. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Shao Y, Gao Z, Marks PA and Jiang X:
Apoptotic and autophagic cell death induced by histone deacetylase
inhibitors. Proc Natl Acad Sci USA. 101:18030–18035. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Li XN, Shu Q, Su JM, et al: Valproic acid
induces growth arrest, apoptosis, and senescence in medulloblastoma
by increasing histone hyperacetylation and regulating expression of
p21Cip1, CDK4, and CMYC. Mol Cancer Ther. 4:1912–1922. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science (Washington DC). 281:1309–1312. 1998. View Article : Google Scholar
|
|
46
|
Schendel SL, Montal M and Reed JC: Bcl-2
family proteins as ion-channels. Cell Death Differ. 5:372–380.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Bharti AC and Aggarwal BB: Nuclear
factor-kappa B and cancer: its role in prevention and therapy.
Biochem Pharmacol. 64:883–888. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Nalepa G and Wade Harper J: Therapeutic
anti-cancer targets upstream of the proteasome. Cancer Treat Rev.
29(Suppl 1): S49–S57. 2003. View Article : Google Scholar
|
|
49
|
Place RF, Noonan EJ and Giardina C: HDAC
inhibition prevents NF-kappa B activation by suppressing proteasome
activity: Down-regulation of proteasome subunit expression
stabilizes I kappa B alpha. Biochem Pharmacol. 70:394–406. 2005.
View Article : Google Scholar
|
|
50
|
Lewis TS, Shapiro PS and Ahn NG: Signal
transduction through MAP kinase cascades. Adv Cancer Res.
74:49–139. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Nishioka C, Ikezoe T, Yang J, et al:
Inhibition of MEK/ERK signaling synergistically potentiates histone
deacetylase inhibitor-induced growth arrest, apoptosis and
acetylation of histone H3 on p21waf1 promoter in acute myelogenous
leukemia cell. Leukemia. 22:1449–1452. 2008. View Article : Google Scholar
|
|
52
|
Zhang QL, Wang L, Zhang YW, et al: The
proteasome inhibitor bortezomib interacts synergistically with the
histone deacetylase inhibitor suberoylanilide hydroxamic acid to
induce T-leukemia/lymphoma cells apoptosis. Leukemia. 23:1507–1514.
2009. View Article : Google Scholar
|
|
53
|
Shelton JG, Blalock WL, White ER, et al:
Ability of the activated PI3K/Akt oncoproteins to synergize with
MEK1 and induce cell cycle progression and abrogate the
cytokine-dependence of hematopoietic cells. Cell Cycle. 3:503–512.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Kawamata N, Chen J and Koeffler HP:
Suberoylanilide hydroxamic acid (SAHA; vorinostat) suppresses
translation of cyclin D1 in mantle cell lymphoma cells. Blood.
110:2667–2673. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yu C, Rahmani M, Dent P and Grant S: The
hierarchical relationship between MAPK signaling and ROS generation
in human leukemia cells undergoing apoptosis in response to the
proteasome inhibitor Bortezomib. Exp Cell Res. 295:555–566. 2004.
View Article : Google Scholar
|
|
56
|
Yu C, Subler M, Rahmani M, et al:
Induction of apoptosis in BCR/ABL+ cells by histone deacetylase
inhibitors involves reciprocal effects on the RAF/MEK/ERK and JNK
pathways. Cancer Biol Ther. 2:544–551. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Yagi Y, Fushida S, Harada S, et al:
Effects of valproic acid on the cell cycle and apoptosis through
acetylation of histone and tubulin in a scirrhous gastric cancer
cell line. J Exp Clin Cancer Res. 29:1492010. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Gatti L, Benedetti V, De Cesare M, et al:
Synergistic interaction between the novel histone deacetylase
inhibitor ST2782 and the proteasome inhibitor bortezomib in
platinum-sensitive and resistant ovarian carcinoma cells. J Inorg
Biochem. 113:94–101. 2012. View Article : Google Scholar
|
|
59
|
Hwang JJ, Kim Ys, Kim T, et al: A novel
histone deacetylase inhibitor, CG200745, potentiates anticancer
effect of docetaxel in prostate cancer via decreasing Mcl-1 and
Bcl-XL. Invest New Drugs. 30:1434–1442. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Fuino L, Bali P, Wittmann S, et al:
Histone deacetylase inhibitor LAQ824 down-regulates Her-2 and
sensitizes human breast cancer cells to trastuzumab, taxotere,
gemcitabine, and epothilone B. Mol Cancer Ther. 2:971–984.
2003.
|
|
61
|
Huang Y and Waxman S: Enhanced growth
inhibition and differentiation of fluorodeoxyuridine-treated human
colon carcinoma cells by phenylbutyrate. Clin Cancer Res.
4:2503–2509. 1998.
|
|
62
|
Kim MS, Blake M, Baek JH, et al:
Inhibition of histone deacetylase increases cytotoxicity to
anticancer drugs targeting DNA. Cancer Res. 63:7291–7300.
2003.PubMed/NCBI
|
|
63
|
Maggio SC, Rosato RR, Kramer LB, et al:
The histone deacetylase inhibitor MS-275 interacts synergistically
with fludarabine to induce apoptosis in human leukemia cells.
Cancer Res. 64:2590–2600. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Witzig TE, Timm M, Stenson M, et al:
Induction of apoptosis in malignant B cells by phenylbutyrate or
phenylacetate in combination with chemotherapeutic agents. Clin
Cancer Res. 6:681–692. 2000.PubMed/NCBI
|
|
65
|
Linares A, Dalenc F, Balaguer P, et al:
Manipulating protein acetylation in breast cancer: a promising
approach in combination with hormonal therapies. J Biomed
Biotechnol. 2011:8569852011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Munster PN, Thurn KT, Thomas S, et al: A
phase II study of the histone deacetylase inhibitor vorinostat
combined with tamoxifen for the treatment of patients with hormone
therapy-resistant breast cancer. Br J Cancer. 104:1828–1835. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Atmaca A, Al-Batran SE, Maurer A, et al:
Valproic acid (VPA) in patients with refractory advanced cancer: a
dose escalating phase I clinical trial. Br J Cancer. 97:177–182.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Duenas-Gonzalez A, Candelaria M,
Perez-Plascencia C, et al: Valproic acid as epigenetic cancer drug:
Preclinical, clinical and transcriptional effects on solid tumors.
Cancer Treat Rev. 34:206–222. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Newland SE: The role of bioethics in
international prescription drug market: economics and global
justice. Penn Bioeth J. 2:8–12. 2006.PubMed/NCBI
|