Introduction
Multiple myeloma (MM) is a type of cancer that
arises from the neoplastic proliferation of plasma cells and is
characterized by the proliferation of malignant plasma cells into
the bone marrow (BM) and the excessive secretion of monoclonal
immunoglobulin or Bence Jones proteins. The major clinical
manifestations include extensive bone destruction, anemia,
hypercalcemia, hyperviscosity and renal dysfunction (1). Of note, the incidence of MM in
Western countries is higher than that in Asian countries (2). Despite advances in the understanding
of the molecular pathogenesis of MM and promising new therapies,
only 25–35% of patients respond to therapy in the relapsed and
refractory setting (3,4).
The boronic dipeptide, bortezomib, a reversible
proteasome inhibitor, has shown marked anticancer activity in
various cancer cell types, including MM cells, which are resistant
to conventional therapies (5,6).
Despite the promising clinical activity, the efficacy of bortezomib
may differ among tumor types, and some patients with MM fail to
respond to bortezomib therapy (7). Moreover, almost one-third of
patients with MM never respond to treatment with bortezomib,
depending on the clinical situation (7). Whether these observations are
related to the common mechanisms of drug resistance frequently
observed for anticancer drugs remains unclear. However, the
understanding of the characteristics of drug resistance and the
enhancement of the sensitivity to bortezomib may help to overcome
drug resistance. In addition, recent evidence has demonstrated that
silencing the expression of oncogenes, such as myeloid cell
leukemia sequence-1 (Mcl-1) and melanoma antigen gene
(MAGE)-C1/CT7, can sensitize MM cells to bortezomib (8,9),
suggesting that RNA interference (RNAi) may be an effective method
for enhancing the sensitivity of MM cells to bortezomib or even
reversing resistance.
B-cell-specific Moloney murine leukemia virus
insertion site-1 (Bmi-1), a member of the polycomb family, was
initially identified as an oncogene that cooperates with c-Myc in
the initiation of lymphoma in murine models (10,11). Over the years, Bmi-1 has been
reported to be involved in axial patterning (12), hematopoiesis (13), the regulation of proliferation and
senescence (14). It has also
been found to be essential for the self-renewal of normal and
malignant stem cells (15).
Several lines of evidence have also indicated that Bmi-1 is
extensively upregulated in a variety of malignancies, including
non-small cell lung cancer (16),
leukemia (17), as well as breast
(18), colorectal (19), pancreatic (20) and prostate cancer (21). Moreover, it has been demonstrated
that Bmi-1 is overexpressed in MM and can regulate the growth and
clonogenic capacity of MM cells both in vitro and in
vivo (22). However, whether
Bmi-1 can affect the sensitivity of MM cells to bortezomib is
largely unknown. Furthermore, it has been reported that the
downregulation of Bmi-1 can result in cancer cell apoptosis and
that the RNAi-mediated depletion of Bmi-1 can sensitize tumor cells
to chemotherapy and radiotherapy (23–25). Thus, we hypothesized that the
abrogation of Bmi-1 expression may be an effective strategy for
sensitizing MM cells to bortezomib.
In this study, we demonstrate that Bmi-1 plays an
important role in the sensitization of MM cells to bortezomib. To
determine the combined effect of RNAi and bortezomib treatment on
MM cells in vitro, we introduced a lentiviral interference
vector expressing short hairpin RNA (shRNA) to silence Bmi-1 in MM
cells. Our results demonstrate that Bmi-1 silencing sensitizes MM
cells to bortezomib by inhibiting cell proliferation and inducing
cell cycle arrest and apoptosis, suggesting that Bmi-1 may serve as
a potential and specific novel therapeutic target in MM.
Materials and methods
Cell culture
HEK-293T cells were cultured in Dulbecco’s modified
Eagle’s medium containing 10% fetal bovine serum. U266 and RPMI8226
cells were cultured in RPMI-1640 containing 10% fetal bovine serum,
50 U/ml penicillin and 50 μg/ml streptomycin.
Lentivirus production and infection
Bmi-1 shRNA was designed and cloned into the pLVTHM
lentiviral vector. The shRNA sequence for Bmi-1 was as follows:
5′-GAGATAATAA GCTTGTCTA-3′ (26).
A shRNA targeting a scrambled sequence (general sequence,
5′-TTCTCCGAACGTGTCACGT-3′) (27)
served as the negative control. The clone identity was verified by
restriction digestion analysis and plasmid DNA sequencing. The
pWPXL vectors were transfected into the HEK-293T cells with the
packaging plasmid, psPAX2, and the VSV-G envelope plasmid, pMD2.G
(a gift from Dr Didier Trono, School of Life Sciences, Ecole
Polytechnique Fédérale de Lausanne, Lausanne, Switzerland), using
Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). The viral
particles were harvested 48 h following transfection. The U266 and
RPMI8226 cells (1×105) were infected with
1×106 recombinant lentivirus-transducing units plus 6
μg/ml of polybrene (Sigma, Natick, MA, USA).
RNA extraction and quantitative reverse
transcription polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the U266 and RPMI8226
cells using TRIzol reagent (Invitrogen) and reverse transcribed to
generate cDNA. cDNA was synthesized using the PrimeScript RT
Reagent kit (Takara, Tokyo, Japan). qPCR analyses were performed
using SYBR Premix Ex Taq (Takara). The primers used for the PCR
amplification were as follows: Bmi-1 forward, 5′-AAATCA
GGGGGTTGAAAAAT CT-3′ and reverse, 5′-GCTAACCA CCAATCTTCCTTTG-3′;
p14 forward, 5′-GCTACTGAGGAG CCAGCGTCTA-3′ and reverse,
5′-AGCACCACCAGCGTGT CCAG-3′; and β-actin forward,
5′-GCTACTGAGGAGCCAG CGTCTA-3′ and reverse,
5′-AGCACCACCAGCGTGTCCAG-3′.
Western blot analysis
The proteins were separated on a 12% SDS-PAGE gels
and then transferred onto a nitrocellulose membrane (Bio-Rad,
Hercules, CA, USA). The membrane was blocked using 5% non-fat milk
and incubated with a mouse anti-Bmi-1, anti-p21 or anti-Bcl-2
monoclonal antibody (mAb) (Cell Signaling Technology, Inc.,
Beverly, MA, USA) or a mouse anti-β-actin mAb (Sigma). The proteins
were visualized and quantified using ECL reagents (Pierce
Biotechnology, Inc., Rockford, IL, USA).
Cell proliferation
Cell proliferation was measured using the cell
counting kit-8 (CCK-8) assay kit (Dojindo Corp., Kumamoto, Japan).
A total of 4,000 cells were plated into each well of a 96-well
plate, wherein 10 μl of the CCK-8 solution was added to 90 μl of
the culture medium. The cells were subsequently incubated for 2 h
at 37°C, and the optical density was measured at 450 and 650 nm.
Three independent experiments were performed.
FACS analysis for determination of cell
cycle progression and apoptosis
The U266 and RPMI8226 cells were collected and fixed
in ice-cold 70% ethanol overnight. The fixed cells were washed with
phosphate-buffered saline and stained with a freshly prepared
solution containing 25 μg/ml propidium iodide (PI; Sigma), 10 μg/ml
RNase A, 0.05 mM ethylene diamine and 0.2% Triton X-100
tetra-acetic acid in phosphate-buffered saline for 30 min in the
absence of light. For each sample, at least 20,000 cells were
analyzed using a flow cytometer (Beckman Coulter EPICS Altra Cell
Sorter; Beckman Coulter Inc., Miami, FL, USA) and Multicycle AV
software for Windows 5.0 (Phoenix Flow Systems, San Diego, CA,
USA).
Cells (2×105) from the control group and
the interference group were collected, stained according to the
instructions provided with the Annexin V-PE/7-AAD Apoptosis
Detection kit (Sigma), and subjected to flow cytometry (FCM) using
a FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA)
for the detection of apoptosis.
Statistical analysis
The results in this study are presented as the means
± SEM. The data were analyzed using the Student’s t-test
(two-tailed) and a value of p<0.05 was considered to indicate a
statistically significant difference, unless otherwise
specified.
Results
Establishment of cell lines stably
transfected with shRNA targeting Bmi-1 (shBmi-1)
To determine the biological role of Bmi-1 in the
survival of MM cells, two cell lines stably transfected with
shBmi-1 were established. The results from RT-qPCR demonstrated
that Bmi-1 mRNA expression in the shBmi-1-transfected cells was
significantly lower than that in the control cells (p<0.05;
Fig. 1A and B). Additionally,
western blot analysis revealed that the Bmi-1 protein levels were
markedly reduced in the shBmi-1-transfected cells (Fig. 1C and D), demonstrating that the
constructed lentivirus-mediated RNAi expression vector
expressingBmi-1 (shBmi-1) inhibited the expression of Bmi-1. Bmi-1
has been shown to inhibit the INK4a/ARF locus, which exerts a
significant negative effect on p14ARF transcriptional regulation
(10). By detecting p14 gene
expression, we established that the reduced expression of Bmi-1
resulted in an increased p14 mRNA and protein expression (Fig. 1). These results demonstrate that
shRNA technology can be used to effectively inhibit Bmi-1 mRNA and
protein expression in myeloma cells, indicating that myeloma cell
lines in which the expression of Bmi-1 was effectively silenced by
RNAi were created successfully.
Bmi-1 knockdown sensitizes U266 and
RPMI8226 cells to bortezomib
To examine the effects of bortezomib on the survival
of cells in which Bmi-1 was knocked down, a CCK-8 assay was used to
detect the proliferation of U266-shBmi-1, RPMI8226-shBmi-1,
U266-control and RPMI8226-control cells. The results indicated that
the silencing of the Bmi-1 gene gradually enhanced the inhibition
of MM cell proliferation in a time-dependent manner compared to the
control (Fig. 2A and B).
Subsequently, the stably transfected cell lines were treated with
various concentrations (0, 10, 20, 40 and 80 nM) of bortezomib for
72 h, and a CCK-8 assay was performed in order to detect the
proliferative ability of the cells. The results indicated a marked
inhibition of cell proliferation in the shBmi-1-transfected cells
compared with the control group (Fig.
2C and D). The 50% inhibitory concentration (IC50) values of
bortezomib in the U266-control and U266-shBmi-1 cells were
24.73±0.375 nM and 18.59±0.286 nM (p<0.05), respectively. The
IC50 values of bortezomib in the RPMI8226-control and
RPMI8226-shBmi-1 cells were 32.99±0.458 nM and 21.56±0.526 nM
(p<0.05), respectively. These results indicated that the
knockdown of Bmi-1 sensitized the U266 and RPMI8226 cells to
bortezomib.
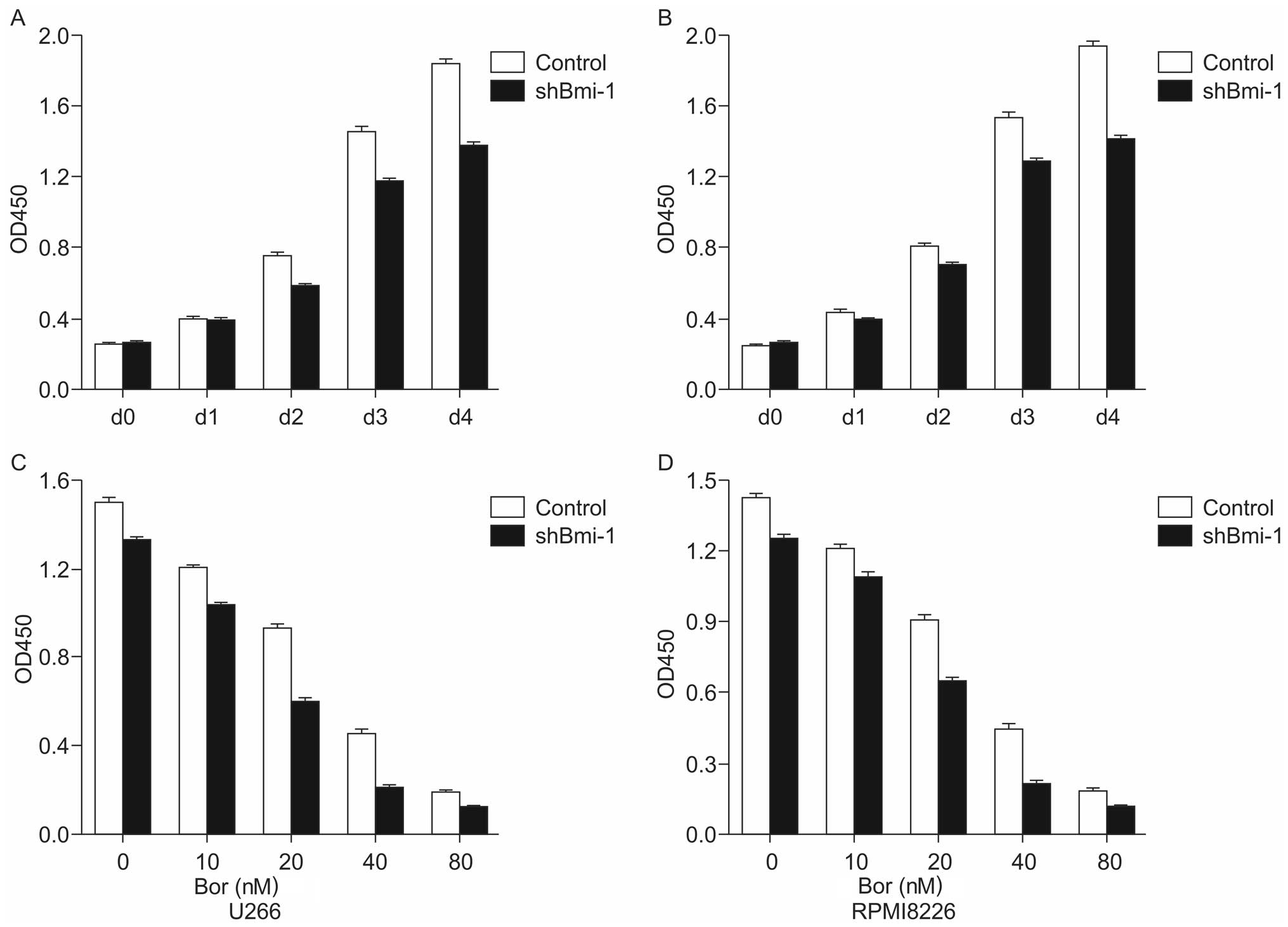 | Figure 2Inhibitory effect of short hairpin RNA
(shRNA) targeting B-cell-specific Moloney murine leukemia virus
insertion site-1 (Bmi-1; shBmi-1) combined with or without
bortezomib treatment on multiple myeloma (MM) cells. (A and B) A
CCK8 cell proliferation assay of U266-control, U266-shBmi-1,
RPMI8226-control and RPMI8226-shBmi-1 cells was performed for 2 h
after 0, 24, 48, 72 and 96 h. (C and D) U266-control, U266-shBmi-1,
RPMI8226-control, and RPMI8226-shBmi-1 cells were exposed to
bortezomib for 72 h, and a CCK8 assay was used to evaluate cell
viability. The data are presented as the means ± SD of three
independent experiments. Bor, bortezomib. |
Bmi-1 silencing induces myeloma cell
cycle arrest in the G1 phase
FCM was used to detect the cell cycle progresion of
the U266 and RPMI8226 cells which were stably transfected with
shBmi-1 followed by treatment with or without 20 or 30 nM of
bortezomib. The G1 distribution rates of the
U266-control and U266-shBmi-1 cells were 39.91±0.26% and
45.65±0.68% (p<0.05), respectively. The G1
distribution rates of the RPMI8226-control and RPMI8226-shBmi-1
cells were 42.30±0.47% and 50.86±0.38% (p<0.05), respectively.
Furthermore, in combination with bortezomib treatment for 48 h, the
silencing of Bmi-1 induced a significant increase in the number of
myeloma cells arrested in the G1 phase (p<0.05;
Fig. 3). The G1
distribution rates of the U266-control and U266-shBmi-1 cells were
45.35±0.77% and 56.81±0.50% (p<0.05), respectively. The
G1 distribution rates of the RPMI8226-control and
RPMI8226-shBmi-1 cells were 49.27±0.90% and 56.94±0.61%
(p<0.05), respectively. These data demonstrate that the
silencing of Bmi-1 enhances the effects of bortezomib on cell cycle
arrest.
Bmi-1 silencing enhances the apoptotic
effects of bortezomib
Annexin V-PE/7-AAD staining was utilized to evaluate
the effects of Bmi-1 knockdown on MM cell apoptosis. The results
indicated that the percentage of U266-control and U266-shBmi-1
apoptotic cells was 9.8 and 12.12% (p<0.05), respectively, and
the percentage of RPMI8226-control and RPMI8226-shBmi-1 apoptotic
cells was 9.6 and 15.69% (p<0.05), respectively (Fig. 4). Furthermore, we examined the
apoptotic rate of the U266-shBmi-1 and U266-control cells as well
as that of the RPMI8226-shBmi-1 and RPMI8226-control cells
following treatment with 20 or 30 nM bortezomib for 72 h. The FCM
data demonstrated that the apoptotic rate of the U266-shBmi-1 and
RPMI8226-shBmi-1 cells was 24.55 and 49.14%, respectively, compared
with 20.83 and 22.50% of the U266-control and RPMI8226-control
cells, respectively (p<0.05; Fig.
4). These observations indicated that the silencing of Bmi-1
enhanced the ability of bortezomib to induce MM cell apoptosis.
Bmi-1 silencing combined with bortezomib
regulates the expression levels of p21, Bax and Bcl-2
To further explore the mechanisms underlying the
enhancement of bortezomib-induced apoptosis in Bmi-1-silenced
cells, we examined the expression levels of p21, Bax and Bcl-2 in
the U266-shBmi-1 and U266-control cells (treated with or without 20
nM bortezomib) and in the RPMI8226-shBmi-1 and RPMI8226-control
cells (treated with or without 30 nM bortezomib). Compared with the
control group, the protein expression levels of p21 and Bax in the
shBmi-1 cells were significantly increased, while the expression of
the anti-apoptotic protein, Bcl-2, was significantly reduced
(p<0.05; Fig. 5). Furthermore,
when combined with the results obtained at 48 h of bortezomib
treatment, the difference was even more evident. These results
suggest that the robust increase in p21 and Bax expression and the
prominent decrease in Bcl-2 expression enhanced the sensitivity of
MM cells to bortezomib.
Discussion
MM is a malignant mature B cell neoplasm
characterized by the clonal proliferation of plasma cells in BM,
which is characterized by a profound genomic instability involving
both numerical and structural chromosomal aberrations of potential
prognostic relevance (28). As an
incurable cancer, MM can occur de novo or proceed from a
monoclonal gammopathy of undetermined significance (MGUS; ~1% per
year) (4). Multilevel molecular
changes, such as the activation of oncogenes, inactivation of tumor
suppressor genes, and alteration of the BM microenvironment, are
involved in the pathogenesis and progression of MM. Due to
comprehensive advances in the understanding of the molecular
pathogenesis of MM, some therapeutic medicines, such as protease
inhibitors, lenalidomide and farnesyl transferase inhibitors, that
target signaling pathways or the BM microenvironment have been
introduced to clinical practice, and have significantly improved
the survival and prognosis of patients with MM (2). Among the therapeutic drugs,
bortezomib, which is a proteasome inhibitor, has been shown to
specifically inhibit the 26S proteasome subunit and I-κB
degradation, prevent NF-κB activation, and induce apoptosis in MM.
It has also been shown to have marked clinical activity in the
relapsed or refractory MM setting (5). In the present study, we also
demonstrated that bortezomib induced a dose- and time-dependent
increase in cell toxicity and a decrease in the viability of MM
cells. Despite a promising clinical activity, some patients with MM
have failed to respond to bortezomib therapy. Moreover, only 25–35%
of patients respond to bortezomib treatment in the relapsed and
refractory setting (3,4). Therefore, to improve the outcome of
this incurable disease, further research is required to overcome
resistance to bortezomib in MM. Studies have demonstrated that the
administration of bortezomib in combination with other chemical
medications can sensitize cancer cells to bortezomib or even
reverse resistance (29,30). Furthermore, it has been reported
that the RNAi-mediated silencing of the oncogenes, Mcl-1 and
MAGE-C1/CT7, can also sensitize MM cells to bortezomib, suggesting
that RNAi may be a potential method for enhancing the sensitivity
of MM cells to bortezomib.
Bmi-1, the first polycomb-group gene to be
identified, was originally isolated as an oncogenic partner of
c-Myc in the initiation of lymphoma in murine models. There is
accumulating evidence confirming that Bmi-1 plays a crucial role in
diverse biological and pathological processes, such as axial
patterning, hematopoiesis, the regulation of proliferation,
senescence and the maintenance of cancer stem cell self-renewal.
Consistent with its role in inhibiting the p16ink4a locus, Bmi-1 is
upregulated extensively in a variety of malignancies, including MM,
and can regulate cell proliferation and carcinogenesis. Previous
studies have also suggested that Bmi-1 is associated with the
protection of tumor cells from apoptosis (31,32). Moreover, the increased expression
of Bmi-1 in certain tumors correlates with a poor prognosis
(20,32,33). Additionally, increasing evidence
indicates that the RNAi-mediated depletion of Bmi-1 can sensitize
tumor cells to chemotherapy or radiotherapy. In an attempt to
enhance sensitivity to bortezomib, these observations prompted us
to investigate the possibility of combining Bmi-1 silencing and
bortezomib treatment as a clinical therapeutic strategy for MM.
In order to evaluate the possibility of developing
Bmi-1 into a novel therapeutic agent for the treatment of MM, two
MM cell lines (U266 and RPMI8226), which overexpress Bmi-1, were
employed in the present study. A lentiviral interference vector
expressing shRNA was introduced into the U266 and RPMI8226 cells,
effectively silencing Bmi-1. We established that the proliferation
of U266 and RPMI8226 cells was markedly inhibited after the
silencing of Bmi-1, and the depletion of Bmi-1 resulted in an
enhanced sensitivity of these cells to bortezomib. FACS
observations revealed that silencing Bmi-1 expression in
combination with bortezomib treatment prompted cell cycle arrest in
the G1-S phase and enhanced bortezomib-induced
apoptosis. These results indicate that the silencing of Bmi-1
sensitizes MM cells to bortezomib by inhibiting cell proliferation
and inducing cell cycle arrest and apoptosis.
Bortezomib induces apoptosis by stabilizing p53 and
activating p53 downstream target genes, such as p21. However,
bortezomib can induce apoptosis in cells that do not contain
wild-type p53 and the effect of bortezomib on p21 may actually
limit its direct cytotoxic activities (34). Furthermore, bortezomib may
selectively induce apoptosis in SV40-transformed, but not in
normal, fibroblasts, although the accumulation of p21 occurs in
both cell types (35). Therefore,
the accumulation of p21 may be involved in, but is not absolutely
required for, the induction of apoptosis. However, p21, a
cyclin-dependent kinase inhibitor, can induce growth arrest by
inhibiting cyclin-dependent kinases that drive cell cycle
progression, sensitizing cancer cells to chemotherapy (36). The Bcl-2 family is comprised of
structurally related proteins that can either inhibit (i.e., Bcl-2)
or promote (i.e., Bax) cell death (37). The ratio of Bcl-2 to Bax is a key
regulator for determining apoptosis following damage to DNA.
Proteasome inhibitors can induce apoptosis by inducing an
accumulation of pro-apoptotic Bcl-2 family members. In addition,
evidence indicates that bortezomib overcomes the protective effects
of Bcl-2 overexpression by inhibiting the degradation of Bax
(38,39). However, alterations in the balance
between Bcl-2 and Bax may be involved in resistance to bortezomib
(40). Therefore, in this study,.
we focused on the expression levels of p21, Bcl-2 and Bax. The
results indicated that the knockdown Bmi-1 resulted in the
upregulation of p21, the downregulation of Bcl-2 and the
accumulation of Bax in the cells treated with bortezomib. These
findings suggest that p21 is involved in the Bmi-1-driven cell
proliferation inhibition and G1 phase arrest in MM
cells. Furthermore, the results indicated that Bmi-1 downregulated
the Bcl-2/Bax ratio and enhanced bortezomib-induced apoptosis.
In conclusion, to the best of our knowledge, this is
the first report of the the anticancer potential of bortezomib
treatment combined with the depletion of Bmi-1 in MM. The present
study demonstrates that the knockdown of Bmi-1 sensitizes MM cells
to bortezomib treatment and that the silencing of Bmi-1 enhances
bortezomib-induced apoptosis. Furthermore, we demonstrate that the
knockdown of Bmi-1 upregulates the expression of p21 and Bcl-2 and
decreases the expression of Bax. The alteration in the Bcl-2/Bax
ratio prompted bortezomib-induced apoptosis in Bmi-1-silenced MM
cells. Taken together, our data suggest that bortezomib treatment
combined with Bmi-1 knockdown may be a potential therapeutic
strategy for MM.
Acknowledgements
We are most grateful to Dr Didier Trono for offering
the pWPXL, psPAX2 and pMD2.G lentivirus plasmids as gifts. The
present study was supported by grants from the National Natural
Science Foundation of China (no. 81201872), the Natural Science
Foundation of Fujian Province (no. 2013J01308), the Foundation of
Fujian Key Laboratory of Hematology (no. 2009J1004), and the Fujian
Provincial Health Bureau Youth Research Projects (no.
2010-1-11).
References
|
1
|
Kyle RA and Rajkumar SV: Multiple myeloma.
N Engl J Med. 351:1860–1873. 2004. View Article : Google Scholar
|
|
2
|
Mitsiades CS, Mitsiades N, Munshi NC and
Anderson KC: Focus on multiple myeloma. Cancer Cell. 6:439–444.
2004. View Article : Google Scholar
|
|
3
|
Bergsagel PL and Kuehl WM: Molecular
pathogenesis and a consequent classification of multiple myeloma. J
Clin Oncol. 23:6333–6338. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hideshima T, Mitsiades C, Tonon G,
Richardson PG and Anderson KC: Understanding multiple myeloma
pathogenesis in the bone marrow to identify new therapeutic
targets. Nat Rev Cancer. 7:585–598. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Richardson PG, Barlogie B, Berenson J, et
al: A phase 2 study of bortezomib in relapsed, refractory myeloma.
N Engl J Med. 348:2609–2617. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kumar SK, Rajkumar SV, Dispenzieri A, et
al: Improved survival in multiple myeloma and the impact of novel
therapies. Blood. 111:2516–2520. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cheriyath V, Jacobs BS and Hussein MA:
Proteasome inhibitors in the clinical setting: benefits and
strategies to overcome multiple myeloma resistance to proteasome
inhibitors. Drugs RD. 8:1–12. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
de Carvalho F, Costa ET, Camargo AA, et
al: Targeting MAGE-C1/CT7 expression increases cell sensitivity to
the proteasome inhibitor bortezomib in multiple myeloma cell lines.
PLoS One. 6:e277072011.PubMed/NCBI
|
|
9
|
Podar K, Gouill SL, Zhang J, et al: A
pivotal role for Mcl-1 in Bortezomib-induced apoptosis. Oncogene.
27:721–731. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Jacobs JJ, Kieboom K, Marino S, DePinho RA
and van Lohuizen M: The oncogene and Polycomb-group gene bmi-1
regulates cell proliferation and senescence through the ink4a
locus. Nature. 397:164–168. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Jacobs JJ, Scheijen B, Voncken JW, Kieboom
K, Berns A and van Lohuizen M: Bmi-1 collaborates with c-Myc in
tumorigenesis by inhibiting c-Myc-induced apoptosis via INK4a/ARF.
Genes Dev. 13:2678–2690. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Leung C, Lingbeek M, Shakhova O, et al:
Bmi1 is essential for cerebellar development and is overexpressed
in human medulloblastomas. Nature. 428:337–341. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Park IK, Qian D, Kiel M, et al: Bmi-1 is
required for maintenance of adult self-renewing haematopoietic stem
cells. Nature. 423:302–305. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Molofsky AV, He S, Bydon M, Morrison SJ
and Pardal R: Bmi-1 promotes neural stem cell self-renewal and
neural development but not mouse growth and survival by repressing
the p16Ink4a and p19Arf senescence pathways. Genes Dev.
19:1432–1437. 2005. View Article : Google Scholar
|
|
15
|
Yang J, Chai L, Liu F, et al: Bmi-1 is a
target gene for SALL4 in hematopoietic and leukemic cells. Proc
Natl Acad Sci USA. 104:10494–10499. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Vonlanthen S, Heighway J, Altermatt HJ, et
al: The bmi-1 oncoprotein is differentially expressed in non-small
cell lung cancer and correlates with INK4A-ARF locus expression. Br
J Cancer. 84:1372–1376. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Larmonie NS, Dik WA, Beverloo HB, van
Wering ER, van Dongen JJ and Langerak AW: BMI1 as oncogenic
candidate in a novel TCRB-associated chromosomal aberration in a
patient with TCRgammadelta+ T-cell acute lymphoblastic leukemia.
Leukemia. 22:1266–1267. 2008. View Article : Google Scholar
|
|
18
|
Choi YJ, Choi YL, Cho EY, et al:
Expression of Bmi-1 protein in tumor tissues is associated with
favorable prognosis in breast cancer patients. Breast Cancer Res
Treat. 113:83–93. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kim JH, Yoon SY, Kim CN, et al: The Bmi-1
oncoprotein is overexpressed in human colorectal cancer and
correlates with the reduced p16INK4a/p14ARF proteins. Cancer
Letters. 203:217–224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Song W, Tao K, Li H, et al: Bmi-1 is
related to proliferation, survival and poor prognosis in pancreatic
cancer. Cancer Sci. 101:1754–1760. 2010. View Article : Google Scholar
|
|
21
|
van Leenders GJ, Dukers D, Hessels D, et
al: Polycomb-group oncogenes EZH2, BMI1, and RING1 are
overexpressed in prostate cancer with adverse pathologic and
clinical features. Eur Urol. 52:455–463. 2007.PubMed/NCBI
|
|
22
|
Jagani Z, Wiederschain D, Loo A, et al:
The Polycomb group protein Bmi-1 is essential for the growth of
multiple myeloma cells. Cancer Res. 70:5528–5538. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Crea F, Duhagon Serrat MA, Hurt EM, Thomas
SB, Danesi R and Farrar WL: BMI1 silencing enhances docetaxel
activity and impairs antioxidant response in prostate cancer. Int J
Cancer. 128:1946–1954. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang E, Bhattacharyya S, Szabolcs A, et
al: Enhancing chemotherapy response with Bmi-1 silencing in ovarian
cancer. PLoS One. 6:e17918
|
|
25
|
Wu J, Hu D, Yang G, et al: Down-regulation
of BMI-1 cooperates with artemisinin on growth inhibition of
nasopharyngeal carcinoma cells. J Cell Biochem. 112:1938–1948.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fasano CA, Dimos JT, Ivanova NB, Lowry N,
Lemischka IR and Temple S: shRNA knockdown of Bmi-1 reveals a
critical role for p21-Rb pathway in NSC self-renewal during
development. Cell Stem Cell. 1:87–99. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Huang BB, Gao QM, Liang W, Xiu B, Zhang WJ
and Liang AB: Down-regulation of SENP1 expression increases
apoptosis of Burkitt lymphoma cells. Asian Pac J Cancer Prev.
13:2045–2049. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Fonseca R, Barlogie B, Bataille R, et al:
Genetics and cytogenetics of multiple myeloma: a workshop report.
Cancer Res. 64:1546–1558. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Briani C, Berno T, Campagnolo M and
Zambello R: Lenalidomide for bortezomib-resistant multiple myeloma.
Nat Rev Clin Oncol. 7: View Article : Google Scholar : 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hideshima T, Bradner JE, Wong J, et al:
Small-molecule inhibition of proteasome and aggresome function
induces synergistic antitumor activity in multiple myeloma. Proc
Natl Acad Sci USA. 102:8567–8572. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Liu L, Andrews LG and Tollefsbol TO: Loss
of the human polycomb group protein BMI1 promotes cancer-specific
cell death. Oncogene. 25:4370–4375. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang H, Pan K, Zhang HK, et al: Increased
polycomb-group oncogene Bmi-1 expression correlates with poor
prognosis in hepatocellular carcinoma. J Cancer Res and Clin Oncol.
134:535–541. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Mihara K, Chowdhury M, Nakaju N, et al:
Bmi-1 is useful as a novel molecular marker for predicting
progression of myelodysplastic syndrome and patient prognosis.
Blood. 107:305–308. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu J, Tiwari S, Steiner P and Zhang L:
Differential apoptotic response to the proteasome inhibitor
Bortezomib [VELCADE, PS-341] in Bax-deficient and p21-deficient
colon cancer cells. Cancer Biol Ther. 2:694–699. 2003.
|
|
35
|
An B, Goldfarb RH, Siman R and Dou QP:
Novel dipeptidyl proteasome inhibitors overcome Bcl-2 protective
function and selectively accumulate the cyclin-dependent kinase
inhibitor p27 and induce apoptosis in transformed, but not normal,
human fibroblasts. Cell Death Differ. 5:1062–1075. 1998. View Article : Google Scholar
|
|
36
|
Lazzarini R, Moretti S, Orecchia S, Betta
PG, Procopio A and Catalano A: Enhanced antitumor therapy by
inhibition of p21waf1 in human malignant mesothelioma. Clin Cancer
Res. 14:5099–5107. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Gross A, McDonnell JM and Korsmeyer SJ:
BCL-2 family members and the mitochondria in apoptosis. Genes Dev.
13:1899–1911. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Herrmann JL, Briones F Jr, Brisbay S,
Logothetis CJ and McDonnell TJ: Prostate carcinoma cell death
resulting from inhibition of proteasome activity is independent of
functional Bcl-2 and p53. Oncogene. 17:2889–2899. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Tan TT, Degenhardt K, Nelson DA, et al:
Key roles of BIM-driven apoptosis in epithelial tumors and rational
chemotherapy. Cancer Cell. 7:227–238. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
McConkey DJ and Zhu K: Mechanisms of
proteasome inhibitor action and resistance in cancer. Drug Resist
Updat. 11:164–179. 2008. View Article : Google Scholar : PubMed/NCBI
|


















