Introduction
Apigenin (4′,5,7,-tirhydoxyflavone) is a dietary
flavone that is naturally found as a dimer, diapigenin (1), apigenin-7-O-glucoside or acetylated
derivative (2). It is abundantly
present in various fruits, plants, vegetables and some herbs
(3–6). Plant-derived beverages, including
herbal tea and red wine, are good sources of apigenin (7). Apigenin has received significant
attention as a preventive, as well as a therapeutic agent due to
its efficacy and low intrinsic toxicity.
The effects of apigenin on growth inhibition, cell
cycle arrest and the induction of apoptosis have been demonstrated
in various types of cancer, such as breast cancer with high levels
of HER2/neu (8), as well as
cervical (8), lung (9), colon (10), hematologic (11) and ovarian cancer (12). In mouse models of prostate cancer,
orally delivered apigenin has been shown to reduce tumor volume and
suppress metastasis (13). The
anti-angiogenic property of apigenin has also been reported in lung
(14), ovarian (15) and prostate cancer (16), which is the result of the
transcriptional repression of vascular endonthelial growth factor
expression through the downregulation of hypoxia-inducible factor
(HIF)1-α expression levels.
In the human genome, 58 receptor tyrosine kinases
(RTKs) are known to date; these are catagorized into 20 subfamilies
and integrated into one large RTK family. Axl RTK, also known as
Ark or Ufo, belongs to the TAM subfamily, which also contains Tyro
3 and Mer. It was initially identified in 1988 as an unidentified
transforming gene (17) from
chronic myelogenous leukemia patients and cloned in 1991 from
primary human leukemia cells (18).
Axl has been shown to be overexpressed in various
types of cancer, including acute leukemia (19), as well as breast (20), colon (21), thyroid (22), esophageal (23) and lung cancer (24). Enhanced Axl expression and
activation through binding with its ligand, growth arrest-specific
6 (a vitamin K-dependent protein), has been shown to promote
downstream signaling for cell proliferation and survival (25–27). Recently, Axl activation has also
been identified as a novel mechanism that renders non-small cell
lung cancer (NSCLC) cells and HER2/neu-positive breast cancer cells
resistant to epidermal growth factor (EGF) RTK inhibitor (TKI)
(28) and dual TKIs, such as
lapatinib (29), respectively.
Therefore, the inhibition of Axl expression and Axl-mediated
signaling may be a potential therapeutic target for cancer
treatment.
In this study, we examined the effects of apigenin
on Axl expression and the subsequent effects on cell proliferation
in human lung cancer cells. Apigenin was found to downregulate Axl
expression, which resulted in the inhibition of cell proliferation
through the induction of p21 protein expression and the suppression
of the expression of X-linked inhibitor of apoptosis protein
(XIAP).
Materials and methods
Reagents and antibodies
A549 and H460 cells were purchased from the American
Type Culture Collection (Manassas, VA, USA). Apigenin was obtained
from Sigma (St. Louis, MO, USA). Primers for Axl were synthesized
by the domestic company, Bioneer Corp. (Daejeon, Korea). TRI
reagent was obtained from Solgent Co., Ltd. (Daejeon, Korea).
AmpliTaq DNA polymerase and Lipofectamine 2000 were obtained from
Roche Diagnostics Corp. (Indianapolis, IN, USA) and Invitrogen
(Carlsbad, CA, USA), respectively. G418 was from Gibco BRL
(Gaithersburg, MD, USA). The plasmid, pGL3-basic vector, and the
Dual-Glo luciferase assay kit were purchased from Promega Corp.
(Madison, WI, USA). For western blot analysis, specific antibodies
against Axl, p21, XIAP and GAPDH, as well as secondary antibodies
were obtained from Santa Cruz Biotechnology (Dallas, TX, USA).
Cell culture
The A549 and H460 cells were grown in RPMI-1640
(Gibco BRL) containing 10% FBS, 2 mM L-glutamine, 10 U/ml
penicillin and 10 g/ml streptomycin at 37°C in 5% CO2 in
a water-saturated atmosphere.
Reverse transcription PCR (RT-PCR) and
quantitative PCR (qPCR)
The A549 and H460 cells (1×106) were
seeded in a 100-mm culture dish and grown overnight. The cells were
then treated with the indicated concentrations (0, 10, 20 and 40
μM) of apigenin for 24 h. Total RNA was extracted using TRI reagent
and subjected to cDNA synthesis and PCR. The specific primers were
as follows: Axl sense, 5′-AACCTTCAACTCC TGCCTTCTCG-3′ and
antisense, 5′-CAGCTTCTCCTTCAGC TCTTCAC-3′; GAPDH sense,
5′-GGAGCCAAAAGGGTCAT CAT-3′ and antisense,
5′-GTGATGGCATGGACTGTGGT-3′. To quantify the Axl mRNA levels in the
A549 and H460 cells before and after treatment with apigenin, qPCR
was performed using SYBR-Green PCR Master mix and the ABI PRISM
7900HT system (Applied Biosystems, Foster City, CA, USA). The mRNA
level of Axl was normalized to that of GAPDH, using the
2−ΔΔCT method.
Western blot analysis
Total cell lysates were prepared from the A549 or
H460 cells treated with the indicated concentrations (0, 10, 20 and
40 μM) of apigenin using lysis buffer [1% Triton X-100, 50 mM Tris
(pH 8.0), 150 mM NaCl, 1 mM PMSF, 1 mM
Na3VO4, and protease inhibitor cocktail].
Untreated cells were used as controls. Protein concentrations were
determined using Bio-Rad protein assays. Proteins from the cell
lysates (20–40 μg) were separated by 12% SDS-PAGE, and
electrotransferred onto nitrocellulose membranes. The membranes
were blocked for 30 min at room temperature in Tris-buffered saline
with 0.05% Tween-20 (TTBS) containing 5% non-fat dry milk, and then
incubated with TTBS containing a primary antibody for 4 h at room
temperature. After 3×10 min washes in TTBS, the membranes were
incubated with peroxidase-conjugated secondary antibody for 1 h.
Following 3 additional 10-min washes with TTBS, the protein bands
of interest were visualized using an enhanced chemiluminescence
detection system (Amersham™ ECL™ Prime Western Blotting Detection
Reagent; GE Healthcare, Piscataway, NJ, USA).
Clonogenic assay
The A549 or H460 cells were seeded in 35-mm culture
dishes (2×103 cells/dish) and cultured for the following
7–10 days under the indicated concentrations (0, 10, 20 and 40 μM)
of apigenin to form colonies. Colonies of >50 cells were stained
with Crystal violet (in 60% methanol; Junsei Chemical Co., Ltd.,
Tokyo, Japan) and images were acqired using the RAS-3000 Image
Analysis System (FujiFilm, Tokyo, Japan).
Cell viability assay
The Cell Counting kit-8 (Dojindo Laboratories,
Kumamoto, Japan) was used to measure the viability of the cells.
Briefly, 1×103 cells were seeded in each well of 96-well
plates and grown overnight at 37°C and then treated with the
indicated concentrations (0, 10, 20 and 40 μM) of apigenin for 24
h. At the end of treatment, 10 μl of CCK-8 solution were added
followed by incubation for a further 4 h. The absorbance at 450 nm
was measured using a microplate reader (Model 680 microplate
reader; Bio-Rad Laboratories, Inc., Hercules, CA, USA). Values are
the means ± SD for triplicate wells and were normalized to those of
the control group to determine the percentage viability.
Ectopic expression of Axl
To ectopically express Axl, the recombinant plasmid,
pcDNA3-Axl, was constructed by cloning the Axl cDNA into the
EcoRI and BamHI sites of the pcDNA3 vector and 2 μg
of purified plasmids were transfected into the A549 or H460 cells
(3×105 cells in a 100-mm dish) using Lipofectamine 2000
(Invitrogen). To establish stable cell lines, which constitutively
express Axl, the transfected cells were cultured in the presence of
400 μg/ml of G418. The RPMI-1640 medium containing G418 was
refreshed every 3 days. After 3–4 weeks, the Axl-expressing cells
were enriched and the Axl expression in these cells was analyzed by
western blot analysis.
Promoter activity test
To construct pGL3-AXL, the Axl promoter
region ranging from −887 to +7 bp of the transcriptional start site
was amplified by PCR and subcloned into the pGL3-basic vector, the
luciferase reporter plasmid. The constructed promoter-reporter
plasmid was co-transfected into cells (3×105 cells in a
100 mm dish) with renilla luciferase vectors, pRL-SV40, as an
internal control. Luciferase activity was measured using a Dual-Glo
luciferase assay system.
siRNA trasnfection
To inhibit Axl expression, RNA interference
(RNAi)-induced gene silencing was performed. The H460 cells
(1×106) were seeded in a 100-mm culture dish, grown
overnight and then transfected with 50 nM siRNA targeting Axl
(sense, 5′-AAGAUUUGGAGAACACACUGA-3′; and antisense,
5′-UCAGUGUGUUCUCCAAAUCUU-3′), as previously described (30), or control siRNA. The cells were
harvested for 24 and 48 h after transfection and used to evaluate
protein expression, as well as in cell proliferation and colony
formation assays.
Statistical analysis
Data are expressed as the means ± SD of triplicate
samples or at least 3 independent experiments. To determine
statistical significance, the Student’s t-test was used with a
p-value threshold of <0.05.
Results
Apigenin suppresses Axl expression in
human lung cancer cells
To determine the effects of apigenin on Axl
expression, the human lung cancer cells, A549 and H460, were
incubated with 10, 20, or 40 μM of apigenin for 24 h and
subsequently, Axl protein expression was measured. As shown in
Fig. 1A and B, the results from
western blot analysis revealed that treatment with apigenin induced
a dose-dependent decrease in the protein expression of Axl in both
cell lines. More specifically, when the cells were exposed to 40 μM
apigenin for 24 h, the Axl protein levels in the A549 and H460
cells were diminished to 57 and 35% compared with the untreated
cells, respectively.
 | Figure 1Apigenin reduces Axl expression in
human lung cancer cells. The A549 and H460 cells were treated with
the indicated concentrations of apigenin for 24 h. (A and B) The
protein level of Axl was determined by western blot analysis in
order to assess the effects of apigenin on its expression. GAPDH
was used as a loading control. Results are from 3 independent
experiments. *P<0.05, apigenin-treated vs. untreated
cells; **P<0.001, apigenin-treated vs. untreated
cells. (C) For RT-PCR, total RNA from the cells was isolated and
used for the analysis of Axl mRNA expression. The level of Axl mRNA
was normalized to that of GAPDH. The data shown are representative
of 3 independent experiments. (D) qPCR (SYBR-Green) was performed
to measure Axl mRNA expression in the A549 and H460 cells treated
with apigenin for 24 h. Each sample was performed in triplicate,
and the Axl mRNA expression was calculated relative to GAPDH. The
asterisks indicate the significant difference compared to the
control value (*P<0.05, apigenin-treated vs.
untreated cells). (E) To determine Axl promoter activity,
the A549 cells (3×103 cells/dish) were transfected with
pGL3 or pGL3-Axl, Axl promoter-luciferase plasmid, using
Lipofectamine 2000. The cells were then incubated with apigenin and
total cell lysates were used to measure luciferase activity. The
data shown are representative of at least 3 independent
experiments. Data are expressed as the means ± SD of triplicate
samples conducted in 3 independent experiments. The asterisks
indicate the significant difference compared to the control value
(*P<0.05, pGL3-Axl/apigenin vs. pGL3; and
**P<0.001, pGL3-Axl vs. pGL3). |
The downregulation of Axl expression in the
apigenin-treated cells was further confirmed by RT-PCR and a
promoter activity test. The results from RT-PCR revealed that in
both cells lines, the mRNA levels of Axl were decreased following
treatment with apigenin (Fig.
1C); these results were consistent with those from western blot
analysis. Additionally, the results from qPCR revealed that the Axl
mRNA levels in the A549 and H460 cells treated with 40 μM apigenin
for 24 h were reduced to 37 and 48% compared with the untreated
cells, respectively (Fig.
1D).
To determine the effects of apigenin on the
transcription of the Axl gene, Axl promoter activity
was measured using luciferase reporter plasmid under the control of
the human Axl promoter plasmid, pGL3-Axl. A549 cells were
transfected with pGL3-Axl and then incubated with 40 μM apigenin
for 24 h. As illustrated in Fig.
1E, luciferase activity significantly declined following
treatment with apigenin. The results of RT-PCR as well as those
from the promoter activity test indicated that apigenin suppressed
Axl expression in the lung cancer cells at the transcriptional
level.
Apigenin-mediated downregulation of Axl
is responsible for its anti-proliferative effects on human lung
cancer cells
Since Axl has been known to transduce cell survival,
growth and proliferation (19,20,25,27,31), we wished to determine whether the
downregulation of Axl by apigenin affects lung cancer cell
viability. The cells were incubated with 10, 20, 40 μM of apigenin
for 24 h, and the number of viable cells was then counted. As shown
in Fig. 2A, treatment with
apigenin reduced cell viability in a dose-dependent manner.
Following treatment of the A549 and H460 cells with 40 μM apigenin,
only 49 and 37% of the cells survived, respectively.
The anti-proliferative effects of apigenin on the
lung cancer cells were further confirmed by colony formation assay.
The cells were cultured for 10 days in the presence of 10, 20, 40
μM of apigenin. The exposure of the cells to apigenin resulted in a
dose-dependent inhibition of colony formation (Fig. 2B). More specifically, the H460
cells failed to grow into a colony under 40 μM apigenin,
demonstrating the cytotoxic effects of apigenin on lung cancer
cells.
To verify the involvement of Axl in the
apigenin-mediated inhibition of cell proliferation, we examined
whether the overexpression of Axl has an impact on the
anti-proliferative effects of apigenin. The Axl-expressing
construct, pcDNA3-Axl, was transiently transfected into the A549
cells and the cells were then cultured with or without apigenin for
24 h. Compared to the control cells transfected with a pcDNA3 empty
vector, the cells transfected with the pcDNA3-Axl plasmid were
found to be slightly less sensitive to apigenin treatment (Fig. 2C), indicating that the
overexpression of Axl protein diminisehd the anti-proliferative
effects of apigenin in the cells transfected with the pcDNA3-Axl
plasmid.
Subsequently, we established Axl-overexpressing
stable cell lines, A549/Axl and H460/Axl, and observed the effects
of apigenin on cell proliferation. Western blot analysis revealed
that the Axl protein level in the Axl-overexpressing cells was
higher than in their parental cells, even following treatment with
apigenin (Fig. 2D). Colony
formation assay also revealed that both the A549/Axl and H460/Axl
cells formed more colonies and were less affected by treatment with
apigenin (Fig. 2E); these results
are consistent with those from western blot analysis.
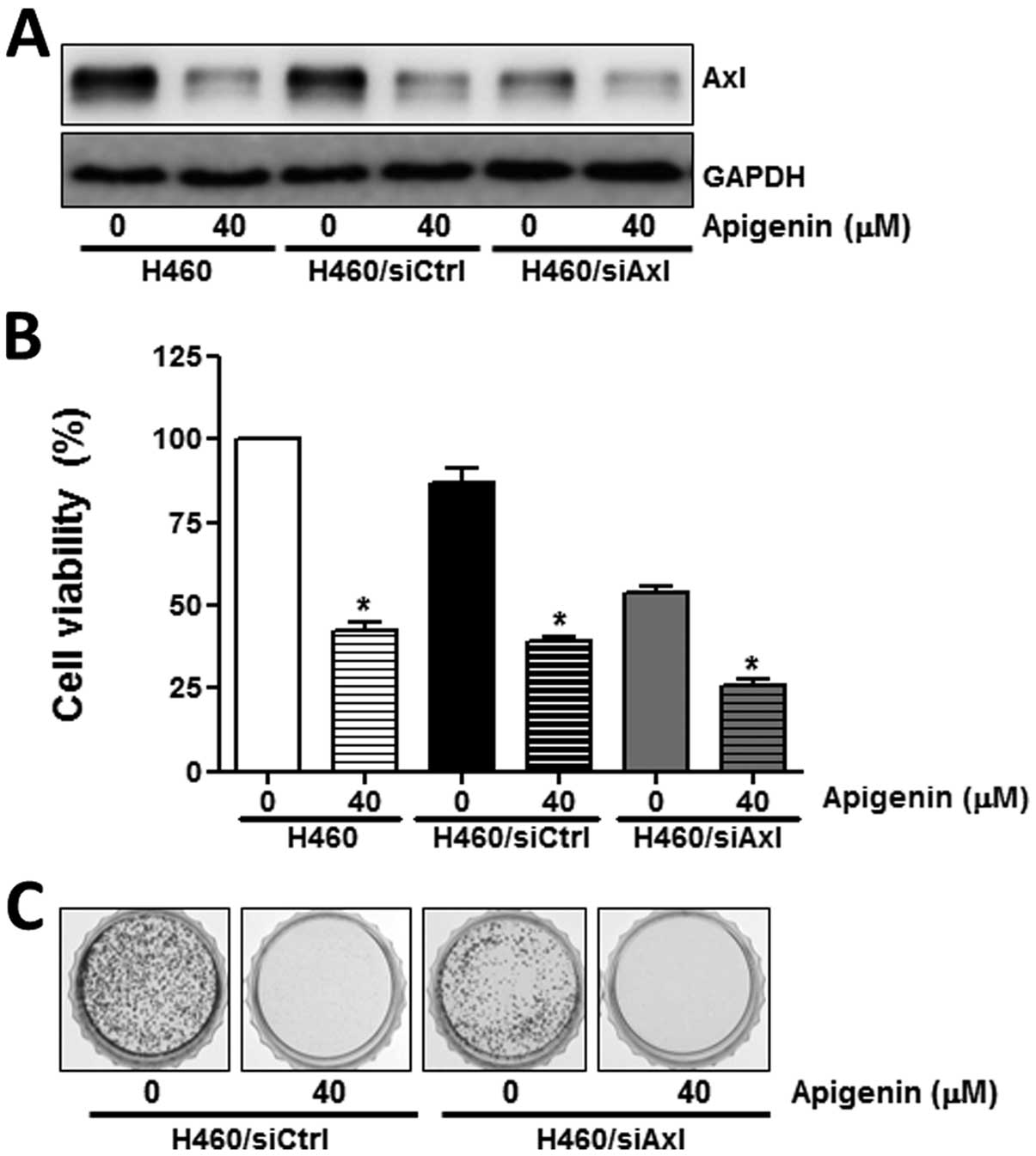
To further demonstrate the role of Axl in the
anti-proliferative effects of apigenin, its expression was
inhibited by specific siRNA. In contrast to the control siRNA,
siCtrl, the Axl targetingsiRNA, siAxl, induced a significant
decrease in Axl expression (Fig.
3A), resulting in a reduction in cell proliferation (Fig. 3B), as well as in colony formation
(Fig. 3C). Taken together, these
results suggest that Axl protein expression correlates with cell
proliferation and that the anti-proliferative effects of apigenin
are mediated by the modulation of Axl expression.
Apigenin-induced downregulation of Axl
expression facilitates the induction of p21 and reduction of XIAP
expression
To elucidate the molecular mechanisms by which the
apigenin-mediated downregulation of Axl expression results in the
inhibition of cell proliferation, we examined the effects of
apigenin on several molecules related to cell cycle regulation and
apoptosis. The A549 and H460 cells were treated with 40 μM apigenin
for 24 h. The results from western bolt analysis revealed that
treatment with apigenin increased the levels of the
cyclin-dependent kinase inhibitor, p21, which induces cell cycle
arrest, but decreased the levels of XIAP, which inhibits apoptosis
(Fig. 4A and B). These effects
(the induction of p21 and the decrease in XIAP expressino) upon
treatment with apigenin were attenuated or augmented in the
Axl-overexpressing A549 and H460 cells (Fig. 4A) or in the siAxl-transfected H460
cells (Fig. 4B), respectively.
These data indicate that the apigenin-induced downregulation of Axl
expression is a prerequisite for the subsequent increase or
reduction in p21 and XIAP expression.
Discussion
The first observation by Birt et al
demonstrated that apigenin is an anti-mutagenic and anti-promotion
bioflavonoid based on its inhibitory effects on ornithine
decarboxylase, which plays an important role in tumor promotion
(32). Subsequently, a number of
other studies further confirmed its antioxidant, anti-inflammatory,
anti-angiogenic and anti-proliferative activities, which revealed
various targets of apigenin simultaneously. For instance, apigenin
has been shown to suppress lipopolysaccharide (LPS)-induced
cyclooxygenase-2 and nitric oxide synthase-2 expression (33) and TNF-α induced nuclear factor-κB
activation (34), which are major
mediators for eliciting inflammation. In addition, apigenin has
been verified as an effective inhibitor of the p34 (cdc2) kinase,
which increases p53 stability (35), and a good inducer of the p21 and
Apaf-1 proteins, which are the main players for cell cycle arrest
and the induction of apoptosis (36,37). Based on these data, apigenin has
evoked particular interest in its potential as a chemopreventive
and chemotherapeutic agent.
In this study, we found a new target of apigenin,
Axl RTK, which is a member of the TAM RTK subfamily and has been
reported to be overexpressed in a various types of cancer and to
have an oncogenic potential (18,23,24). The exposure of NSCLC cells (A549
and H460) to apigenin resulted in a decrease in Axl mRNA and
protein expression (Fig. 1A–D).
Furthermore, Axl promoter activity was also reduced
following treatment with apigenin (Fig. 1E), indicating that apigenin
suppresses Axl expression at the transcriptional level.
Specific targeting of Axl with RNAi or monoclonal
antibodies, which causes the downregulation of Axl expression, has
been reported to reduce the proliferation of NSCLC cells in
vitro and in vivo (using tumor xenografts) (38,39). In addition, the overexpression
and/or activation of Axl has been shown to be a mechanism that
confers acquired resistance to various chemotherapeutic drugs,
including gefitinib or erlotinib that are EGF receptor (EGFR)
inhibitors in NSCLC (40–42) and head and neck cancer (31), imatinib that is a TKI in chronic
myelogenous leukemia (43) and
gastrointestinal stromal tumors (44), and tumor necrosis factor-related
apoptosis-inducing ligand in esophageal adenocarcinoma (45). Therefore, we examined whether the
inhibition of Axl expression by apigenin affects the proliferation
of NSCLC cells. In agreement with previous studies, we also found
that treatment with apigenin inhibited the proliferation and
clonogenic ability of the A549 and H460 cells in a dose-dependent
manner (Fig. 2A and B).
Furthermore, the anti-proliferative effects of apigenin were
decreased or augmented by the induction (Fig. 2C and E) or inhibition of Axl
expression (Fig. 3B),
respectively. We also found that the ectopic expression of Axl
diminished the apigenin-induced increase in p21 protein expression
and the reduction in XIAP expression (Fig. 4A), which facilitates cell cycle
arrest and apoptosis, respectively. By contrast, the inhibition of
Axl expression using specific siRNA was found to augment both the
induction of p21 and the reduction of XIAP upon treatment with
apigenin (Fig. 4B). These results
suggest that the Axl protein level correlates with cell
proliferation, and the anti-proliferative effects of apigenin are
mediated by the modulation of Axl expression.
In conclusion, our observations indicate that the
treatment of NSCLC cells with apigenin suppresses Axl expression at
the transcriptional level, which subsequently induces the
inhibition of cell proliferation through the induction of cell
cycle arrest and/or apoptosis. These results suggest that Axl is a
novel target of apigenin through which it exerts its
anti-proliferative effects on cancer cells.
Acknowledgements
This study was supported by the Basic Science
Research Program through the, National Research Foundation of Korea
(NRF) funded by the Ministry of Education, Science and Technology
(grant no. 2006-2005303).
References
|
1
|
Liu C, Tu FX and Chen X: Neuroprotective
effects of apigenin on acute transient focal cerebral
ischemia-reperfusion injury in rats. Zhong Yao Cai. 31:870–873.
2008.(In Chinese).
|
|
2
|
Svehliková V, Bennett RN, Mellon FA, et
al: Isolation, identification and stability of acylated derivatives
of apigenin 7-O-glucoside from chamomile (Chamomilla
recutita [L.] Rauschert). Phytochemistry. 65:2323–2332.
2004.PubMed/NCBI
|
|
3
|
Birt DF, Hendrich S and Wang W: Dietary
agents in cancer prevention: flavonoids and isoflavonoids.
Pharmacol Ther. 90:157–177. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Surh YJ: Cancer chemoprevention with
dietary phytochemicals. Nat Rev Cancer. 3:768–780. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Manach C, Scalbert A, Morand C, Rémésy C
and Jiménez L: Polyphenols: food sources and bioavailability. Am J
Clin Nutr. 79:727–747. 2004.PubMed/NCBI
|
|
6
|
Yang CS, Landau JM, Huang MT and Newmark
HL: Inhibition of carcinogenesis by dietary polyphenolic compounds.
Annu Rev Nutr. 21:381–406. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Bevilacqua L, Buiarelli F, Coccioli F and
Jasionowska R: Identification of compounds in wine by HPLC-tandem
mass spectrometry. Ann Chim. 94:679–689. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zheng PW, Chiang LC and Lin CC: Apigenin
induced apoptosis through p53-dependent pathway in human cervical
carcinoma cells. Life Sci. 76:1367–1379. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lu HF, Chie YJ, Yang MS, et al: Apigenin
induces apoptosis in human lung cancer H460 cells through caspase-
and mitochondria-dependent pathways. Hum Exp Toxicol. 30:1053–1061.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhong Y, Krisanapun C, Lee SH, et al:
Molecular targets of apigenin in colorectal cancer cells:
involvement of p21, NAG-1 and p53. Eur J Cancer. 46:3365–3374.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ruela-de-Sousa RR, Fuhler GM, Blom N,
Ferreira CV, Aoyama H and Peppelenbosch MP: Cytotoxicity of
apigenin on leukemia cell lines: implications for prevention and
therapy. Cell Death Dis. 1:e192010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li ZD, Hu XW, Wang YT and Fang J: Apigenin
inhibits proliferation of ovarian cancer A2780 cells through Id1.
FEBS Lett. 583:1999–2003. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shukla S, MacLennan GT, Flask CA, et al:
Blockade of beta-catenin signaling by plant flavonoid apigenin
suppresses prostate carcinogenesis in TRAMP mice. Cancer Res.
67:6925–6935. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu LZ, Fang J, Zhou Q, Hu X, Shi X and
Jiang BH: Apigenin inhibits expression of vascular endothelial
growth factor and angiogenesis in human lung cancer cells:
implication of chemoprevention of lung cancer. Mol Pharmacol.
68:635–643. 2005.PubMed/NCBI
|
|
15
|
Fang J, Xia C, Cao Z, Zheng JZ, Reed E and
Jiang BH: Apigenin inhibits VEGF and HIF-1 expression via
PI3K/AKT/p70S6K1 and HDM2/p53 pathways. FASEB J. 19:342–353. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mirzoeva S, Kim ND, Chiu K, Franzen CA,
Bergan RC and Pelling JC: Inhibition of HIF-1 alpha and VEGF
expression by the chemopreventive bioflavonoid apigenin is
accompanied by Akt inhibition in human prostate carcinoma PC3-M
cells. Mol Carcinog. 47:686–700. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu E, Hjelle B and Bishop JM:
Transforming genes in chronic myelogenous leukemia. Proc Natl Acad
Sci USA. 85:1952–1956. 1988. View Article : Google Scholar
|
|
18
|
O’Bryan JP, Frye RA, Cogswell PC, et al:
axl, a transforming gene isolated from primary human myeloid
leukemia cells, encodes a novel receptor tyrosine kinase. Mol Cell
Biol. 11:5016–5031. 1991.PubMed/NCBI
|
|
19
|
Rochlitz C, Lohri A, Bacchi M, et al: Axl
expression is associated with adverse prognosis and with expression
of Bcl-2 and CD34 in de novo acute myeloid leukemia (AML): results
from a multicenter trial of the Swiss Group for Clinical Cancer
Research (SAKK). Leukemia. 13:1352–1358. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Berclaz G, Altermatt HJ, Rohrbach V,
Kieffer I, Dreher E and Andres AC: Estrogen dependent expression of
the receptor tyrosine kinase axl in normal and malignant human
breast. Ann Oncol. 12:819–824. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Craven RJ, Xu LH, Weiner TM, et al:
Receptor tyrosine kinases expressed in metastatic colon cancer. Int
J Cancer. 60:791–797. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ito T, Ito M, Naito S, et al: Expression
of the Axl receptor tyrosine kinase in human thyroid carcinoma.
Thyroid. 9:563–567. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Nemoto T, Ohashi K, Akashi T, Johnson JD
and Hirokawa K: Overexpression of protein tyrosine kinases in human
esophageal cancer. Pathobiology. 65:195–203. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Shieh YS, Lai CY, Kao YR, et al:
Expression of axl in lung adenocarcinoma and correlation with tumor
progression. Neoplasia. 7:1058–1064. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hutterer M, Knyazev P, Abate A, et al: Axl
and growth arrest-specific gene 6 are frequently overexpressed in
human gliomas and predict poor prognosis in patients with
glioblastoma multiforme. Clin Cancer Res. 14:130–138. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sun WS, Fujimoto J and Tamaya T:
Coexpression of growth arrest-specific gene 6 and receptor tyrosine
kinases Axl and Sky in human uterine endometrial cancers. Ann
Oncol. 14:898–906. 2003. View Article : Google Scholar
|
|
27
|
Gustafsson A, Martuszewska D, Johansson M,
et al: Differential expression of Axl and Gas6 in renal cell
carcinoma reflecting tumor advancement and survival. Clin Cancer
Res. 15:4742–4749. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Byers LA, Diao L, Wang J, et al: An
epithelial-mesenchymal transition gene signature predicts
resistance to EGFR and PI3K inhibitors and identifies Axl as a
therapeutic target for overcoming EGFR inhibitor resistance. Clin
Cancer Res. 19:279–290. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Liu L, Greger J, Shi H, et al: Novel
mechanism of lapatinib resistance in HER2-positive breast tumor
cells: activation of AXL. Cancer Res. 69:6871–6878. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Holland SJ, Powell MJ, Franci C, et al:
Multiple roles for the receptor tyrosine kinase axl in tumor
formation. Cancer Res. 65:9294–9303. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Giles KM, Kalinowski FC, Candy PA, et al:
Axl mediates acquired resistance of head and neck cancer cells to
the epidermal growth factor receptor inhibitor erlotinib. Mol
Cancer Ther. 12:2541–2558. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Birt DF, Walker B, Tibbels MG and Bresnick
E: Anti-mutagenesis and anti-promotion by apigenin, robinetin and
indole-3-carbinol. Carcinogenesis. 7:959–963. 1986. View Article : Google Scholar
|
|
33
|
Liang YC, Huang YT, Tsai SH, Lin-Shiau SY,
Chen CF and Lin JK: Suppression of inducible cyclooxygenase and
inducible nitric oxide synthase by apigenin and related flavonoids
in mouse macrophages. Carcinogenesis. 20:1945–1952. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Choi JS, Choi YJ, Park SH, Kang JS and
Kang YH: Flavones mitigate tumor necrosis factor-alpha-induced
adhesion molecule upregulation in cultured human endothelial cells:
role of nuclear factor-kappa B. J Nutr. 134:1013–1019. 2004.
|
|
35
|
Plaumann B, Fritsche M, Rimpler H,
Brandner G and Hess RD: Flavonoids activate wild-type p53.
Oncogene. 13:1605–1614. 1996.PubMed/NCBI
|
|
36
|
Lepley DM and Pelling JC: Induction of
p21/WAF1 and G1 cell-cycle arrest by the chemopreventive agent
apigenin. Mol Carcinog. 19:74–82. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Shukla S and Gupta S: Molecular mechanisms
for apigenin-induced cell-cycle arrest and apoptosis of hormone
refractory human prostate carcinoma DU145 cells. Mol Carcinog.
39:114–126. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li Y, Ye X, Tan C, et al: Axl as a
potential therapeutic target in cancer: role of Axl in tumor
growth, metastasis and angiogenesis. Oncogene. 28:3442–3455. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Ye X, Li Y, Stawicki S, et al: An anti-Axl
monoclonal antibody attenuates xenograft tumor growth and enhances
the effect of multiple anticancer therapies. Oncogene.
29:5254–5264. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gusenbauer S, Vlaicu P and Ullrich A: HGF
induces novel EGFR functions involved in resistance formation to
tyrosine kinase inhibitors. Oncogene. 32:3846–3856. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Zhang Z, Lee JC, Lin L, et al: Activation
of the AXL kinase causes resistance to EGFR-targeted therapy in
lung cancer. Nat Genet. 44:852–860. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Postel-Vinay S and Ashworth A: AXL and
acquired resistance to EGFR inhibitors. Nat Genet. 44:835–836.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Dufies M, Jacquel A, Belhacene N, et al:
Mechanisms of AXL overexpression and function in Imatinib-resistant
chronic myeloid leukemia cells. Oncotarget. 2:874–885.
2011.PubMed/NCBI
|
|
44
|
Mahadevan D, Cooke L, Riley C, et al: A
novel tyrosine kinase switch is a mechanism of imatinib resistance
in gastrointestinal stromal tumors. Oncogene. 26:3909–3919. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hong J and Belkhiri A: AXL mediates TRAIL
resistance in esophageal adenocarcinoma. Neoplasia. 15:296–304.
2013.PubMed/NCBI
|