Introduction
Atherosclerosis (AS) is a chronic inflammatory
disease and has been identified as the leading cause of mortality
in the industrialized world. Cell apoptosis is a key event in the
progression of advanced AS (1).
Most importantly, the apoptosis of macrophage-derived foam cells,
particularly in advanced lesions where the phagocytic clearance of
apoptotic macrophages is defective, leads to the development of a
necrotic core of atherosclerotic plaque, which may contribute to
plaque instability and the majority of cardiovascular complications
(2). A number of
sero-epidemiological studies have demonstrated that human
atherosclerotic diseases are related to previous exposure to
pathogens, such as those of the human immunodeficiency virus (HIV)
(3) and Helicobacter
pylori (4). Influenza virus
RNA (5) and antigen (6) have been found in human
atherosclerotic plaques. A meta-analysis revealed that the
influenza virus triggered acute myocardial infarction (AMI) and
cardiovascular death (7), which
was consistent with the results of our previous study (8); however, the mechanisms behind this
remain elusive.
Influenza virus is a negative-stranded RNA virus,
which is recognized by Toll-like receptor (TLR)7 and retinoic
acid-inducible gene-I (RIG-I) (9,10).
TLRs comprise an evolutionarily conserved receptor family that is
capable of detecting and responding to microbial challenge
(11). Single-stranded RNA
(ssRNA) has been identified as a ligand for TLR7 (9). In fact, TLR7 has been verified to be
expressed in macrophages (12).
Imiquimod (IMQ) is a synthetic small nucleotide-like compound of
imidazoquinoline, a well known TLR7 agonist. It was first
identified as a compound that has antiviral activity and has been
successfully used for the treatment of genital warts caused by
human papilloma virus in clinical practice (13). In recent years, IMQ has been
reported to exert direct pro-apoptotic effects against various
tumor cell populations, such as oral squamous carcinoma cells
(14) and basal cell carcinoma
cells (15) by activating the
caspase-dependent mitochondrial pathway.
The endoplasmic reticulum (ER) is a membranous
organelle that plays a crucial role in cell homeostasis. Under
certain conditions, such as those caused by chemical agents,
adverse metabolic conditions induce protein misfolding and protein
assembly in the ER, leading to ER stress. Ample evidence implicates
prolonged ER stress in the progression of a number of diseases,
including AS (16). It has been
considered that ER stress is linked to cellular processes with
multiple risk factors in all stages of AS (17), and it is believed to play a
critical role in cellular apoptosis (18). The majority of research on ER
stress has focused on the unfolding protein response (UPR), a
collection of intracellular signal transduction reactions designed
to restore protein homeostasis. In UPR, ER-resident chaperones
prevent ER stress and promote cell survival (19). Among these proteins,
glucose-regulated protein (GRP)78, a member of the ER heat-shock
protein (HSP)70 family that is the best characterized ER-resident
chaperone, is indicative of UPR activation (20) and is used to determine whether the
ER stress is activated (21). On
the other hand, some mediators in ER stress have pro-apoptotic
effects; the first of these is the transcriptional activation of
the gene for C/EBP homologous protein (CHOP), which is ubiquitously
expressed at very low levels, but robustly expressed by intense
stimulation, ultimately leading to apoptosis.
In the present study, we investigated the
association between influenza virus A (IVA) infection and
macrophage viability, as well as the underlying mechanisms of cell
apoptosis. Furthermore, we investigated the synergistic role of
TLR7 activation with lipid loading in cell apoptosis. Our results
revealed that TLR7 activation either by IVA infection or IMQ
promoted apoptosis and increased cytokine secretion. In addition,
it was found that ER stress plays an important role in IMQ-induced
cell apoptosis. Pre-treatment oxidized low-density lipoprotein
(oxLDL) had a synergistic pro-apoptotic effect. These data suggest
that a high lipid not only plays a crucial role in foam cell
formation, but also acts as a ‘second hit’ to promote cell
apoptosis with other detrimental stimulations.
Materials and methods
Reagents
RPMI 1640 medium and fetal bovine serum (FBS) were
obtained from Gibco-BRL (Gaithersburg, MD, USA). Tunicamycin (TM),
dexamethasone (DEX),
3-(4,5-dimethylthiazol-2-y-1)-2,5-diphenyl-2H-tetrazolium bromide
(MTT), phorbol 12-myristate 13-acetate (PMA) and monoclonal
antibody to β-actin were from Sigma-Aldrich (St. Louis, MO, USA).
IMQ was obtained from InvivoGen (San Diego, CA, USA). Monoclonal
antibodies to GRP78 and CHOP were from Cell Signaling Technology
(Beverly, MA, USA). The horseradish peroxidase (HRP)-conjugated
anti-rabbit/mouse secondary antibodies were acquired from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Polyvinylidene
fluoride (PVDF) membranes were purchased from Millipore (Bedford,
MA, USA). TRIzol reagent was from Invitrogen (Carlsbad, CA, USA).
Interleukin (IL)-6 and monocyte chemotactic protein (MCP)-1
enzyme-linked immunosorbent assay (ELISA) kits were from R&D
Systems (Minneapolis, MN, USA). Enhanced chemiluminescence (ECL)
kits were from Thermo Scientific Pierce (Rockford, IL, USA). The
reagent kits for cDNA synthesis and quantitative reverse
transcription PCR (qRT-PCR) were from Bioneer Corp. (Daejeon,
Korea); the reagents for reverse trancription PCR (RT-PCR) reagents
were from Tiangen Biotech (Beijing, China). The Annexin V/PI
apoptosis kit was purchased from BD Biosciences (Franklin Lakes,
NJ, USA). oxLDL was obtained from Union Biological Chemistry
(Beijing, China). The reagents for western blot analysis were from
Beyotime Institute of Biotechnology (Nantong, China). The other
reagents were of analytical grade and were obtained from commercial
sources.
Cell culture and differentiation
The THP-1 monocyte cell line was purchased from the
Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China). The cells were cultured in RPMI 1640 medium
supplemented with 10% heat-inactivated FBS, 2 mM L-glutamine, 100
U/ml penicillin and 100 U/ml streptomycin, and were maintained at
(3–5)x105 cells/ml at 37°C in a humidified incubator
containing 5% CO2. The THP-1 cells were seeded at
5×105 cells/ml in tissue culture dishes, and
differentiation was induced by 5 ng/ml PMA for 48 h with
maintenance medium (2% FBS). Our observation that 5 ng/ml of PMA
stimulated the THP-1 cells to differentiate into macrophages and
did not induce undesirable gene upregulation was in accordance with
the results presented in the sudy by Park et al (22). Subsequently, the THP-1-derived
macrophages were pre-incubated with or without oxLDL (5 μg/ml) for
24 h prior to the addition of the TLR7 agonist, IMQ, for 24 h at 1
μg/ml or infected with IVA at the indicated concentrations in the
presence or absence of 1 μg/ml dexamethasone (DEX) for 24 h. TM was
used as a positive control (10 μg/ml) for 24 h. The IVA infection
model was as follows: cells were prepared in dishes and inoculated
with the indicated viral titers (1×103 TCID50/ml –
1×104 TCID50/ml) for 1 h. The inoculums were removed
after 1 h of viral adsorption, followed by washing the monolayer 3
times with PBS. The monolayer cells were maintained in a virus
isolation medium for the indicated periods of time and used for the
subsequent experiments.
Virus preparation
The influenza A strain PR/8/34 (H1N1) was provided
by the Department of Epidemiology of Harbin Medical University,
Harbin, China. The virus was propagated in MDCK cells and stored in
a −80°C freezer, following the standard procedure. To evaluate the
presence of the virus in the cell culture, we used RT-PCR and a
hemagglutination assay as previously described by Mehrbod et
al (23) and tested the
median (50%) tissue culture infective dose (TCID50).
ELISA
The cells were plated in 48-well plates at
2×104 cells per well overnight and treated with IVA or
IMQ as described above. The supernatant was harvested to detect the
levels of IL-6 and MCP-1 by ELISA. All assays were performed
according to the manufacturer’s instructions. The absorbance of the
reaction solution was measured at 450 nm using a microplate
autoreader (LUmo microplate luminometer; PHOmo microplate reader;
Autobio Diagnostics, Henan, China).
Isolation of cell proteins and western
blot analysis
The cells were seeded in 6-well plates, and after
being subjected to the different treatments, whole cell protein
extracts were prepared using RIPA lysis buffer following the
manufacturer’s instructions. The lysates were sonicated for 20 sec
and kept at 4°C for 15 min. Following 10 min of centrifugation
(12,000 × g, 4°C), the supernatant was saved as a whole-cell lysate
and measured using the Micro BCA™protein assay reagent kit (Thermo
Scientific Pierce). Loading buffer was added to adjust the lysate
followed by boiling for 10 min. The samples were resolved on
SDS-polyacrylamide gels and transferred onto PVDF membranes.
Following blocking with 5% non-fat dry milk for 1 h at room
temperature and washing with Tris-buffered saline with 0.05%
Tween-20 (TBST), the PVDF membranes were incubated overnight at 4°C
with primary antibody in TBST containing 5% non-fat dry milk. The
HRP-conjugated secondary antibody (1:5,000 dilution) was
subsequently incubated with the membranes for 1 h at room
temperature and washed extensively 3 times, 8–10 min each time with
TBST at room temperature. The blots were probed using the ECL
western blot detection system and visualized using X-ray film.
RNA extraction and RT-PCR, and
qRT-PCR
Total RNA was extracted using TRIzol reagent. Equal
amounts of total RNA were reverse transcribed using a cDNA kit
following the manufacturer’s instructions. The primers used for PCR
are listed in Table I. For
RT-PCR, initial denaturation was performed at 95°C for 3 min,
followed by 40 cycles of denaturation at 94°C for 20 sec, then
annealing at 60°C for 30 sec and extension at 72°C for 30 sec, and
a final extension at 72°C for 10 min. The PCR products were
visualized after electrophoresis on a 2% agarose gel containing
ethidium bromide. qPCR was performed using Bioneer Corporation
SYBR-Green qRT-PCR Master Mix kits. The incubation conditions were
as follows: 95°C for 20 sec, followed by 40 cycles of 15 sec at
95°C, annealing/extension for 45 sec, at 60°C. The data were
calculated using the 2−ΔΔCt method and normalized
against GAPDH. For each sample, qRT-PCR was performed in triplicate
for both the target genes and GAPDH control.
 | Table IPrimers used for RT-PCR and
qRT-PCR. |
Table I
Primers used for RT-PCR and
qRT-PCR.
| Target mRNA | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
| M1 |
TTCTAACCGAGGTCGAAAC |
AAGCGTCTACGCTGCAGTCC |
| GRP78 (RT-PCR) |
GAACGTCTGATTGGCGATGC |
TCTTTGGTTGCTTGGCGTTG |
| GRP78
(qRT-PCR) |
CCCGAGAACACGGTCTTTGA |
TTCAACCACCTTGAACGGCA |
| CHOP (RT-PCR) |
CACCACTCTTGACCCTGCTT |
CTTTCTCCTTCATGCGCTGC |
| CHOP (qRT-PCR) |
GCTCAGGAGGAAGAGGAGGA |
CTCCTTCATGCGCTGCTTTC |
| TLR7 |
ATTGCCCTCGTTGTTATA |
TTCCTGGAGTTTGTTGAT |
| GAPDH |
GACCTGACCTGCCGTCTA |
AGGAGTGGGTGTCGCTGT |
| β-actin |
CCCAGCACAATGAAGATCAAGATCAT |
ATCTGCTGGAAGGTGGACAGCGA |
MTT assay
The THP-1 cells were seeded at 5×103
cells per well in 96-well flat-bottomed plates and incubated in 2%
FBS-supplemented 1640 medium and treated with 5 ng/ml PMA for 48 h.
Following treatment, MTT solution was added to a final
concentration of 0.5 mg/ml, and the cells were incubated in a
CO2 incubator at 37°C for 4 h. After discarding the
supernatant, the reduced MTT was solubilized in 150 μl per well of
dimethylsulfoxide (DMSO), and the absorbance was measured at 490 nm
using a PHOmo microplate reader. Cell viability was expressed as
the percentage of the untreated controls.
Detection of apoptosis by flow
cytometry
Annexin V-FITC/PI double-staining was used to
quantify the number of apoptotic cells by flow cytometry. Following
treatment as described above, the cells harvested and washed 3
times with PBS. The concentration was adjusted to 1×106
cells/ml, and 100 μl of cell suspension were stained with 5 μl
Annexin V and 5 μl propidium iodide (PI) for 15 min in the dark at
room temperature; the cells were then re-suspended in 400 μl
calcium-binding solution and 1×104 cells per sample were
analyzed immediately using a BD FACSCantoII flow cytometer (BD
Biosciences, Oxford, UK) and relevant software (BD FACSDiva). The
identification of the different cell population patterns was as
follows: live cells (FITC− PI−), early
apoptotic cells (FITC+ PI−), late apoptotic
cells (FITC+ PI+) and necrotic cells
(FITC− PI+).
Statistical analysis
All data are presented as the means ± SD. One-way
ANOVA was applied in order to detect statistically significant
differences. Statistically significant differences between the
treatment groups were calculated using SPSS 17.0 software for
Windows. Values of P<0.05 were considered to indicate
statistically significant differences.
Results
IVA or IMQ increases TLR7 mRNA expression
and induces IL-6 and MCP-1 secretion
The mRNA expression of the influenza matrix protein
(M1) was detected after the cells were infected with growing
concentrations of IVA for 24 h to prove that IVA can infect
THP-1-derived macrophages in our cell model of IVA infection. M1 is
a conservative protein among the influenza viruses. The results of
RT-PCR revealed that IVA infected the THP-1-derived macrophages as
M1 mRNA expression was observed in the IVA-infected groups and not
in the untreated controls (Fig.
1A).
To confirm the effects of IVA or IMQ on TLR7
expression, we examined the mRNA expression of TLR7 in macrophages
co-incubated with IVA or IMQ by RT-PCR. As shown in Fig. 1B, the mRNA expression of TLR7 was
detected in all groups. In addition, both IVA infection
(1×103 TCID50/ml) and IMQ treatment (1 μg/ml) for 24 h
increased TLR7 mRNA expression. At the same time, the production of
IL-6 and MCP-1 was determined by ELISA following treatment with IVA
or IMQ. The expression of IL-6 and MCP-1 was significantly
increased following treatment with IVA or IMQ (Fig. 1C and D). These findings indicate
that TLR7 is actually activated during IVA infection or IMQ
treatment.
Pro-apoptotic effect of IVA infection or
IMQ treatment
The THP-1 macrophages were co-incubated with
increasing IVA concentrations (1×103, 2×103,
5×103, 1×104 TCID50/ml) as described above
and collected at 8, 24 and 48 h post-inoculation (pi). The results
revealed a pronounced decrease in cell viability in the infected
groups, as determined by MTT assay (Fig. 2A). In early infection (8 h pi),
cell viability fluctuated slightly, and the difference was not
statistically significant (P>0.05). The decline in viability was
more pronounced at 24 h pi. For further confirmation of this
phenomenon, we detected the percentage of apoptotic cells
(FITC+ PI− and FITC+
PI+) by flow cytometry at 24 h pi. Similar results were
observed, except that the number of apoptotic cells following
treatment with 1×104 TCID50/ml IVA was significantly
higher compared with the apoptosis observed at the other IVA
concentrations (Fig. 2B),
suggesting that flow cytometry was more sensitive in detecting
apoptotic cells than MTT assay.
A diversity of concentrations of IMQ (0.1, 1, 10
μg/ml) incubated with the THP-1 macrophages for 24 h induced cell
apoptosis in a dose-dependent manner (Fig. 2C and D). These results suggest
that TLR7 activation is important in cell apoptosis, which
represents a host defense mechanism to limit viral replication.
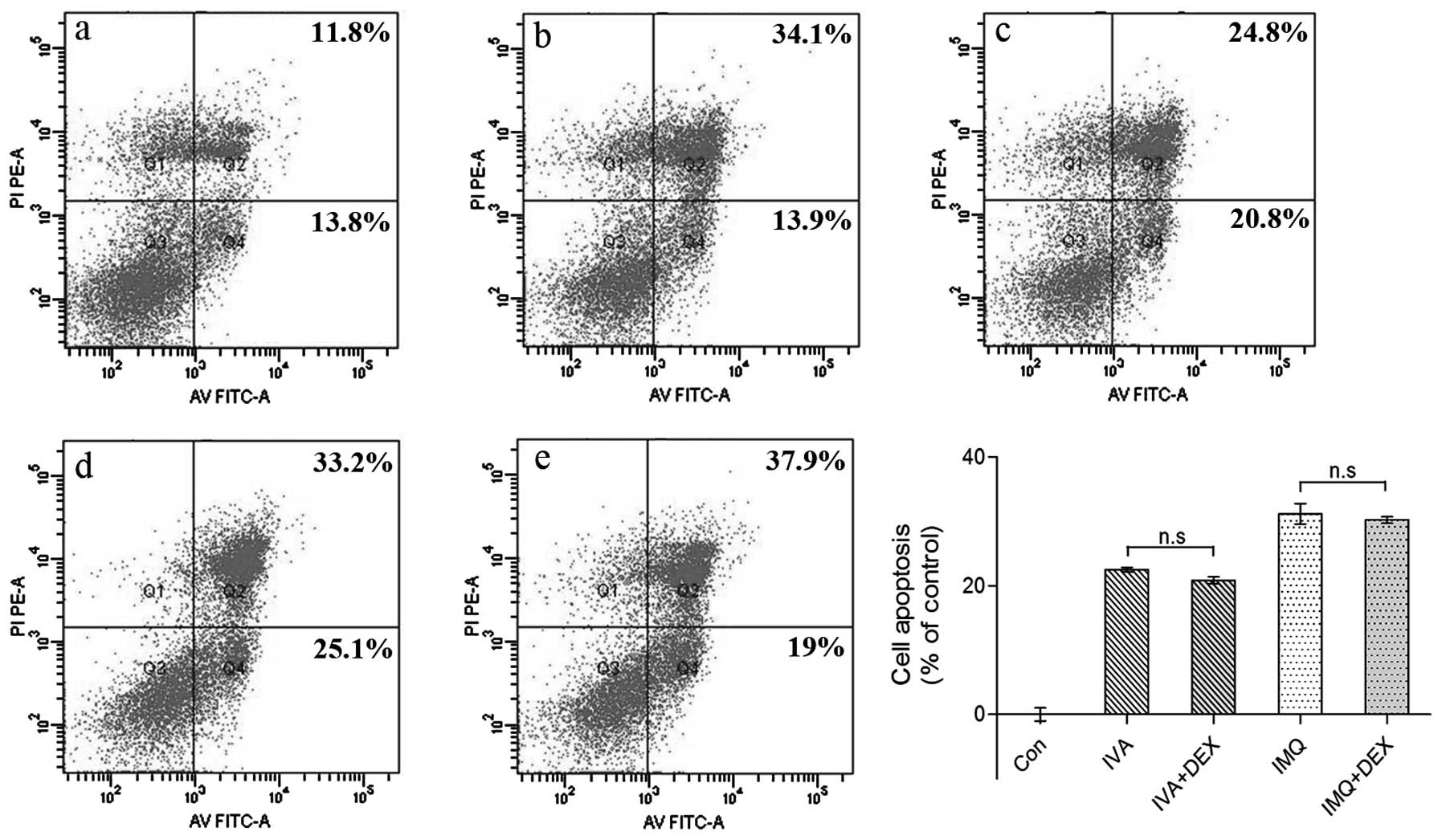
Cell apoptosis is independent of
pro-inflammatory cytokine secretion
It has been proven that IMQ has no direct antiviral
activity in vitro and is not virucidal; rather, the
antiviral activity is indirect through cytokine induction and
immune activation (24). To
investigate whether pro-inflammatory cytokines play a role in cell
apoptosis, we used DEX to decrease inflammation as described above.
As shown in Fig. 1C and D, DEX
inhibited pro-inflammatory cytokine secretion to a large extent,
although apoptosis did not decline (Fig. 3). These data indicate that cell
apoptosis is independent of pro-inflammatory cytokine secretion
in vitro.
oxLDL plays a synergistic role in cell
apoptosis with TLR7 activation
oxLDL is a potent atherogenic factor in the
progression of AS. It has been proven that a high concentration of
oxLDL (200 μg ApoB/ml) triggers a prolonged ER stress activation
that switched towards apoptosis, as supported by the increased
expression of the pro-apoptotic mediator, CHOP, and JNK (25). Our results, which suggest that
oxLDL had contradictory pro-apoptotic effects that depended on the
oxLDL concentrations, are consistent with those of Hundal et
al (26). In our study, oxLDL
at 5 μg/ml did not influence cell viability and foam cells formed
from macrophages (data not shown). To mimic IVA infection in obese
individuals who are in a sensitive pro-atherogenic situation, we
pre-treated the THP-1 macrophages with oxLDL (5 μg/ml) prior to
treatment with IVA or IMQ. As shown in Fig. 4, oxLDL pre-treatment increased
cell apoptosis as compared with IVA or IMQ treatment alone. This
phenomenon demonstrates that oxLDL has a potential pro-apoptotic
effect and acts as a ‘second hit’ to promote cell apoptosis with
other detrimental stimulations.
Upregulation of GRP78 and CHOP in IVA
infection
Previous studies have produced controversial results
on ER stress occurring in influenza infection (27–29). To investigate this, in this study,
GRP78 and CHOP mRNA were detected by RT-PCR. As shown in Fig. 5A, the mRNA level of GRP78 and CHOP
markedly increased. In agreement with this finding, the results
produced by qRT-PCR (Fig. 5B)
showed that the GRP78 levels markedly increased at first. It is
interesting to note however that the GRP78 levels declined
following treatment with 1×104 TCID50/ml IVA. In
contrast to GRP78, CHOP mRNA expression was induced in a
dose-dependent manner and paralleled with the number of apoptotic
cells (Fig. 2B). Collectively,
these data verified that IVA infection induced ER stress and that
CHOP played a role in IVA-induced apoptosis. Additionally, it is
generally accepted that GRP78 expression suppresses cell apoptosis.
In the present study, we found that GRP78 was inhibited when the
cells were exposed to strong stimuli (1×104 TCID50/ml);
this resulted in the ratio of CHOP/GRP78 becoming much higher. The
same phenomenon was observed in the cells treated with TM, a strong
inducer of ER stress. It faintly induced GRP78 (10-fold)
expression, but significantly increased CHOP (>2,000-fold) mRNA
expression (data not shown). This phenomenon indicated that
CHOP/GRP78 was a more sensitive index in predicting cell apoptosis.
We also observed an induction in GRP78 and CHOP protein levels in
the IVA-infected cell lysates, thus confirming the qRT-PCR results
(Fig. 5C).
ER stress plays a crucial role in the IMQ
pro-apoptotic effects
Having established that the caspase-dependent
mitochondrial pathway was activated in IMQ-induced cell apoptosis
(30,31), we wished to determine whether ER
stress also participates in IMQ-induced apoptosis. To examine this
hypothesis, GRP78 and CHOP expression was detected by RT-PCR,
qRT-PCR and western blot analysis. As shown in Fig. 6A and B, the levels of GRP78 and
CHOP mRNA were increased following treatment with IMQ, particularly
at a high concentration (10 μg/ml). The protein levels of GRP78 and
CHOP were consistent with the mRNA levels (Fig. 6C). These results suggest that ER
stress plays a pro-apoptotic role in IMQ treatment, particularly
with a potent stimulation.
Discussion
AS and its complications are a major threat to human
health, and there is a strong correlation between apoptotic
macrophages and plaque rupture (32). In this study, we demonstrated that
TLR7 activation aggravated ER stress and promoted apoptosis in
THP-1-derived macrophages. Collectively, cell apoptosis was
independent of pro-inflammatory cytokines. Pre-treatment with oxLDL
at a dose of 5 μg/ml aggravated apoptosis along with TLR7
activation. The mechanism responsible for this phenomenon involved
the increase in the epxression of the pro-apoptotic sensor, CHOP,
in ER stress.
As already mentioned in the ‘Introduction’, certain
pathogens, including IVA, correlate with the development of AS. In
clinical practice, during influenza epidemics, there are many
deaths and serious complications in vulnerable populations,
particularly in cold wheather (33). The American Heart Association and
American College of Cardiology recommend influenza immunization
with inactivated vaccine as part of a comprehensive secondary
prevention in individuals with coronary and other atherosclerotic
vascular diseases (34). These
findings led to the hypothesis that influenza infection may be a
predisposing factor for cardiovascular events; however, the
underlying mechanisms remain elusive. Van Lenten et al
proved that the influenza infection contributed to high-density
lipoprotein losing its anti-inflammatory properties and promoting
macrophage traffic into the arteries of mice (35,36). On the other hand, our study
demonstrated that IVA infection induced pro-inflammatory cytokine
secretion and cell apoptosis. The greatest number of apoptotic
cells was observed at 24 h pi; this result was in accordance with
the results presented in the study by Mitchell et al
(37). Therefore, the early
treatment of IVA infection is an important measure to reduce
serious cardiovascular incidents.
Several lines of evidence have suggested that IVA
infection activates the distinct ER stress pathway in different
cell types (27,28). Our data confirmed that IVA
infection induced GRP78 and CHOP expression in a dose-dependent
manner (Fig. 5). Furthermore, it
accelerated macrophage apoptosis by increasing CHOP expression.
However, the pro-inflammatory cytokines did not affect cell
apoptosis in vitro (Fig.
3). IMQ had the same effect on macrophages (e.g., pro-apoptotic
effects and pro-inflammatory cytokine secretion) as IVA. As
expected, ER stress also played a role in IMQ-induced cell
apoptosis in addition to the mitochondrial-dependent death pathway,
particularly in potent stimulation (Fig. 6). It is vague whether IMQ induced
cell apoptosis via the TLR7-dependent pathway (12,38). Our results showed that TLR7
activation promoted cell apoptosis; however, cannot reach a final
conclusion regarding this matter as we did not inhibit TLR7
activation.
Generally speaking, oxLDL is a potent atherogenic
factor in AS progression. Cells incubated with a lower cytotoxic
concentration of oxLDL (5 μg/ml), followed by TLR7 activation,
showed an increase in cell apoptosis which was more significant
than that of the cells not treated with oxLDL (Fig. 4). Two reviews have suggested that
obesity is a risk factor for influenza infection (39) and impaired immune function
(40). Smith et al
demonstrated that obesity increased the risk of death from
influenza infection in vivo (41). As mentioned above, obesity is not
only a cause of AS but also has pleiotropic effects on the
progression of AS.
As mentioned above, TLR7 activation can promote cell
apoptosis by CHOP expression, and oxLDL pre-treatment aggravates
apoptosis. Therefore, the control of influenza infection and
obesity is an important precaution for preventing AS. Our study is
not without limitations. First, we did not demonstrate the concrete
pathway of ER stress in IVA infection or IMQ treatment. Secondly,
we did not knock down TLR7 to elucidate its effect. These, however,
are our main aims for future studies.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (81241006), the China
Postdoctoral Science Foundation project of Heilongjiang Province
(LBH-Q12033) and the Postgraduate Innovation Foundation of
Heilongjiang Province (YJSC2012-200HLJ).
References
|
1
|
Laufer EM, Winkens HM, Corsten MF,
Reutelingsperger CP, Narula J and Hofstra L: PET and SPECT imaging
of apoptosis in vulnerable atherosclerotic plaques with
radiolabeled Annexin A5. Q J Nucl Mol Imaging. 53:26–34.
2009.PubMed/NCBI
|
|
2
|
Seimon T and Tabas I: Mechanisms and
consequences of macrophage apoptosis in atherosclerosis. J Lipid
Res. 50:S382–S387. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Badiou S, Thiebaut R and
Aurillac-Lavignolle V: Association of non-HDL cholesterol with
subclinical atherosclerosis in HIV-positive patients. J Infect.
57:47–54. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Oshima T, Ozono R and Yano Y: Association
of Helicobacter pylori infection with systemic inflammation
and endothelial dysfunction in healthy male subjects. J Am Coll
Cardiol. 45:1219–1222. 2005.
|
|
5
|
Gurevich VS, Pleskov VM, Levaia MV,
Bannikov AI, Mitrofanova LB and Urazgil’deeva SA: Influenza virus
infection in progressing atherosclerosis. Kardiologiia. 42:21–24.
2002.PubMed/NCBI
|
|
6
|
Haidari M, Wyde PR, Litovsky S, Vela D,
Ali M, Casscells SW and Madjid M: Influenza virus directly infects,
inflames, and resides in the arteries of atherosclerotic and normal
mice. Atherosclerosis. 208:90–96. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Warren-Gash C, Smeeth L and Hayward AC:
Influenza as a trigger for acute myocardial infarction or death
from cardiovascular disease: a systematic review. Lancet Infect
Dis. 9:601–610. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Guan X, Yang W, Sun X, Wang L, Ma B, Li H
and Zhou J: Association of influenza virus infection and
inflammatory cytokines with acute myocardial infarction. Inflamm
Res. 61:591–598. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Diebold SS, Kaisho T, Hemmi H, Akira S and
Reis e Sousa C: Innate antiviral responses by means of
TLR7-mediated recognition of single-stranded RNA. Science.
303:1529–1531. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Fukuyama S and Kawaoka Y: The pathogenesis
of influenza virus infections: the contributions of virus and host
factor. Curr Opin Immunol. 23:481–486. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cluff CW, Baldridge JR and Stöver AG:
Synthetic toll-like receptor 4 agonists stimulate innate resistance
to infectious challenge. Infect and Immun. 5:3044–3052. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
De Meyer I, Martinet W, Schrijvers DM,
Timmermans JP, Bult H and De Meyer GR: Toll-like receptor 7
stimulation by imiquimod induces macrophage autophagy and
inflammation in atherosclerotic plaques. Basic Res Cardiol.
107:2692012.PubMed/NCBI
|
|
13
|
von Krogh G, Lacey CJ, Gross G, Barrasso R
and Schneider A: European course on HPV associated pathology:
guidelines for primary care physicians for the diagnosis and
management of anogenital warts. Sex Transm Infect. 76:162–168.
2000.PubMed/NCBI
|
|
14
|
Ahn MY, Kwon SM and Cheong HH: Toll like
receptor 7 agonist, Imiquimod, inhibits oral squamous carcinoma
cells through apoptosis and necrosis. J Oral Pathol Med.
41:540–546. 2012.PubMed/NCBI
|
|
15
|
Huang SW, Chang CC and Lin CC: Mcl-1
determines the imiquimod-induced apoptosis but not
imiquimod-induced autophagy in skin cancer cells. J Dermatol Sci.
7:170–178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ozcan L and Tabas I: Role of endoplasmic
reticulum stress in metabolic disease and other disorder. Annu Rev
Med. 63:317–328. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hotamisligil GS: Endoplasmic reticulum
stress and atherosclerosis. Nat Med. 16:396–399. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kedi X, Ming Y, Yongping W, Yi Y and
Xiaoxiang Z: Free cholesterol overloading induced smooth muscle
cells death and activated both ER-and mitochondrial-dependent death
pathway. Atherosclerosis. 207:123–130. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yao S, Zong C, Zhang Y, Sang H, Yang M,
Jiao P, Fang Y, Yang N, Song G and Qin S: Activating transcription
factor 6 mediates oxidized LDL-induced cholesterol accumulation and
apoptosis in macrophages by up-regulating CHOP expression. J
Atheroscler Thromb. 20:94–107. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hoozemans JJ, Veerhuis R, Van Haastert ES,
Rozemuller JM, Baas F, Eikelenboom P and Scheper W: The unfolded
protein response is activated in Alzheimer’s disease. Acta
Neuropathol. 110:165–172. 2005.
|
|
21
|
Lin WC, Chuang YC and Chang YS:
Endoplasmic reticulum stress stimulates p53 expression through
NF-κB activation. PLoS One. 7:e391202012.PubMed/NCBI
|
|
22
|
Park EK, Jung HS, Yang HI, Yoo MC, Kim C
and Kim KS: Optimized THP-1 differentiation is required for the
detection of responses to weak stimuli. Inflamm Res. 56:45–50.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mehrbod P, Ideris A, Omar AR, Hair-Bejo M,
Tan SW, Kheiri MT and Tabatabaian M: Attenuation of influenza virus
infectivity with herbal-marine compound (HESA-A): an in vitro study
in MDCK cells. Virol J. 9:442012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Miller RL, Meng TC and Tomai MA: The
antiviral activity of Toll-like receptor 7 and 7/8 agonists. Drug
News Perspect. 21:69–87. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Muller C, Salvayre R, Nègre-Salvayre A and
Vindis C: HDLs inhibit endoplasmic reticulum stress and autophagic
response induced by oxidized LDLs. Cell Death Differ. 18:817–828.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hundal RS, Gómez-Muñoz A, Kong JY, Salh
BS, Marotta A, Duronio V and Steinbrecher UP: Oxidized low density
lipoprotein inhibits macrophage apoptosis by blocking ceramide
generation, thereby maintaining protein kinase B activation and
Bcl-XL level. J Biol Chem. 278:24399–24408. 2003. View Article : Google Scholar
|
|
27
|
Roberson EC, Tully JE and Guala AS:
Influenza induces endoplasmic reticulum stress, caspase-12
dependent apoptosis, and c-Jun N-terminal kinase-mediated
transforming growth factor-β release in lung epithelial cells. Am J
Respir Cell Mol Biol. 46:573–581. 2012.PubMed/NCBI
|
|
28
|
Hassan IH, Zhang MS and Powers LS:
Influenza A viral replication is blocked by inhibition of the
inositol-requiring enzyme (IRE-1) stress pathway. J Biol Chem.
287:4679–4689. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Numajiri Haruki A, Naito T, Nishie T,
Saito S and Nagata K: Interferon-inducible antiviral protein MxA
enhances cell death triggered by endoplasmic reticulum stress. J
Interferon Cytokine Res. 31:847–856. 2011.PubMed/NCBI
|
|
30
|
Kim CH, Ahn JH, Kang SU, Hwang HS, Lee MH,
Pyun JH and Kang HY: Imiquimod induces apoptosis of human
melanocytes. Arch Dermatol Res. 302:301–306. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Schön M, Bong AB, Drewniok C, Herz J,
Geilen CC, Reifenberger J, Benninghoff B, Slade HB, Gollnick H and
Schön MP: Tumor-selective induction of apoptosis and the
small-molecule immune response modifier imiquimod. J Natl Cancer
Inst. 95:1138–1149. 2003.PubMed/NCBI
|
|
32
|
Halasiddappa LM, Koefeler H, Futerman AH
and Hermetter A: Oxidized phospholipids induce ceramide
accumulation in RAW 264.7 macrophages: role of ceramide synthases.
PLoS One. 8:e700022013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
The Eurowinter Group. Cold exposure and
winter mortality from ischaemic heart disease, cerebrovascular
disease, respiratory disease, and all causes in warm and cold
regions of Europe. Lancet. 349:1341–1346. 1997. View Article : Google Scholar
|
|
34
|
Davis MM, Taubert K and Benin AL:
Influenza vaccination as secondary prevention for cardiovascular
disease: a science advisory from the American Heart
Association/American College of Cardiology. Circulation.
114:1549–1553. 2006. View Article : Google Scholar
|
|
35
|
Van Lenten BJ, Wagner AC, Nayak DP, Hama
S, Navab M and Fogelman AM: High-density lipoprotein loses its
anti-inflammatory properties during acute influenza a infection.
Circulation. 103:2283–2288. 2001.PubMed/NCBI
|
|
36
|
Van Lenten BJ, Wagner AC, Anantharamaiah
GM, et al: Influenza infection promotes macrophage traffic into
arteries of mice that is prevented by D-4F, an apolipoprotein A-I
mimetic peptide. Circulation. 106:1127–1132. 2002.PubMed/NCBI
|
|
37
|
Mitchell H, Levin D, Forrest S, Beauchemin
CA, Tipper J, Knight J, Donart N, Layton RC, Pyles J, Gao P, Harrod
KS, Perelson AS and Koster F: Higher level of replication
efficiency of 2009 (H1N1) pandemic influenza virus than those of
seasonal and avian strains: kinetics from epithelial cell culture
and computational modeling. J Virol. 85:1125–1135. 2011. View Article : Google Scholar
|
|
38
|
Han JH, Lee J and Jeon SJ: In vitro
and in vivo growth inhibition of prostate cancer by the
small molecule imiquimod. Int J Oncol. 42:2087–2093. 2013.
|
|
39
|
Hur SJ, Kim DH, Chun SC and Lee SK: Effect
of adenovirus of influenza virus infection on obesity. Life Sci.
93:531–535. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Karlsson EA and Beck MA: The burden of
obesity on infectious disease. Exp Biol Med (Maywood).
235:1412–1424. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Smith AG, Sheridan PA, Harp JB and Beck
MA: Diet-induced obese mice have increased mortality and altered
immune responses when infected with influenza virus. J Nutr.
137:1236–1243. 2007.PubMed/NCBI
|