Introduction
Osteoarthritis (OA) is the most common degenerative
disease of the human articular cartilage, and a major cause of
physical disability due to symptoms, such as pain, stiffness and
loss of mobility, characterized by the progressive destruction of
articular cartilage, subchondral bone alterations, the formation of
osteophytes and synovitis (1).
Although multiple patient-specific variables contribute to the risk
of OA, such as aging, failure of nutrient supply and genetic
predisposition, joint injuries increase the risk of developing OA
by as much as 10-fold, or in some injuries, even by much more than
20-fold (2). It has been
suggested that excessive acute impact energy or chronic mechanical
overload causes damage to chondrocytes and is thus responsible for
OA (3). Yet, the mechanisms
through which excessive mechanical force causes OA remain unknown.
A recent study on cartilage injury in vitro demonstrated
that chondrocyte damage and death caused by mechanical injury
releases the effector molecules that activate chondroprogenitor
cells in vitro that propogate and migrate to regions of
damaged cartilage (4).
Another study revealed that these mechanically
injured chondrocytes produce chemokines and cytokines that can
cause joint inflammation and progressive cartilage loss (5). Blocking the expression of the
effector molecules in mechanically injured joints may have the
potential to prevent destructive inflammation. Therefore, the
identification of the effector molecules and the molecular
mechanisms responsible for regulating their expression is crucial
for improving the effectiveness of current treatments for
osteoarthritis.
Mechanical injury is a major cause of OA in humans.
The pathological process of joint injury has 2 phases: the primary
injury caused by a sudden mechanical force, and the secondary
injury. The delayed, secondary injury is considered to be due to a
cascade of biochemical reactions that are important in the
mechanisms of OA. As cellular and micron-sized structural changes
in vivo are hard to detect immediately post-injury, research
on joint mechanical injury has focused on understanding the
mechanisms of the secondary step that accounts for chondrocytes
damaged by mechanical injury in vitro (6–8).
Apoptosis, or programmed cell death, is a
physiological process responsible for maintaining homeostasis in
articular cartilage (9). The OA
cartilage contains a higher percentage of chondrocytes undergoing
apoptosis than normal cartilage. Previous studies have demonstrated
that chondrocyte apoptosis is related to the progression of OA.
Chondrocytes undergo apoptosis in response to mechanical injury
in vitro (10,11). Chondrocyte apoptosis and necrosis
have been reported in response to bovine and human cartilage
wounding, following the mechanical injury loading of cartilage
explants (12,13). Mechanical injury as an important
inducer of apoptosis plays a considerable role in the pathogenetic
mechanisms of OA (11). It may be
an important mediator of chronic articular lesions in OA.
MicroRNAs (miRNAs or miRs) are a family of
~22-nucleotide endogenous non-coding small RNAs that regulate the
expression of multiple genes involved in a number of physiological
functions and disease processes, including OA (14). Typically, they bind to the
3′-untranslated region (3′-UTR) of their target mRNAs and repress
protein expression by affecting mRNA translation and/or
destabilization (15).
Previous studies have indicated that miRNAs are
known to play a key role in mediating the effects of the main risk
factors for OA, such as innate and adaptive immune responses
(16–18), aging, chronic pain and
inflammation (19–22), through the control of target genes
(23–25). The functions of regulated genes
involve almost all aspects of cellular process, such as
proliferation, differentiation, motivation, communication,
senescence and apoptosis (26–28). Three previous studies (29–31) identified a signature of 17, 16 and
7 miRNAs, respectively that distinguishes normal from
osteoarthritic cartilage tissue, by microarray and real-time PCR
assays. miR-146a is one of these identified miRNAs associated with
OA cartilage (32).
Recent evidence suggests that miR-146a is markedly
expressed in OA cartilage compared with normal cartilage, and its
expression is induced by the stimulation of interleukin (IL)-1β
(33). However, miR-146a
expression levels in OA synovial tissue are low (29,34). miRNA-146a expression is induced in
response to lipopolysaccharide (LPS) and pro-inflammatory mediators
in THP-1 cells and this induction is regulated by nuclear factor
(NF)-κB, which in turn downregulates inflammatory cascades by
decreasing the expression of IL-1 receptor-associated kinase-1
(IRAK1) and tumor necrosis factor (TNF) receptor-associated factor
6 (TRAF6). IRAK1 and TRAF6 impair the NF-κB activation pathway and
suppress the expression of NF-κB target genes, such as IL-6, IL-8,
IL-1β and TNF-α (16,23,25,35). These findings suggest that
miR-146a plays a role in the damage of chondrocytes due to
mechanical injury; miR-146a thus has potential as a novel
therapeutic target in OA.
In this study, we used a self-designed, mechanical
pressure-controlled cellular injury unit [utility model patent of
China granted (36)] to precisely
generate 10 MPa of pressure on normal human chondrocytes inside a
high-pressure container, which was placed in a 37°C thermostatic
apparatus for 60 min. The results indicated that mechanical
pressure affected the viability of the chondrocytes and induced the
early apoptosis of chondrocytes. We used miRNA and gene microarray
analysis to screen the differentially expressed genes and miRNAs
between normal chondrocytes and mechanically injured chondrocytes.
The results of the miRNA microarray and gene microarray
demonstrated that the expression of mir-146a was upregulated in the
mechanically injured chondrocytes. Bioinformatics analysis revealed
that Smad4 is a potential target gene of miR-146a. Combined with
the gene microarray report that the expression levels of Smad4 were
downregulated and the expression levels of vascular endothelial
growth factor (VEGF) were upregulated, we put forward the
hypothesis that Smad4 regulates the expression of VEGF and mediates
the damaging effects of miR-146a in the process of mechanical
injury responsible for the pathogenesis of OA. We then evaluated
the alterations in miR-146a, Smad4 and VEGF expression in normal
chondrocytes following mechanical injury in vitro, and
confirmed that Smad4 is a direct target of miR-146a. We found that
in chondrocytes subjected to mechanical pressure injury, the
expression levels of miR-146a and VEGF increased and the levels of
Smad4 decreased.
Furthermore, we demonstrate that the increase in
miR-146a expression downregulated Smad4 and upregulated VEGF
expression, and induced the apoptosis of the mechanically injured
chondrocytes. Conversely, the inhibition of miR-146a or the
overexpression of Smad4 reduced VEGF expression in the mechanically
injured chondrocytes. Taken together, these findings suggest that
the dysregulation of miR-146a may contribute to the pathogenesis of
OA by inhibiting Smad4, a key component in the anabolic forming
growth factor (TGF)-β pathway, by stimulating VEGF in the
angiogenesis, chondrocyte hypertrophy and extracellular matrix
degradation pathways, and by inducing chondrocyte death.
Materials and methods
Ethics statement
All human tissue samples were obtained in accordance
with the World Medical Association Declaration of Helsinki Ethical
Principles for Medical Research Involving Human Subjects, and were
approved by the Ethics Review Committee of the Fourth Military
Medical University, Xi’an, China (approval ID: 2013023) and written
informed consent was obtained from all participating patients.
Isolation and culture of normal human
chondrocytes
Normal knee cartilage samples were obtained from
subjects undergoing post-traumatic above-knee amputation. Primary
chondrocytes were isolated from the femoral condyles and tibial
plateau of these human knee cartilage samples and cultured. The
tissue samples were minced into small fragments, followed by
digestion first with 0.25% trypsin (Gibco/Invitrogen, Carlsbad, CA,
USA) for 30 min at 37°C and then with 0.2% collagenase (Sigma, St.
Louis, MO, USA) for 5 h at 37°C. The dissociated cell suspension
was filtered through a 40-μm cell strainer (BD Falcon, Bedford, MA,
USA), and thecells were collected by centrifugation at 800 × g for
10 min. The chondrocytes were then cultured in DMEM/F-12 medium
(Gibco/Invitrogen) supplemented with 10% fetal bovine serum (FBA,
Gibco/Invitrogen). Following overnight culture, non-adherent cells
were removed, and the adherent cells were further incubated in
fresh medium. Primary chondrocytes were cultured according to
previously described methods (37–39). The chondrocytes used in the
experiments were all first sub-culture cells. The results of
quantitative reverse transcription PCR (RT-qPCR) demonstrated that
all chondrocytes expressed type II collagen and did not express
type I collagen.
Mechanical pressure injury to normal
human chondrocytes
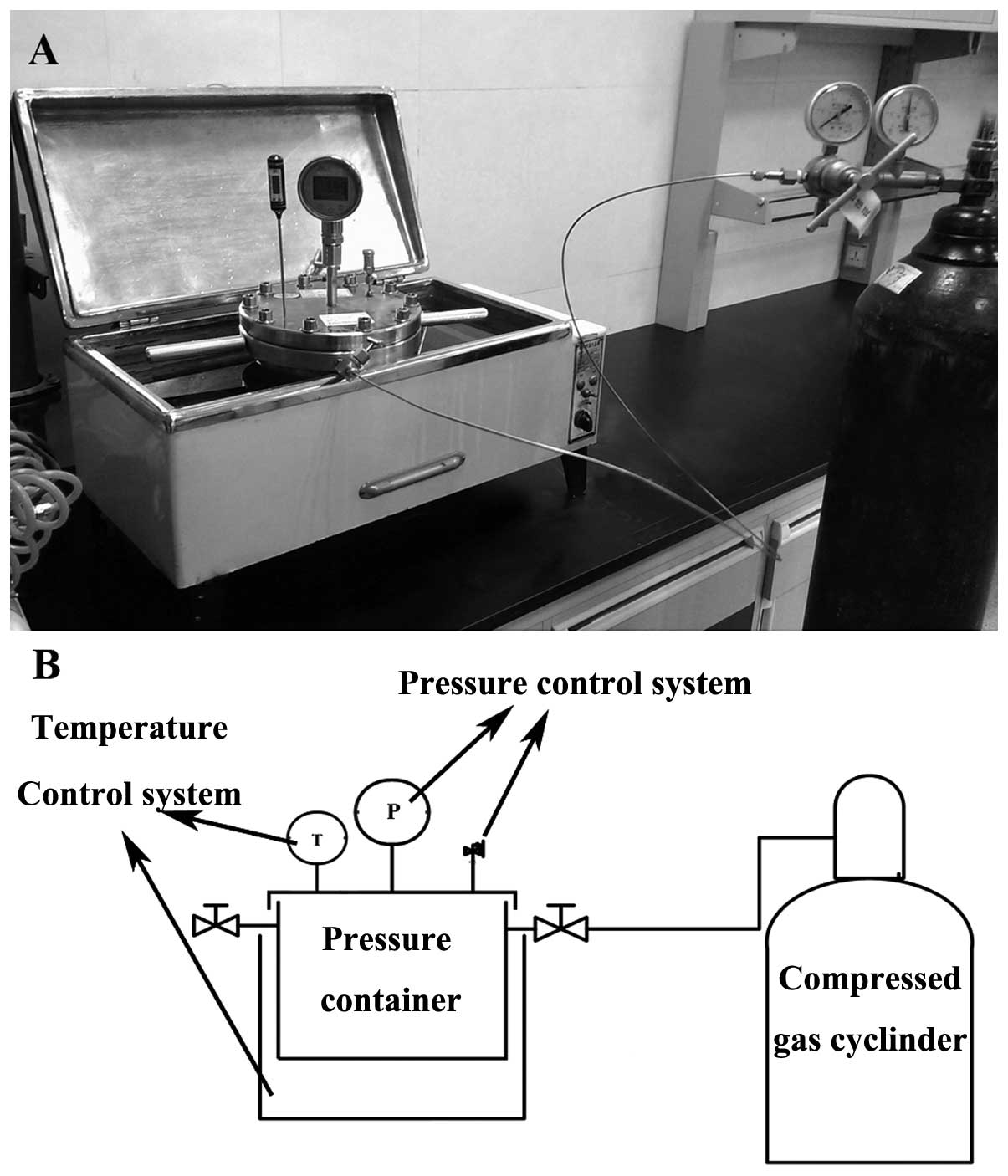
We used a self-designed, mechanical
pressure-controlled cellular injury unit to precisely generate 10
MPa of pressure on normal human chondrocytes which were grown to
70–80% confluence inside a high-pressure container, which was
placed in a 37°C incubator for 60 min. The control groups were
placed in the unit but not loaded. This unit [utility model patent
of China granted (36)], consists
of 4 parts: the mechanical pressure container, the pressure control
system, the temperature control system and the compressed gas
cylinder (Fig. 1). The
chondrocytes were cultured immediately following injury and
maintained in culture for 8, 12, 24 or 48 h to provide time for
metabolic changes to occur before the subsequent experimental
procedures.
MTT assay
The viability of the human chondrocytes was assessed
at 8, 12, 24 or 48 h following mechanical injury by MTT assay, as
previously described (40). Human
chondrocytes were seeded at a density of 1×105
cells/well in a 96-well plate and were grown to 70–80% confluence
before mechanical pressure injury. The chondrocytes were cultured
immediately after injury and maintained in culture for 4 h to
provide time for metabolic changes to occur before the subsequent
experimental procudures. Subsequently, each well was supplemented
with 10 μl of 5 mg/ml MTT and incubated for an additional 4 h at
37°C. The medium was then removed, and 150 μl DMSO (Sigma-Aldrich,
Shanghai, China) were added to solubilize the MTT formazan. The
optical density was read with a spectrometer at a wavelength of 490
nm. Wells without cells were used as blanks, and their values were
considered the background values and subtracted from each
sample.
Apoptosis assay by flow cytometry
The apoptotic rate of the chondrocytes was detected
and quantified by flow cytometry following staining with Annexin
V-FITC and propidium iodide (PI; both from Roche, Mannheim,
Germany). The chondrocytes (1×104) were harvested,
washed and incubated with Annexin V-PI for 15 min at room
temperature in the dark. The chondrocytes were analyzed by flow
cytometry using a Becton Dickinson fluorescence-activated cell
sorter (BD Biosciences, San Jose, CA, USA) with emission filters of
530/30 nm (FITC) and 585/42 nm (PI). The percentage of apoptosis
was analyzed using Becton Dickinson Cell Quest software. Each test
was repeated in triplicate.
Microarray analysis
The miRNA expression profiles of the normal human
chondrocytes and the human chondrocytes subjected to mechanical
pressure injury were determined by miRNA microarray analysis using
the GeneChip miRNA 3.0 Array (Affymetrix, Santa Clara, CA, USA),
based on Sanger miRBase Release 17.0 (41). The transcription profiles of the
normal human chondrocytes and the human chondrocytes subjected to
mechanical pressure injury were determined by gene microarray
analysis using the GeneChip Human Genome U133 Plus 2.0 Array
(Affymetrix). The method was employed to identify differentially
expressed miRNAs and genes between the normal chondrocytes and the
chondrocytes subjected to mechanical pressure injury, as previously
described (23,31,42).
Target prediction
Putative target genes regulated by the miRNAs
differentially expressed in normal chondrocytes and those subjected
to mechanical pressure injury were predicted bioinformatically and
combining the prediction of their supposed targets with the
different genes expression of chondrocytes. Bioinformatics analysis
was performed using these specific programs: miRanda (http://www.microrna.org), Pictar (http://pictar.mdc-berlin.de/) and Targetscan
(http://www.targetscan.org/), as
previously described (31).
RNA oligonucleotides, plasmids, siRNA and
transfection
The FAM modified 2′-O-me-oligonucleotides were
synthesized by GenePharma (Shanghai, China). The sequences of the
2′-O-me-miR-146a mimics and 2′-O-me-miR-221 inhibitor, as well as a
negative control of miRNA mimics (negative mimics) or inhibitors
(negative inhibitors), were as follows:
5′-UGAGAACUGAAUUCCAUGGGUU-3′, 5′-AACCCAUGGAAUUCAGUUCUCA-3′ and
5′-UUGUACUACACAAAAGUACUG-3′. Smad4 siRNA (Smad4 siRNA: sc-29484;
control siRNA: sc-37007) was purchased from Santa Cruz
Biotechnology (Santa Cruz, CA, USA). When the cells were grown to
70–80% confluence, transfection was performed using the
Lipofectamine™ 2000 transfection reagent (Invitrogen) according to
the manufacturer’s instructions. After 4 h of transfection, the
medium was replaced with fresh medium (DMEM/F12) containing 10%
fetal bovine serum.
RNA extraction and RT-qPCR
Total RNA (miRNA and mRNA) was extracted using
TRIzol reagent (Invitrogen) according to the manufacturer’s
instructions. Subsequently, 1 μg total RNA was reverse transcribed
with a specific stem-loop primer for miRNA and with a random primer
for mRNA using the AMV First-Strand cDNA Synthesis kit (Fermentas,
Pittsburgh, PA, USA). After RT reaction, real-time PCR was
performed on a Light Cycler 480 (Roche, Indianapolis, IN, USA)
using ABI SYBR-Green PCR Master mix (Applied Biosystems, Bedford,
MA, USA). β-actin and small nuclear RNA U6 were used as an internal
normalized reference for cDNA and miRNA, respectively. The primers
used were as follows: miR-146a forward,
5′-ACACTCCAGCTGGGTGAGAACTGAATTCC-3′ and reverse,
5′-CTCAACTGGTGTCGTGGAGTCGGCAATTCAGTTGAGAACCCATGG -3′; Smad4
forward, 5′-CTCTAAACCTCAGGCCACATC-3′ and reverse,
5′-CAATACCTCCTCCATCAAAGC-3′; VEGF forward,
5′-ATGAACTTTCTGCTGTCTTGG-3′ and reverse,
5′-TCACCGCCTCGGCTTGTCACA-3′; β-actin forward,
5′-CTCTTCCAGCCTTCCTTCCT-3′ and reverse, 5′-TCATCGTACTCCTGCTTGCT-3′;
U6 forward, 5′-CTCGCTTCGGCAGCACA-3′ and reverse,
5′-AACGCTTCACGAATTTGCGT-3′. The RT-qPCR results were analyzed and
expressed as the relative miRNA levels of the Ct (cycle threshold)
value, which was then calculated to fold change by the value of
each control sample set at 1.
Western blot analysis
Chondrocyte total protein was washed with
pre-chilled PBS and then subjected to whole-cell ice-cold lysis
buffer with 50 mmol/l Tris-HCl, pH 7.4; 1% NP-40; 150 mmol/l NaCl;
0.1% sodium dodecyl sulfate (SDS); and supplemented with proteinase
inhibitor (one tablet per 10 ml; Roche, Indianapolis, IN, USA). The
concentration of proteins in the chondrocyte lysate was quantified
using the DC protein assay kit (Bio-Rad Laboratories, Hercules, CA,
USA), and diluted to an equal concentration with hypotonic buffer.
A total of 40 μg of the protein lysates was size-fractionated by
4–20% SDS-PAGE and then transblotted electrically on to
nitrocellulose membranes (Invitrogen). The membranes were blocked
with TBST containing 5% non-fat dry milk for 1 h and hybridized
with primary antibody against Smad4 (1:1,000; Santa Cruz
Biotechnology), VEGF (1:1,000; Santa Cruz Biotechnology), and GAPDH
(1:5,000; Abcam, Shanghai, China) overnight at 4°C. After washing 3
times with TBST, the membranes were hybridized with horseradish
peroxidase (HRP)-conjugated anti-mouse or rabbit secondary antibody
(Santa Cruz Biotechnology) for 2 h. After washing 3 times with TBST
again, the specific protein was detected by chemiluminescence using
the enhanced chemiluminescence reagent, Pierce ECL Western Blotting
Substrate (Thermo Fisher Scientific, Waltham, MA, USA). GAPDH was
used as an internal control. The optical density of the immunoblots
was quantified using Quantity One software (Bio-Rad
Laboratories).
Luciferase reporter assay
The sequence of the human Smad4 3′-UTR containing
the miR-146a binding side was amplified by PCR from genomic DNA
using the following primers: forward,
5′-CCGCTCGAGTGAAGGAATCATTCCAGTGCTAG-3′ and reverse,
5′-TGCTCTAGACTTGGTAAAATTAACTCACCCACA-3′, and the PCR products were
cloned into the pMIR-Report vector (Ambion, Austin, TX, USA)
between the HindIII and SpeI sites downstream of the
firefly luciferase gene present to develop the wild-type 3′-UTR
luciferase reporter vector. The mutant 3′-UTR luciferase reporter
vector was generated by site-directed mutagenesis using the
QuikChange Mutagenesis kit (Stratagene, La Jolla, CA, USA) using
the following primers: forward,
5′-TTAAAGGCAGAGAACAAGAGAAAGTTAATTCACC-3′ and reverse,
5′-GGTGAATTAACTTTCTCTTGTTCTCTGCCTTTAA-3′. All sequences of the
amplified products were confirmed by DNA sequencing. Human
chondrocytes were passaged on 24-wells plate the day prior to
transfection to achieve 70–80% confluence on the following day.
Human chondrocytes were transiently transfected using Lipofectamine
2000 (Invitrogen) according to the manufacturer’s instructions,
with wild-type or mutant-type pMIR-report-Smad4 vector in which the
putative miR-146a binding site was mutated, and co-transfected with
miR-146a mimics, miR-146a inhibitor or their negative control (NC)
and inhibitor NC (GenePharma), individually. The human chondrocytes
were also transfected with with Renilla luciferase reporter
(pRL-TK) vector as an internal standard to determine and normalize
the luciferase activity. At 24 h after transfection, the human
chondrocyte lysates were extracted and luciferase activity was
measured using a Dual Luciferase Reporter Assay System (Promega,
Madison, WI, USA) on a Berthold AutoLumat LB9507 rack luminometer
(Berthold Technologies China, Shanghai, China). The results were
expressed as relative luciferase activity (firefly
Luc/Renilla Luc). All experiments were repeated at least 3
times.
Statistical analysis
The results are expressed as the means ± standard
deviation unless otherwise indicated, and all error bars represent
the standard deviation of the mean. Statistical analysis was
carried out using the Student’s t-test between 2 groups or one-way
analysis of variance followed by Student-Newman-Kuels test for
multiple comparisons with SPSS 13.0 statistical software (SPSS Inc,
Chicago, IL, USA). A value of P<0.05 was considered to indicate
a statistically significant difference.
Results
Effects of mechanical pressure injury on
chondrocyte viability
In a preliminary experiment, the human chondrocytes
were exposed to 5, 8, 10 or 12 MPa mechanical pressure for 10, 30
or 60 min, and chondrocyte viability was determined at 8, 12, 24 or
48 h after the injury was sustained by MTT assay. The experiment
demonstrated that loads below 8 MPa did not result in any
measurable cell death and loads above 12 MPa resulted in extensive
cell death. The OD values of the chondrocytes significantly
decreased in a force-dependent manner at 8, 10, 12 Mpa after
loading of mechanical pressure for 60 min (P<0.05) (Fig. 2A). The OD values of the
chondrocytes significantly decreased in a time-dependent manner
after loading of 10 Mpa mechanical pressure (P<0.05) (Fig. 2B). Based on the preliminary
results, the mechanical pressure was set at 10 MPa for 60 min in
the main experiment. The OD values of the chondrocytes loaded with
10 MPa of pressure for 60 min were reduced in a time-dependent
manner (P<0.05) (Fig. 2C).
Compared with the non-loaded chondrocytes, the OD values of the
chondrocytes loaded with 10 MPa of pressure for 60 min were reduced
in a time-dependent manner at 12, 24 and 48 h after pressure injury
was sustained (P<0.05) (Fig.
2D).
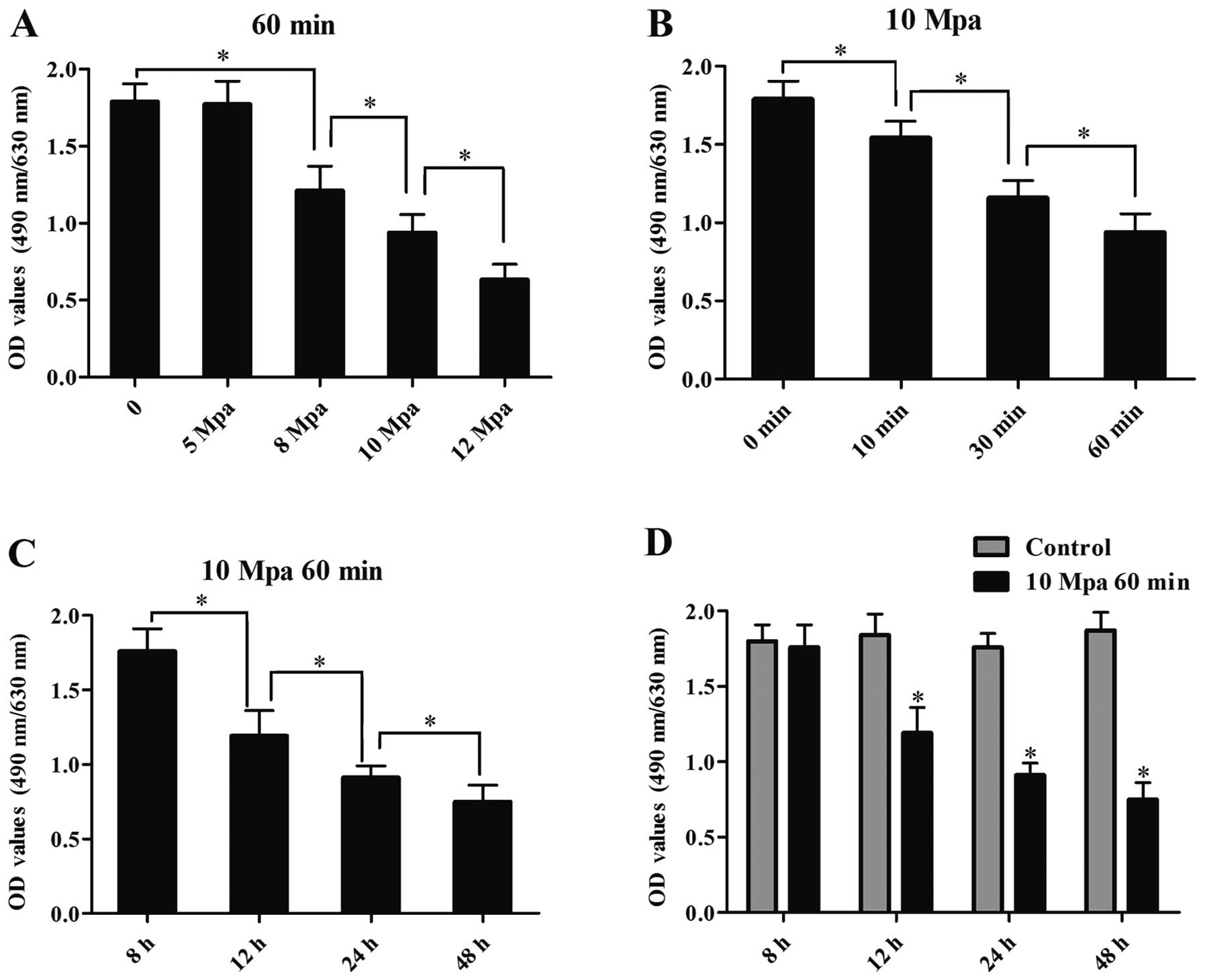 | Figure 2Effects of mechanical injury on the
viability of human chondrocytes. (A) The OD values of normal
chondrocytes exposed to 5, 8, 10 and 12 Mpa of mechanical pressure
for 60 min, at 24 h following exposure to mechanical pressurey
(*P<0.05; bars, means ± SD; n=3). (B) The OD values
of normal chondrocytes exposed to 10 Mpa of mechanical pressure for
10, 30 or 60 min, at 24 h following exposure to mechanical pressure
(*P<0.05; bars, means ± SD; n=3). (C) The OD values
of normal chondrocytes exposed to 10 Mpa mechanical pressure for 60
min, at 8, 12, 24 and 48 h following exposure to mechanical
pressure (*P<0.05; bars, means ± SD; n=3). (D) The OD
values of the normal chondrocytes exposed to 10 Mpa mechanical
pressure for 60 min, at 8, 12, 24 and 48 h following exposure to
mechanical pressure (*P<0.05; bars, means ± SD; n=3).
Control, non-loaded cells. |
Mechanical pressure injury increases the
expression levels of miR-146a and VEGF and decreases the levels of
Smad4 in human chondrocytes
To identify the miRNAs involved in the process of
injury sustained by chondrocytes due to mechanical pressure and
their role in the pathogenesis of OA, we screened for the miRNAs
which responded to exposure to mechanical pressure (10 Mpa, 60 min)
in the human chondrocytes. This is a novel cell injury model to
mimic mechanical pressure injury substained by chondrocytes related
to the progression of OA in vitro. The expression profiles
of miRNAs and the transcription profiles in mechanically injured
chondrocytes at 48 h following exposure to mechanical pressure were
investigated by miRNA and gene microarray analysis. A series of
miRNAs and genes was found to have altered expression levels in
response to mechanical pressure injury. The results revealed that
there was a significant difference in the expression of miR-146a
between the mechanically injured and normal human chondrocytes with
a 406 fold change in expression (data not shown); this is in
accordance with the results of a previous study which demonstrated
that miR-146a mediates inflammatory response (16). Its expression is higher in OA
cartilage than in normal cartilage (33). Bioinformatics analysis revealed
that Smad4 is a potential target gene of miR-146a. The gene
microarray report indicated that the expression levels of Smad4
were downregulated and the expression levels of VEGF were
upregulated (data not shown). Thus, we selected miR-146a for
further investigation.
Mechanical pressure injury (10 Mpa, 60 min) rapidly
induced miR-146a and VEGF expression and inhibited Smad4 expression
in human chondrocytes, and their expression gradually increased and
decreased, respectively over a 48-h time course following the
exposure of the chondrocytes to mechanical pressure (Fig. 3A–C), which was consistent with the
microarray results (data not shown). Mechanical pressure injury
stimulated the VEGF protein levels and inhibited the Smad4 protein
levels (Fig. 3D) in a
time-dependent manner.
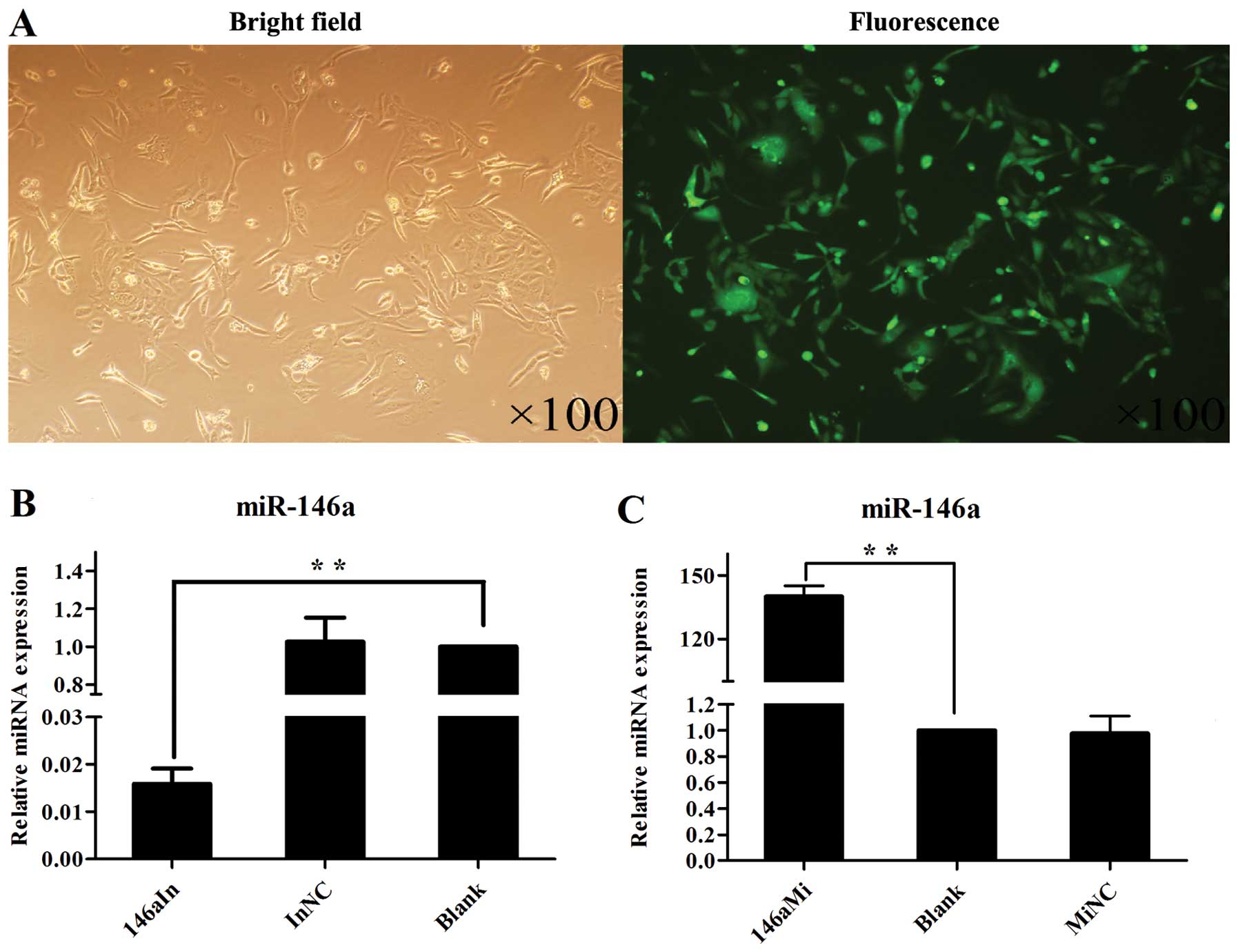
Upregulation of miR-146a and validation
for miR-146a oligonucleotide transfection in human
chondrocytes
To investigate the function of miR-146a, we
transfected the human chondrocytes with miR-146a mimics, negative
control mimics, miR-146a inhibitors or negative control inhibitors.
These oligonucleotides could be observed with a fluorescence
microscope (Olympus China, Beijing, China) as there was a FAM
fluorescent label in their 5′ oligonucleotide structure. At 24 h
after transfection, fluorescence microscopy revealed that the
transfection efficiency of the miR-146a mimics in the human
chondrocytes was >80% (Fig.
4A), and the transfection efficiency of the other groups was
similar (data not shown). miR-146a was significantly knocked down
or overexpressed by transfection with miRNA inhibitors or miRNA
mimics compared with the negative control roups (P<0.01)
(Fig. 4B and C).
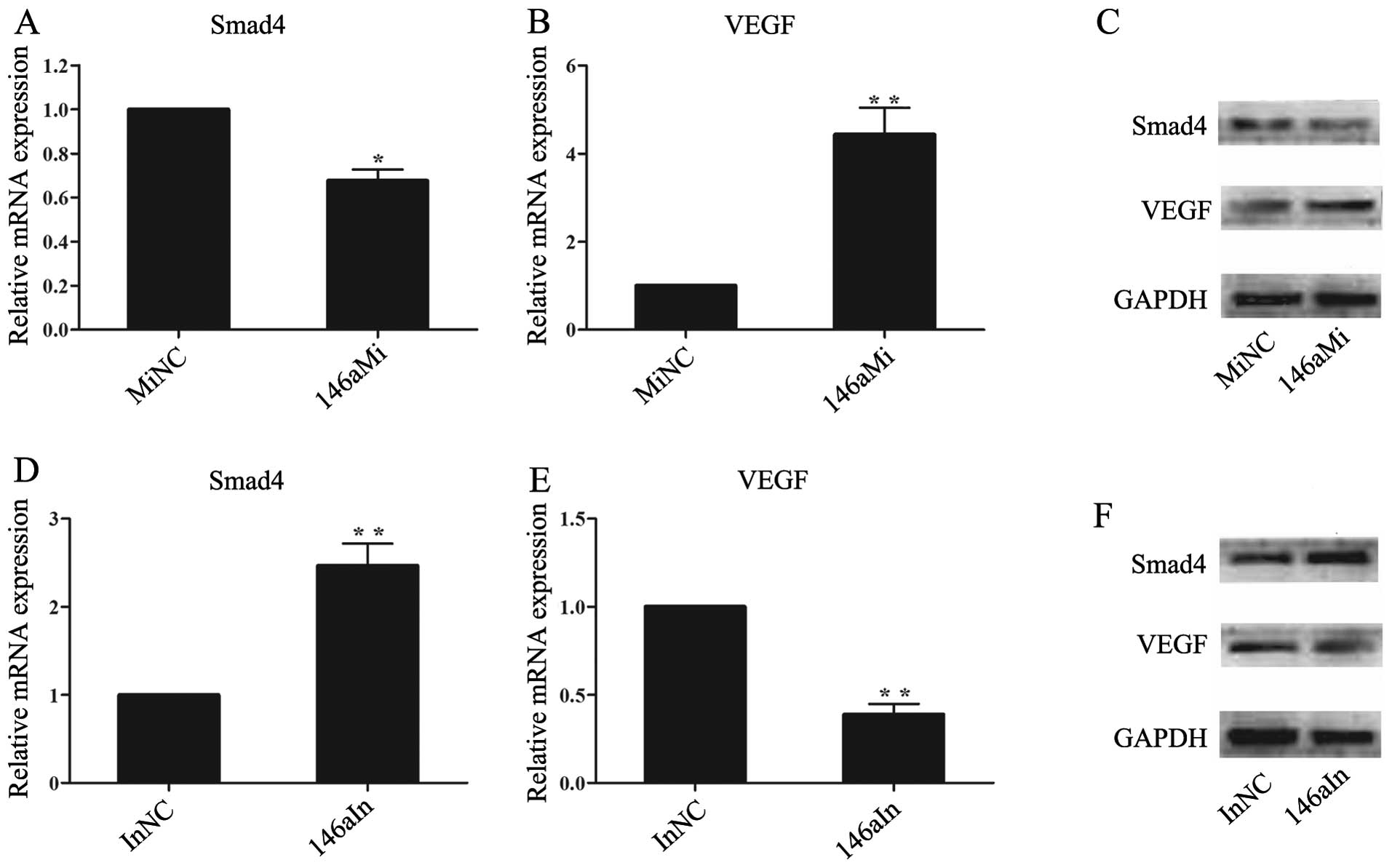
miR-146a targets Smad4 through a seed
site in the 3′-UTR of Smad4 mRNA
To determine whether miR-146a regulates the
expression of Smad4 and VEGF, we transfected the human chondrocytes
with miR-146a mimics (146aMi), negative control mimics (MiNC),
miR-146a inhibitors (146aIn) or negative control inhibitors (InNC).
The results indicated that miR-146a regulates the expression of
Smad4 and VEGF in an opposite manner. The overexpression of
miR-146a inhibited Smad4 expression and stimulated the VEGF mRNA
and protein levels (Fig. 5A–C).
By contrast, the knockdown of miR-146a by miR-146a inhibitor
increased Smad4 expression and inhibited the VEGF mRNA and protein
levels in the human chondrocytes (Fig. 5D–F).
Using miRNA target prediction software as previously
described (43), we examined the
potential targets of miR-146a by searching the PicTar and miRanda,
as well as the TargetScan databases. After consulting the
microarray results (data not shown), among the candidate targets,
we identified a potential miR-146a binding sequence in the 3′-UTR
of Smad4, which contains a putative region that matches the seed
sequence of miR-146a (Fig. 6A).
Furthermore, to determine whether Smad4 is indeed the target of
miR-146a through this seed sequence, we constructed luciferase
reporter plasmids harboring the wild-type 3′-UTR and the mutant
3′-UTR (Fig. 6A). While
luciferase activity of the mutant 3′-UTR reporter was not
statistically significant between the miR-146a mimic and the
negative control group (MiNC), the reporter luciferase activity of
the wild-type 3′-UTR was significantly inhibited by miR-146a in the
miR-146a mimic group (146aMi) compared with the negative control
group (MiNC) (Fig. 6B). The
luciferase activity of the wild-type 3′-UTR reporter was
significantly increased in the miR-146a inhibitor
(146aIn)-transfected cells compared with the inhibitor negative
control group (InNC)-transfected cells, and this increase was
markedly reduced in the mutant 3′-UTR (Fig. 6C). In summary, Smad4 is a direct
target of miR-146a.
Effects of mechanical pressure injury and
miR-146a on apoptosis of human chondrocytes
The number of apoptotic cells was counted and
compared between the non-loaded normal human chondrocytes and the
chondrocytes which were exposed to 10 MPa of mechanical pressure
for 60 min by flow cytometry at 8, 12, 24 or 48 h after the injury
was sustained. The percentage of apoptotic chondrocytes loaded with
10 MPa of pressure for 60 min significantly increased in a
time-dependent manner (P<0.01) (Fig. 7A).
Since mechanical pressure injury stimulates
apoptosis and the expression levels of miR-146a in chondrocytes, we
examined whether the expression of miR-146a affects chondrocyte
apoptosis. The overexpression of miR-146a in the chondrocytes
induced a significant increase in the percentage of apoptotic
chondrocytes at 24 h after transfection (P<0.01) (Fig. 7B), and markedly increased the
percentage of apoptotic chondrocytes exposed to mechanical pressure
(10 Mpa, 60 min, injured at 12 h after transfection) at 12 h after
the injury was sustained (P<0.05) (Fig. 7B). These results indicate that
miR-146a plays a role in mediating mechanical injury-induced
apoptosis in human chondrocytes.
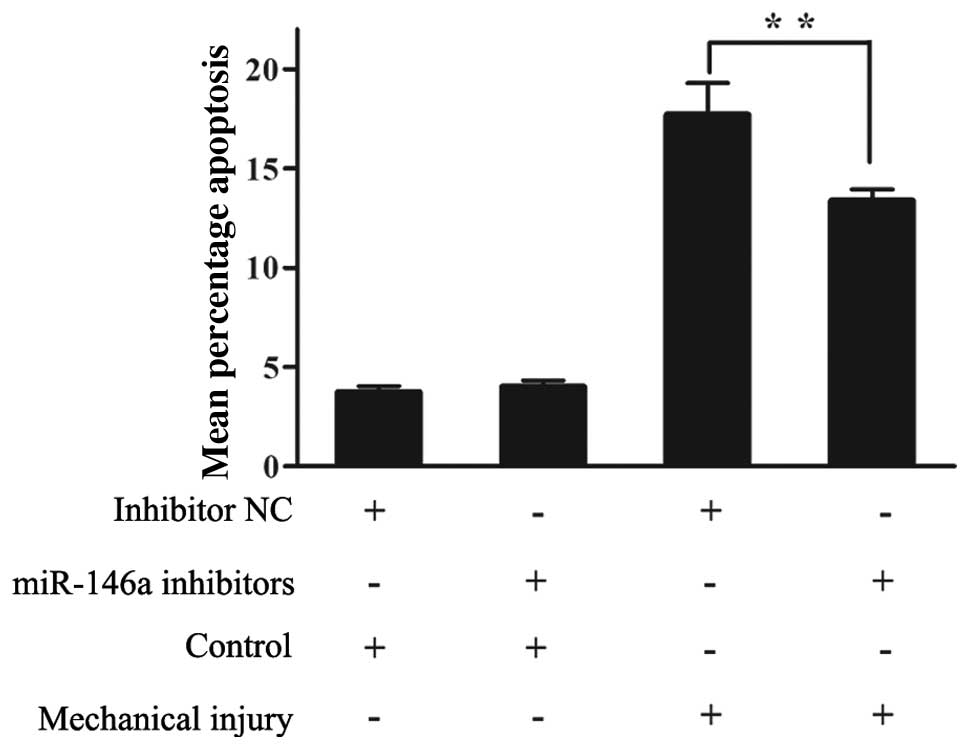
Mechanical pressure injury regulates
Smad4 and VEGF expression and chondrocyte apoptosis through
miR-146a in human chondrocytes
To demonstrate the role of miR-146a in mediating
mechanical injury sustained by chondrocytes, we used miR-146a
inhibitor to block its expression in human chondrocytes. Human
chondrocytes were loaded with mechanical pressure (10 Mpa, 60 min)
12 h following transfection with miR-146a inhibitor. The expression
levels of miR-146a, Smad4 and VEGF were monitored by RT-qPCR and
western blot analysis at 12 h after the injury was sustained. The
knockdown of miR-146a with the inhibitor significantly suppressed
the upregulation of miR-146a expression induced by mechanical
injury (Fig. 8A). Transfection
with the miR-146a inhibitor significantly increased Smad4 mRNA
expression, while the Smad4 mRNA levels were inhibited following
mechanical injury (Fig. 8B).
While mechanical injury markerly increased the VEGF mRNA levels,
transfection with miR-146a inhibitor reversed this effect (Fig. 8C). The knockdown of endogenous
miR-146a induced similar effects on Smad4 and VEGF protein levels
as on their mRNA levels following mechanical injury (Fig. 8D). Compared with the mechanically
injured chondrocytes transfected with the negative control
inhibitor, the percentage of apoptotic chondrocytes transfected
with the miR-146a inhibitor decreased 12 h after the injury was
sustained (Fig. 9). These results
indicte that miR-146a is involved in human chondrocyte apoptosis in
response to mechanical injury by regulating Smad4 and VEGF
expression.
 | Figure 8The inhibition of miR-146a attenuates
the downregulation of Smad4, the upregulation of VEGF, and the
increase in chondrocyte apoptosis after mechanical injury.
Chondrocytes were transfected with miR-146a inhibitors and loaded
with or without mechanical pressure (10 Mpa, 60 min). (A) Compared
with mechanically injured chondrocytes transfected with inhibitor
negative control, miR-146a inhibitors decreased the upregulation of
miR-146a after mechanical injury (**P<0.01; bars,
means ± SD; n=3). (B) Compared with mechanically injured
chondrocytes transfected with inhibitor negative control, the
downregulation of Smad4 mRNA expression was markedly increased by
miR-146a inhibitors after mechanical injury
(**P<0.01; bars, means ± SD; n=3). (C) Compared with
mechanically injured chondrocytes transfected with inhibitor
negative control, VEGF expression was markerdly reduced by miR-146a
inhibitors after mechanical injury (**P<0.01; bars,
means ± SD; n=3). (D) Compared with mechanically injured
chondrocytes transfected with inhibitor negative control, miR-146a
inhibitors counteracted the downregulation of Smad4 and
upregulation of VEGF at the protein level after mechanical
injury. |
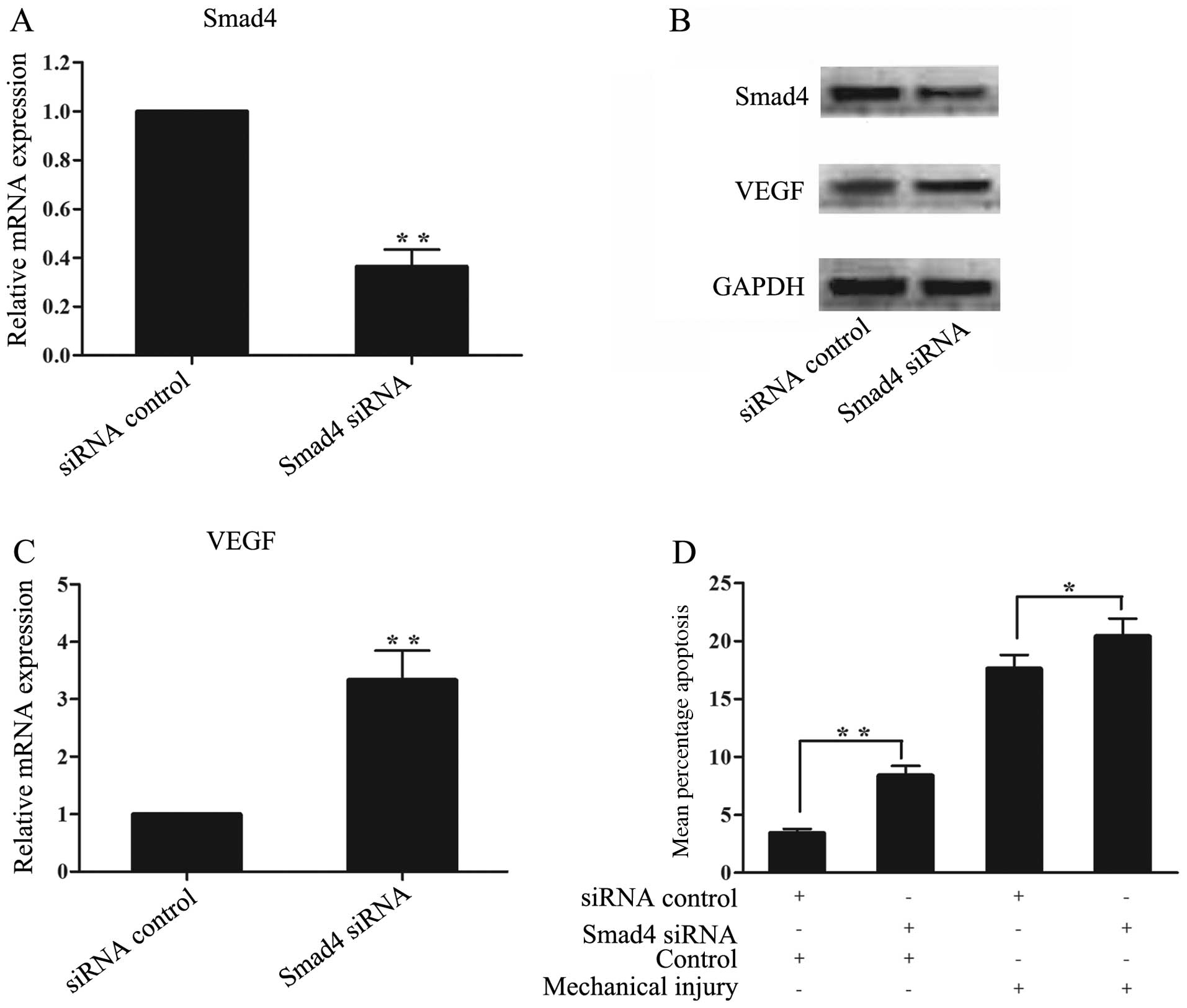
Mechanical pressure injury upregulates
VEGF expression and chondrocyte apoptosis through Smad4 in human
chondrocytes
To determine whether miR-146a mediates the
upregulation of VEGF and chondrocyte apoptosis through Smad4
following mechanical pressure injury, the chondrocytes were
transfected with Smad4 siRNA to inhibit Smad4 expression.
Transfection with Smad4 siRNA reduced the levels of both Smad4 mRNA
(Fig. 10A) and protein (Fig. 10B). The knockdown of Smad4
significantly increased the VEGF mRNA and protein levels (Fig. 10B and C), and induced a marked
increase in the percentage of apoptotic cells in the non-loaded
chondrocytes, at 24 h after transfection (P<0.01) (Fig. 10D). Compared with the
mechanically injured chondrocytes transfected with control siRNA,
the inhibition of Smad4 markedly increased the percentage of
apoptotic cells (10 Mpa, 60 min, injured at 12 h after
transfection) at 12 h after the injury was sustained (P<0.05)
(Fig. 10D). These results
indicate that Smad4 mediates the upregulation of VEGF and
chondrocyte apoptosis.
Discussion
Blunt mechanical force injury sustained by the
articular cartilage, often results from accidents or sports
injuries (44). Mechanical force
injury sustained by chondrocytes is associated with local
inflammatory reactions and represents a major risk factor for the
development of OA (45). In the
present study, we developed a novel chondrocyte model of
biomechanically defined mechanical pressure-induced human
mechanical injury for in vitro studies, and demonstrate for
the first time that miR-146a is upregulated by
experimentally-induced chondrocyte mechanical injury in this model.
To the best of our knowledge, this is the first time that miR-146a
is identified as a mechano-responsive miRNA in chondrocytes.
However, it remains to be determined whether miR-146a is responsive
to the overload of mechanical stimuli in addition to the gene
expression of catabolic and anabolic markers and the release of
proinflammatory mediators.
In response to overloading external stimuli, cells
are forced to die in different ways (46,47). In animal and human cartilage
injury models, mechanical stimuli represent important regulators of
chondrocyte function and induce mediators of inflammation and
chondrocyte death (48–51). In the pathological process of
chondrocyte mechanical injury, parts of damaged chondrocytes
undergo necrosis in the early post-injury phase. Parts of the
remaining demaged chondrocytes are prone to apoptosis (12,52). A number of studies have shown that
the rate of chondrocyte apoptosis is increased in OA cartilage and
have authenticated the role of apoptosis in the pathogenesis of OA
(9,11,52–55). In the present study, chondrocyte
viability significantly decreased and the percentage of apoptotic
chondrocytes increased, separately, in a time-dependent manner
following the exposure of chondrocytes to mechanical pressure.
However, the process of programmed cell death occurs
through the activation of the caspase cascade, and may be blocked
if one of the proteins involved in executing apoptosis is
genetically impaired or chemically inhibited, or if the apoptotic
machinery is not properly operated under specific conditions, such
as ischemia and microbial infection, which suggests possible
targets for novel therapeutic strategies (56). Thus, modulation of the mechanisms
mediated by substances inducing apoptosis is being considered as a
novel strategy for the treatment of OA. Our study focused on
miR-146a, Smad4 and VEGF following the screening of differentially
expressed genes and miRNAs. Mechanical pressure injury increased
the expression levels of miR-146a and VEGF and decreased the levels
of Smad4 in the chondrocytes in a time-dependent manner. The
results revealed that the expression levels of Smad4 were inversely
related to the miR-146a and VEGF levels. Our follow-up experiments
suggested that Smad4 is a direct target of miR-146a for
post-transcriptional regulation. Furthermore, our data suggest that
miR-146a regulates chondrocyte apoptosis by inhibiting Smad4. Yet,
the mechanisms through which Smad4 reduces chondrocyte apoptosis
remain unknown. It has been shown previously that Smad4 is a key
mediator in transmitting signals from TGF-β (57). Furthermore, extracellular
signal-regulated protein kinases 1 and 2 (ERK1/2) are cental
members of the mitogen-activated protein kinase superfamily that
can mediate cell proliferation and apoptosis (58,59). Moreover, previous studies have
shown that the TGF-β stimulation of ERK1/2 phosphorylation is
independent of Smad4 (60). The
knockdown of Smad4 by miR-146a may thus block the activation of
ERK1/2 to regulate chondrocyte apoptosis.
Mechanical signals are important for normal
cartilage to maintain tissue integrity and homeostasis (61,62). Chondrocytes respond to changes in
the levels of pro-inflammatory mediators and mechanical signals in
OA (63,64). Pro-inflammatory mediators inhibit
homeostatic mechanisms and supress cartilage repair and chondrocyte
viability. However, the physiological levels of mechanical forces
induce matrix synthesis and chondrocyte proliferation (65). Previous studies have suggested
that VEGF, an important synovial and cartilage vascularization
factor (66), appears to also be
involved in the process of OA (67), and can be induced by mechanical
forces to ERK1/2 activation for sustaining the effects of
mechanical signals for mechanisms underlying reparative actions. In
our further observation, the data indicated that the upregulation
of VEGF induced by miR-146a overexpression is mediated by Smad4 in
mechanically injured chondrocytes. These reuslts are consistent
with those of previous studies, showing that Smad4 inhibits VEGF
expression and suppresses tumorigenicity through the inhibition of
angiogenesis in human pancreatic and gastrointestinal carcinoma
cells (68–70). Of note, while the miR-146a
inhibitor or Smad4 siRNA markerly affected the mechanical injury
regulation of VEGF, the inhibition of miR-146a or Smad4 did not
completely counteract the induction of VEGF in response to the
overloading mechanical. This suggests that, in addition to miR-146a
and Smad4, other factors are involved in mediating the mechanical
injury regulation of VEGF and Smad4. Evidence suggests that
chondrocytic mechanosensing is competent of recognizing and
esponding to signals of various intensities to differentially
mediate cartilage repair and pathologies (71). Furthermore, mechanical injury
stimuli includes plural mechanical signals. Moreover, singleness
mechanical signal-stimulates activation of cells is a complex rapid
process and leads to the activation of multiple intracellular
signaling cascades, flow channels and genes (72–74). We speculate that the induction of
VEGF by mechanical injury may partially depend on an unknown
activation of cells by some mechanical signal.
The results of this study demonstrate that miR-146a
is overexpressed in an experimental chondrocyte model of human
mechanical injury, accompanied by the upregulation of VEGF and the
downregulation of Smad4 in vitro. miR-146a is involved in
human chondrocyte apoptosis in response to mechanical injury, and
may contribute to the mechanical injury sustained by chondrocytes
and the pathogenesis of OA by increasing the levels of VEGF and
damaging the TGF-β signaling pathway through the targeted
inhibition of Smad4 in human chondrocytes. These data may provide a
novel signaling cascade that links miR-146a-mediated mechanical
injury stimuli to Smad4-dependent cell apoptosis in human
chondrocytes through a mechanism involving TGF-β, ERK1/2 and VEGF,
and raise the possibility that miR-146a may be a therapeutic target
for the treatment of OA.
Acknowledgements
The authors wish to thank Yunyan Liu, Yanhua Wen and
Qiong Ma for providing excellent technical assistance.
References
|
1
|
Ashford S and Williard J: Osteoarthritis:
A review. Nurse Pract. 39:1–8. 2014. View Article : Google Scholar
|
|
2
|
Anderson DD, Chubinskaya S, Guilak F, et
al: Post-traumatic osteoarthritis: improved understanding and
opportunities for early intervention. J Orthop Res. 29:802–809.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Martin JA and Buckwalter JA:
Post-traumatic osteoarthritis: the role of stress induced
chondrocyte damage. Biorheology. 43:517–521. 2006.PubMed/NCBI
|
|
4
|
Seol D, McCabe DJ, Choe H, et al:
Chondrogenic progenitor cells respond to cartilage injury.
Arthritis Rheum. 64:3626–3637. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Conde J, Scotece M, Gomez R, Lopez V,
Gomez-Reino JJ and Gualillo O: Adipokines and osteoarthritis: novel
molecules involved in the pathogenesis and progression of disease.
Arthritis. 2011:2039012011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hogrefe C, Joos H, Maheswaran V, Durselen
L, Ignatius A and Brenner RE: Single impact cartilage trauma and
TNF-alpha: interactive effects do not increase early cell death and
indicate the need for bi-/multidirectional therapeutic approaches.
Int J Mol Med. 30:1225–1232. 2012.
|
|
7
|
Joos H, Hogrefe C, Rieger L, Durselen L,
Ignatius A and Brenner RE: Single impact trauma in human
early-stage osteoarthritic cartilage: implication of prostaglandin
D2 but no additive effect of IL-1β on cell survival. Int J Mol Med.
28:271–277. 2011.PubMed/NCBI
|
|
8
|
Leucht F, Durselen L, Hogrefe C, et al:
Development of a new biomechanically defined single impact rabbit
cartilage trauma model for in vivo-studies. J Invest Surg.
25:235–241. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Heraud F, Heraud A and Harmand MF:
Apoptosis in normal and osteoarthritic human articular cartilage.
Ann Rheum Dis. 59:959–965. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Colwell CW Jr, D’Lima DD, Hoenecke HR, et
al: In vivo changes after mechanical injury. Clin Orthop Relat Res.
391(Suppl): S116–S123. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
D’Lima DD, Hashimoto S, Chen PC, Colwell
CW Jr and Lotz MK: Human chondrocyte apoptosis in response to
mechanical injury. Osteoarthritis Cartilage. 9:712–719.
2001.PubMed/NCBI
|
|
12
|
Tew SR, Kwan AP, Hann A, Thomson BM and
Archer CW: The reactions of articular cartilage to experimental
wounding: role of apoptosis. Arthritis Rheum. 43:215–225. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
D’Lima DD, Hashimoto S, Chen PC, Lotz MK
and Colwell CW Jr: Cartilage injury induces chondrocyte apoptosis.
J Bone Joint Surg Am. 83-A(Suppl 2): 19–21. 2001.PubMed/NCBI
|
|
14
|
Saito Y, Saito H, Liang G and Friedman JM:
Epigenetic alterations and microRNA misexpression in cancer and
autoimmune diseases: a critical review. Clin Rev Allergy Immunol.
Dec 21–2013.(Epub ahead of print).
|
|
15
|
Bartel DP: MicroRNAs: target recognition
and regulatory functions. Cell. 136:215–233. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Taganov KD, Boldin MP, Chang KJ and
Baltimore D: NF-kappaB-dependent induction of microRNA miR-146, an
inhibitor targeted to signaling proteins of innate immune
responses. Proc Natl Acad Sci USA. 103:12481–12486. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Taganov KD, Boldin MP and Baltimore D:
MicroRNAs and immunity: tiny players in a big field. Immunity.
26:133–137. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tsai CY, Allie SR, Zhang W and Usherwood
EJ: MicroRNA miR-155 affects antiviral effector and effector Memory
CD8 T cell differentiation. J Virol. 87:2348–2351. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Chiyomaru T, Enokida H, Tatarano S, et al:
miR-145 and miR-133a function as tumour suppressors and directly
regulate FSCN1 expression in bladder cancer. Br J Cancer.
102:883–891. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Murphy AJ, Guyre PM and Pioli PA:
Estradiol suppresses NF-kappa B activation through coordinated
regulation of let-7a and miR-125b in primary human macrophages. J
Immunol. 184:5029–5037. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Tili E, Michaille JJ, Cimino A, et al:
Modulation of miR-155 and miR-125b levels following
lipopolysaccharide/TNF-alpha stimulation and their possible roles
in regulating the response to endotoxin shock. J Immunol.
179:5082–5089. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Williams AE, Perry MM, Moschos SA,
Larner-Svensson HM and Lindsay MA: Role of miRNA-146a in the
regulation of the innate immune response and cancer. Biochem Soc
Trans. 36:1211–1215. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Miyaki S and Asahara H: Macro view of
microRNA function in osteoarthritis. Nat Rev Rheumatol. 8:543–552.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ceribelli A, Nahid MA, Satoh M and Chan
EK: MicroRNAs in rheumatoid arthritis. FEBS Lett. 585:3667–3674.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ammari M, Jorgensen C and Apparailly F:
Impact of microRNAs on the understanding and treatment of
rheumatoid arthritis. Curr Opin Rheumatol. 25:225–233. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Goldring MB and Marcu KB: Epigenomic and
microRNA-mediated regulation in cartilage development, homeostasis,
and osteoarthritis. Trends Mol Med. 18:109–118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Okuhara A, Nakasa T, Shibuya H, et al:
Changes in microRNA expression in peripheral mononuclear cells
according to the progression of osteoarthritis. Mod Rheumatol.
22:446–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu C, Chen WP and Wang XH: MicroRNA in
osteoarthritis. J Int Med Res. 39:1–9. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jones SW, Watkins G, Le Good N, et al: The
identification of differentially expressed microRNA in
osteoarthritic tissue that modulate the production of TNF-alpha and
MMP13. Osteoarthritis Cartilage. 17:464–472. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Iliopoulos D, Malizos KN, Oikonomou P and
Tsezou A: Integrative microRNA and proteomic approaches identify
novel osteoarthritis genes and their collaborative metabolic and
inflammatory networks. PLoS One. 3:e37402008. View Article : Google Scholar
|
|
31
|
Diaz-Prado S, Cicione C, Muinos-Lopez E,
et al: Characterization of microRNA expression profiles in normal
and osteoarthritic human chondrocytes. BMC Musculoskelet Disord.
13:1442012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wang JH, Shih KS, Wu YW, Wang AW and Yang
CR: Histone deacetylase inhibitors increase microRNA-146a
expression and enhance negative regulation of interleukin-1beta
signaling in osteoarthritis fibroblast-like synoviocytes.
Osteoarthritis Cartilage. 21:1987–1996. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Yamasaki K, Nakasa T, Miyaki S, et al:
Expression of microRNA-146a in osteoarthritis cartilage. Arthritis
Rheum. 60:1035–1041. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Nakasa T, Miyaki S, Okubo A, et al:
Expression of microRNA-146 in rheumatoid arthritis synovial tissue.
Arthritis Rheum. 58:1284–1292. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bhaumik D, Scott GK, Schokrpur S, Patil
CK, Campisi J and Benz CC: Expression of microRNA-146 suppresses
NF-kappaB activity with reduction of metastatic potential in breast
cancer cells. Oncogene. 27:5643–5647. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ma Baoan and Jin Lei: Multifunctional
constant-temperature high pressure hydrostatic pressure loading
device in in-vitro cell culture. China, utility model patent No. CN
203229539 U. Filed May 16, 2013; issued October 9, 2013.
|
|
37
|
Hashimoto S, Nishiyama T, Hayashi S, et
al: Role of p53 in human chondrocyte apoptosis in response to shear
strain. Arthritis Rheum. 60:2340–2349. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Moon MH, Jeong JK, Lee YJ, Seol JW and
Park SY: Sphingosine-1-phosphate inhibits interleukin-1β-induced
inflammation in human articular chondrocytes. Int J Mol Med.
30:1451–1458. 2012.
|
|
39
|
Takebe K, Nishiyama T, Hayashi S, et al:
Regulation of p38 MAPK phosphorylation inhibits chondrocyte
apoptosis in response to heat stress or mechanical stress. Int J
Mol Med. 27:329–335. 2011.PubMed/NCBI
|
|
40
|
Storch A, Burkhardt K, Ludolph AC and
Schwarz J: Protective effects of riluzole on dopamine neurons:
involvement of oxidative stress and cellular energy metabolism. J
Neurochem. 75:2259–2269. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Griffiths-Jones S, Saini HK, van Dongen S
and Enright AJ: miRBase: tools for microRNA genomics. Nucleic Acids
Res. 36:D154–D158. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bottoni A, Zatelli MC, Ferracin M, et al:
Identification of differentially expressed microRNAs by microarray:
a possible role for microRNA genes in pituitary adenomas. J Cell
Physiol. 210:370–377. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Lewis BP, Burge CB and Bartel DP:
Conserved seed pairing, often flanked by adenosines, indicates that
thousands of human genes are microRNA targets. Cell. 120:15–20.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Weatherall JM, Mroczek K, McLaurin T, Ding
B and Tejwani N: Post-traumatic ankle arthritis. Bull Hosp Jt Dis.
2013. 71:104–112. 2013.
|
|
45
|
Lee JH, Fitzgerald JB, Dimicco MA and
Grodzinsky AJ: Mechanical injury of cartilage explants causes
specific time-dependent changes in chondrocyte gene expression.
Arthritis Rheum. 52:2386–2395. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Douville NJ, Zamankhan P, Tung YC, et al:
Combination of fluid and solid mechanical stresses contribute to
cell death and detachment in a microfluidic alveolar model. Lab
Chip. 11:609–619. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Levin A, Burton-Wurster N, Chen CT and
Lust G: Intercellular signaling as a cause of cell death in
cyclically impacted cartilage explants. Osteoarthritis Cartilage.
9:702–711. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Honda K, Ohno S, Tanimoto K, et al: The
effects of high magnitude cyclic tensile load on cartilage matrix
metabolism in cultured chondrocytes. Eur J Cell Biol. 79:601–609.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Fermor B, Weinberg JB, Pisetsky DS,
Misukonis MA, Banes AJ and Guilak F: The effects of static and
intermittent compression on nitric oxide production in articular
cartilage explants. J Orthop Res. 19:729–737. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Millward-Sadler SJ, Wright MO, Davies LW,
Nuki G and Salter DM: Mechanotransduction via integrins and
interleukin-4 results in altered aggrecan and matrix
metalloproteinase 3 gene expression in normal, but not
osteoarthritic, human articular chondrocytes. Arthritis Rheum.
43:2091–2099. 2000. View Article : Google Scholar
|
|
51
|
D’Lima DD, Hashimoto S, Chen PC, Colwell
CW Jr and Lotz MK: Impact of mechanical trauma on matrix and cells.
Clin Orthop Relat Res. (391 Suppl): S90–S99. 2001.PubMed/NCBI
|
|
52
|
Wenger R, Hans MG, Welter JF, Solchaga LA,
Sheu YR and Malemud CJ: Hydrostatic pressure increases apoptosis in
cartilage-constructs produced from human osteoarthritic
chondrocytes. Front Biosci. 11:1690–1695. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Islam N, Haqqi TM, Jepsen KJ, et al:
Hydrostatic pressure induces apoptosis in human chondrocytes from
osteoarthritic cartilage through up-regulation of tumor necrosis
factor-alpha, inducible nitric oxide synthase, p53, c-myc, and
bax-alpha, and suppression of bcl-2. J Cell Biochem. 87:266–278.
2002. View Article : Google Scholar
|
|
54
|
Sharif M, Whitehouse A, Sharman P, Perry M
and Adams M: Increased apoptosis in human osteoarthritic cartilage
corresponds to reduced cell density and expression of caspase-3.
Arthritis Rheum. 50:507–515. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Loening AM, James IE, Levenston ME, et al:
Injurious mechanical compression of bovine articular cartilage
induces chondrocyte apoptosis. Arch Biochem Biophys. 381:205–212.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Cho YS: Perspectives on the therapeutic
modulation of an alternative cell death, programmed necrosis
(Review). Int J Mol Med. 33:1401–1406. 2014.PubMed/NCBI
|
|
57
|
Liang W, Lin M, Li X, et al: Icariin
promotes bone formation via the BMP-2/Smad4 signal transduction
pathway in the hFOB 1.19 human osteoblastic cell line. Int J Mol
Med. 30:889–895. 2012.PubMed/NCBI
|
|
58
|
Zhang XM, Huang GW, Tian ZH, Ren DL and
Wilson JX: Folate stimulates ERK1/2 phosphorylation and cell
proliferation in fetal neural stem cells. Nutr Neurosci.
12:226–232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Mebratu Y and Tesfaigzi Y: How ERK1/2
activation controls cell proliferation and cell death: Is
subcellular localization the answer? Cell Cycle. 8:1168–1175. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Imamichi Y, Waidmann O, Hein R,
Eleftheriou P, Giehl K and Menke A: TGF beta-induced focal complex
formation in epithelial cells is mediated by activated ERK and JNK
MAP kinases and is independent of Smad4. Biol Chem. 386:225–236.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Zuscik MJ, Hilton MJ, Zhang X, Chen D and
O’Keefe RJ: Regulation of chondrogenesis and chondrocyte
differentiation by stress. J Clin Invest. 118:429–438. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Chiquet M, Gelman L, Lutz R and Maier S:
From mechanotransduction to extracellular matrix gene expression in
fibroblasts. Biochim Biophys Acta. 1793:911–920. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Agarwal S, Deschner J, Long P, et al: Role
of NF-kappaB transcription factors in antiinflammatory and
proinflammatory actions of mechanical signals. Arthritis Rheum.
50:3541–3548. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Dossumbekova A, Anghelina M, Madhavan S,
et al: Biomechanical signals inhibit IKK activity to attenuate
NF-kappaB transcription activity in inflamed chondrocytes.
Arthritis Rheum. 56:3284–3296. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Perera PM, Wypasek E, Madhavan S, et al:
Mechanical signals control SOX-9, VEGF, and c-Myc expression and
cell proliferation during inflammation via integrin-linked kinase,
B-Raf, and ERK1/2-dependent signaling in articular chondrocytes.
Arthritis Res Ther. 12:R1062010. View
Article : Google Scholar
|
|
66
|
Ferrara N: Vascular endothelial growth
factor: basic science and clinical progress. Endocr Rev.
25:581–611. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Carlevaro MF, Cermelli S, Cancedda R and
Descalzi Cancedda F: Vascular endothelial growth factor (VEGF) in
cartilage neovascularization and chondrocyte differentiation:
auto-paracrine role during endochondral bone formation. J Cell Sci.
113:59–69. 2000.PubMed/NCBI
|
|
68
|
Chen C, Sun MZ, Liu S, et al: Smad4
mediates malignant behaviors of human ovarian carcinoma cell
through the effect on expressions of E-cadherin, plasminogen
activator inhibitor-1 and VEGF. BMB Rep. 43:554–560. 2010.
View Article : Google Scholar
|
|
69
|
Schwarte-Waldhoff I and Schmiegel W: Smad4
transcriptional pathways and angiogenesis. Int J Gastrointest
Cancer. 31:47–59. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Schwarte-Waldhoff I, Volpert OV, Bouck NP,
et al: Smad4/DPC4-mediated tumor suppression through suppression of
angiogenesis. Proc Natl Acad Sci USA. 97:9624–9629. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Chowdhury TT, Bader DL and Lee DA: Dynamic
compression counteracts IL-1 beta-induced release of nitric oxide
and PGE2 by superficial zone chondrocytes cultured in agarose
constructs. Osteoarthritis Cartilage. 11:688–696. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Nugent GE, Aneloski NM, Schmidt TA,
Schumacher BL, Voegtline MS and Sah RL: Dynamic shear stimulation
of bovine cartilage biosynthesis of proteoglycan 4. Arthritis
Rheum. 54:1888–1896. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Nam J, Aguda BD, Rath B and Agarwal S:
Biomechanical thresholds regulate inflammation through the
NF-kappaB pathway: experiments and modeling. PLoS One. 4:e52622009.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
McNulty AL, Estes BT, Wilusz RE, Weinberg
JB and Guilak F: Dynamic loading enhances integrative meniscal
repair in the presence of interleukin-1. Osteoarthritis Cartilage.
18:830–838. 2010. View Article : Google Scholar : PubMed/NCBI
|