Introduction
Myocardial ischemia/reperfusion (I/R) injury refers
to cardiac dysfunction induced by the restoration of blood flow
following a period of ischemia or lack of oxygen (1). Myocardial I/R injury is a common
clinical problem and frequently found in the process of coronary
thrombolysis, coronary artery bypass graft and heart
transplantation (2–4). Cardiomyocyte apoptosis is
well-established as one of the major mechanisms for I/R-induced
injury (5,6). Currently, the majority of studies
supported that I/R-induced cardiomyocyte apoptosis was closely
associated with excessive production of reactive oxygen species
(ROS) (7,8).
ROS are chemically-reactive molecules containing the
element oxygen, including superoxide anion
(O2−), hydroxyl radical (•OH) and hydrogen
peroxide (H2O2) (9). Under physiological conditions, there
is a low concentration of ROS in the heart. They are mainly
produced as a by-product of mitochondrial oxidative
phosphorylation. However, multiple pathological conditions,
including I/R, can result in the accumulation of ROS (10,11). Mitochondrion has been reported as
the main target of ROS damage (12). By contrast, increased ROS can
directly lead to the oxidative damage of mitochondrial DNA,
membrane phospholipids and respiratory chain proteins (13,14). Increased ROS are able to enhance
the opening degree of mitochondrial permeability transition pore
(mPTP) (15). The opening of mPTP
leads to the increase in mitochondrial permeability, mitochondrial
swelling, mitochondrial membrane potential collapse and cytochrome
c release from mitochondria to cytosol, which ultimately
results in cardiomyocyte apoptosis (16). The above studies suggested that
ROS-induced mitochondrial dysfunction plays an important role in
cardiomyocyte apoptosis and a decrease in ROS production may be a
promising strategy for the inhibition of I/R-induced cardiomyocyte
apoptosis.
Picroside II (β-D-glucopyranoside,
1a,1b,2,5a,6,6a-hexahydro-6-[(4-hydroxy-3-methoxybenzoyl)oxyl]-1a-(hydroxy
methyl)oxireno[4,5]cyclopenta[1,2-c]pyran-2-yl) is a primary active
ingredient extracted from Picrorhiza scrophulariiflora
Pennell (Scrophulariaceae), which is a traditional Chinese herbal
medicine and has been extensively used in China for >1000 years
(17). In recent years, it has
been reported that picroside II has an antioxidant property. The
study by Li et al (18)
demonstrated that pretreatment of PC12 cells with picroside II
significantly prevented glutamate-induced cell apoptosis by
inhibition of ROS production. In addition, treatment with picroside
II evidently ameliorated liver damage induced by carbon
tetrachloride, D-galactosamine and acetaminophen. The
hepatoprotective effect of picroside II was associated with
scavenging free radicals and protecting normal constructions of
mitochondria membrane (19).
Recently, it has been reported that picroside II has the protective
effect on H/R-induced cardiomyocyte apoptosis (20), but the exact mechanism is not
fully understood.
The present study aimed to investigated the
important role of ROS-induced mitochondrial dysfunction in
cardiomyocyte apoptosis during I/R and the antioxidant property of
picroside II. Using the hypoxia/reoxygenation (H/R) model in the
H9c2 cardiomyocyte cell line to simulate I/R injury, the study
explored whether picroside II has a protective effect on
H/R-induced cardiomyocyte apoptosis, and whether the protective
effect is associated with ameliorating mitochondrial function by
inhibition of ROS production.
Materials and methods
Materials
H9c2 embryonic rat heart-derived cells were obtained
from Academia Sinica (Shanghai, China). Picroside II
(purity>99%) was purchased from the Chinese National Institute
for the Control of Pharmaceutical and Biological Products (Beijing,
China), and dissolved in sterile distilled water. Dulbecco’s
modified Eagle medium (DMEM) and fetal bovine serum (FBS) were
provided from Gibco RBL (Grand Island, NY, USA). Trizol reagent was
a product of Invitrogen Life Technologies (Carlsbad, CA, USA). The
First Strand cDNA Synthesis kit was purchased from MBI Fermentas,
Inc. (Vilnius, Lithuania). Cytochrome c and β-actin
antibodies were from Abcam (Cambridge, UK). The
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide
(MTT), Hoechst 33342, caspase-3 activity assay kits and reactive
oxygen species (ROS) detection kit were from Beyotime Biotechnology
(Jiangsu, China). Lactate dehydrogenase (LDH) and creatine kinase
(CK) assay kits were obtained from Nanjing Jiancheng Bioengineering
Institute (Nanjing, China).
Cells culture and treatment
H9c2 cells were cultured in DMEM supplemented with
15% (v/v) FBS, 100 U/ml penicillin and 100 μg/ml
streptomycin at 37°C in a humidified atmosphere of 5%
CO2, with medium changed every 2–3 days. The cells were
passaged regularly. When they reached 80% confluence, cells were
made quiescent by serum starvation (0.5% FBS) for 12 h, and
subsequently were treated with picroside II (100 μg/ml) for
48 h prior to H/R. For the H/R experiment, a hypoxic incubator was
used to produce an in vitro hypoxia challenge. Cells in the
hypoxic incubator were subjected to hypoxia (<0.5%
O2) using pre-conditioned hypoxic medium [8 g NaCl, 0.4
g KCl, 0.14 g CaCl2, 0.2 g
MgSO4·7H2O, 1.2 g
K2HPO4·12H2O, 0.08 g
KH2PO4 (pH 7.4)] at 37°C. Hypoxic medium was
changed to fresh DMEM medium upon initiation of reoxygenation. The
cells were subsequently cultured in the incubator under an
atmosphere of 5% (v/v) CO2 at 37°C. The selected period
of hypoxia and reoxygenation time was based on the preliminary time
course studies. The most suitable period of H/R was found to be 3 h
hypoxia followed by 12 h reoxygenation. Control cells were cultured
in normoxic conditions.
Evaluation of cell injury
Cardiomyocyte viability was measured by the MTT
quantitative colorimetric assay. After picroside II treatment and
H/R, the medium was removed and the cardiomyocytes were washed
twice with phosphate-buffered saline (PBS), subsequently 10
μl MTT solution was added to each well and incubated for an
additional 4 h at 37°C. Following this, 100 μl
dimethysulfoxide was added to dissolve the formazan product. The
absorbance at 490 nm was read on a microplate reader and the data
were expressed as a percentage of the control, which was considered
100% viable. CK and LDH, the indicators of cardiomyocyte injury,
were detected by the commercially available colorimetric assay kits
according to the manufacturer’s instructions.
Apoptosis analysis
Apoptotic cells were determined by fluorescent
microscopy using the fluorescent dye Hoechst 33342. Briefly, H9c2
cells were seeded at a density of 1×105 cells/well in
12-well plates and cultured as described above. Following picroside
II treatment and H/R, the medium was aspirated and the cells were
washed twice with PBS. Subsequently, Hoeshst 33342 solution (0.1
mg/ml) was added into each well of the 12-well plate for 20 min at
37°C in dark, followed by another three washes with PBS. Nuclear
DNA staining was observed by a fluoresence microscope at 521 nm
emission wavelength. The percent of apoptosis was expressed as a
ratio of apoptotic to total cells. Caspase-3 is a key enzyme in the
apoptosis cell-signaling cascade. The activity of caspase-3 was
measured by the commercially available colorimetric assay kit
according to the manufacturer’s instructions. The data were
expressed as a percentage of the control.
Quantitative polymerase chain reaction
(qPCR)
Following picroside II treatment and H/R, total RNA
was isolated from the H9c2 cells by the Trizol reagent and
quantified by measuring the absorbance at 260 nm. A 1 μg RNA
aliquot from each sample was reverse-transcripted to cDNA using the
M-MLV Reverse transcriptase kit. The cDNA was used for qPCR.
Quantitative analysis of mRNA expression was performed by the ABI
7300 real-time PCR system with the Power SYBR Green PCR Master Mix
kit. PCR primers were as follows: caspase-3 primers (sense,
5′-CAAGTCGATGGACTCTGGAA-3′ and anti-sense,
5′-GTACCATTGCGAGCTGACAT-3′); GAPDH primers (sense,
5′-TGGCCTCCAAGGAGTAAGAAAC-3′ and anti-sense,
5′-GGCCTCTCTCTTGCTCTCAGTATC-3′). The PCR amplification profiles
consisted of denaturation at 95°C for 10 min, followed by 40 cycles
of denaturation at 95°C for 15 sec and annealing at 60°C for 60
sec. All the amplification reactions for each sample were carried
out 4 times, and the relative expression values were normalized to
the expression value of GAPDH.
Preparation of subcellular fractions
H9c2 cells were cultured as described above.
Following picroside II treatment and H/R, the cells were collected
and the preparation of subcellular fractions was isolated using the
Cell Mitochondria Isolation kit. The method was carried out
according to the manufacturer’s instructions. Briefly, cells were
washed in PBS and centrifuged at 600 × g for 10 min at 4°C. The
supernatant was removed and the cells were resuspended in the
mitochondria isolation reagent. Following a 15 min incubation on
ice, lysates were homogenized and centrifuged at 600 × g for 10 min
at 4°C. The supernatant was subsequently centrifuged at 11,000 × g
for 10 min to isolate the mitochondria fraction in the pellet,
while the supernatant was used to isolate the cytosol. The
supernatant was subjected to centrifugation at 12,000 × g for 10
min. The resulting supernatant was designated as the cytosolic
fraction, which was used for the detection of cytochrome c.
Measurement of intracellular ROS
The levels of ROS were measured by oxidative
conversion of cell permeable 2′,7′-dichlo-rofluorescein diacetate
to fluorescent dichlorofluorescein. 2′,7′-dichlorofluorescein was
added at a final concentration of 10 μmol/l and incubated
for 20 min at 37°C. Fluorescence intensity was detected using a
fluorescence plate reader at an excitation wavelength at 488 nm and
an emission wavelength at 525 nm. The results were expressed as a
percentage of the control.
Measurement of mPTP opening
The mPTP opening of cardiomyocytes was measured with
calcein-acetoxymethylester (calcein-AM) in the presence of cobalt
chloride using the mPTP fluorescence assay kit according to the
manufacturer’s instructions. In brief, cardiomyocytes were washed
with Reagent A, and subsequently the cells were incubated with
Reagents B and C for 20 min at 37°C, prior to washing twice with
Reagent A again. Fluorescence intensity was determined by a
SprctraMax M5 microplate reader at an excitation wavelength at 488
nm and an emission wavelength at 505 nm. Fluorescence intensity was
normalized to total protein concentration in the corresponding cell
and the data were expressed as normalized relative fluorescence
units (U/mg protein).
Determination of mitochondrial membrane
potential
Mitochondrial membrane potential was determined by
the tetramethylrhodamine ethyl ester (TMRE) fluorescent dye. TMRE
is positively charged and highly permeable across the mitochondrial
membrane. Mitochondrial depolarization is able to result in the
spread of TMRE from mitochondria to cytoplasm, and enhance the
whole-cell fluorescence intensity. TMRE was added at a final
concentration of 1×10−7 mol/l and incubated in the dark
for 20 min at room temperature. Fluorescence intensity was
determined (excitation wavelength at 514 nm/emission wavelength at
590 nm). The data were expressed as a percentage of the
control.
Western blotting analysis
Protein concentration was determined by a Bradford
protein assay. Equal amounts of protein were separated by 10%
sodium dodecyl sulfate-polyacryl-amide gel electrophoresis and
transferred to a nitrocellulose membrane. After being blocked with
5% milk powder in TBST for 1 h at room temperature, the membranes
were incubated with the primary antibody for cytochrome c
(diluted 1 μg/ml, Cat. no. ab90529) and β-actin (diluted
1:1,000, Cat. no. ab1801) (both rabbit polyclonal antibodies;
Abcam) at 4°C overnight, and followed by incubation with the
corresponding horseradish peroxidase-conjugated secondary antibody
(goat anti-rabbit; (diluted 1:5,000, Cat. no. ab175773; Abcam) at
room temperature for 1 h. Detection was performed using an ECL kit
according to the manufacturer’s instructions. The results were
normalized to β-actin expression.
Statistical analysis
Data are expressed as the means ± standard error of
the mean. All the values were analyzed using analysis of variance
and the Newman-Keuls Student’s t-test. P<0.05 was
considered to indicate a statistically significant difference.
Results
Effect of picroside II on H/R-induced
cell injury
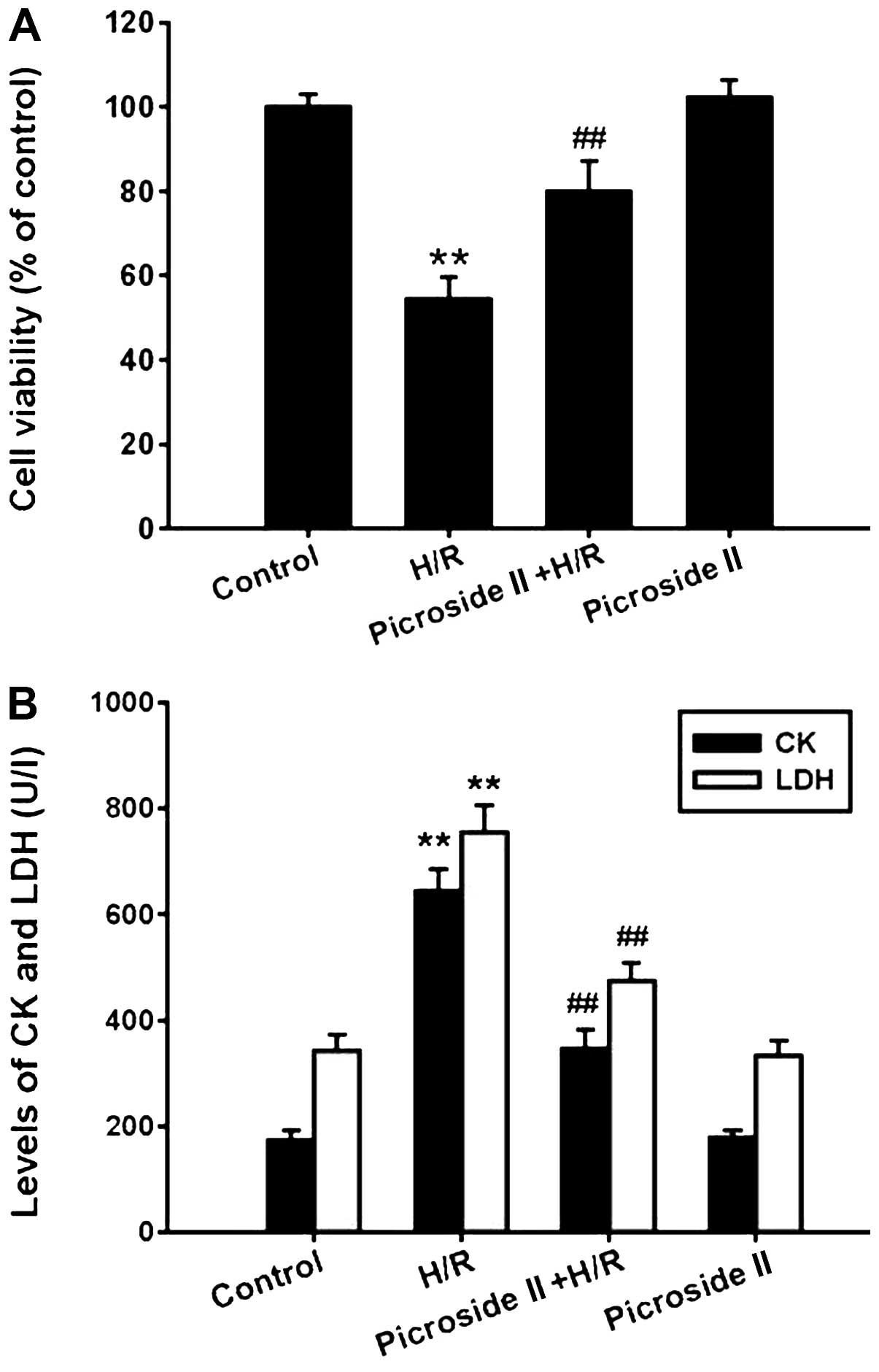
The MTT assay showed that the cell viability was
significantly decreased after hypoxia for 3 h and reoxygenation for
12 h. Pretreatment with picroside II for 48 h prior to H/R was able
to markedly inhibit the decrease in cell viability (Fig. 1A). In line with the MTT assay, the
levels of CK and LDH, which are the indicators of cardiomyocyte
injury, were significantly increased in the H/R group, which was
also inhibited by pretreatment of picroside II (Fig. 1B). However, picroside II alone had
no effect on cell viability and the production of CK and LDH.
Effect of picroside II on H/R-induced
cell apoptosis
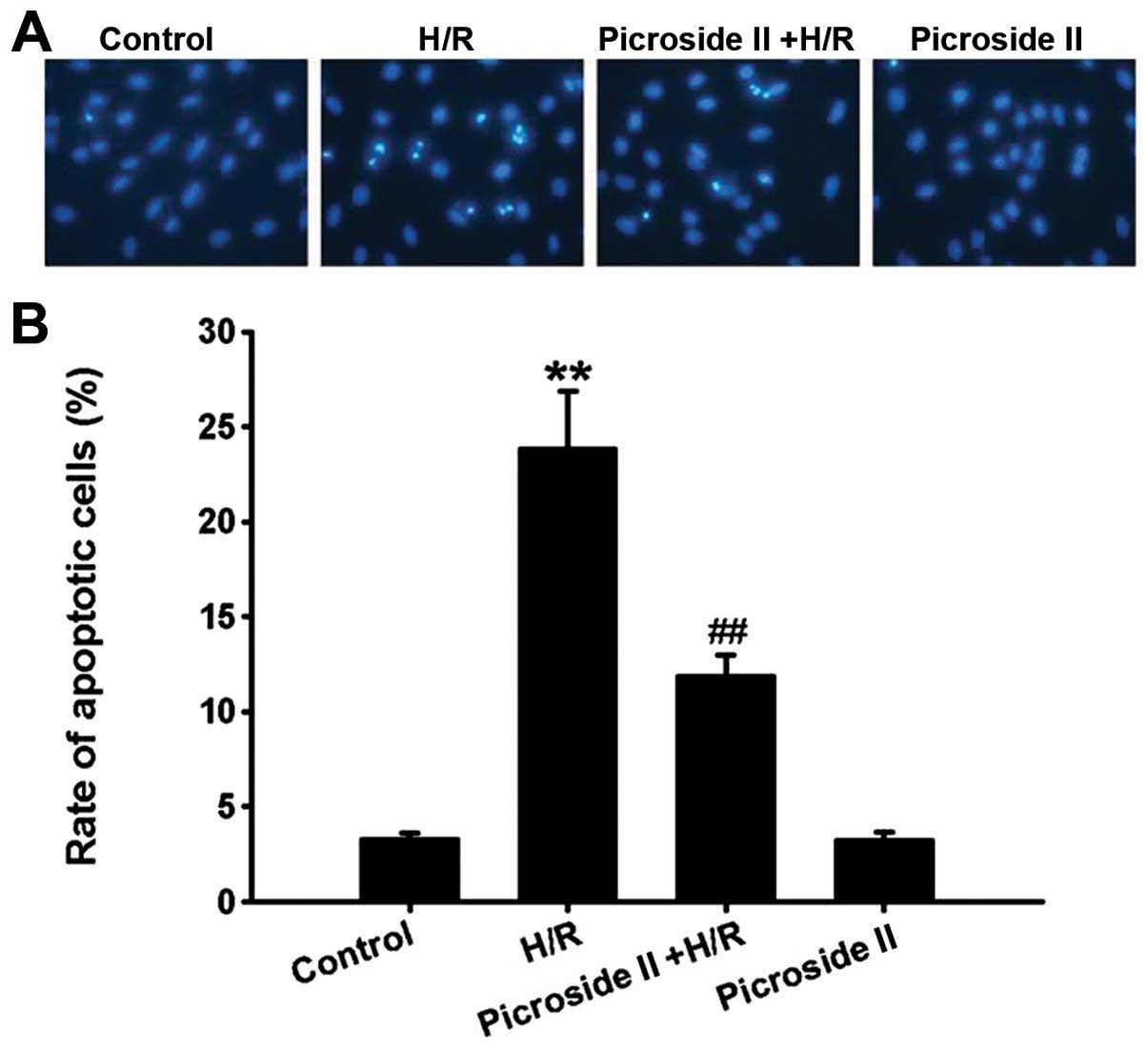
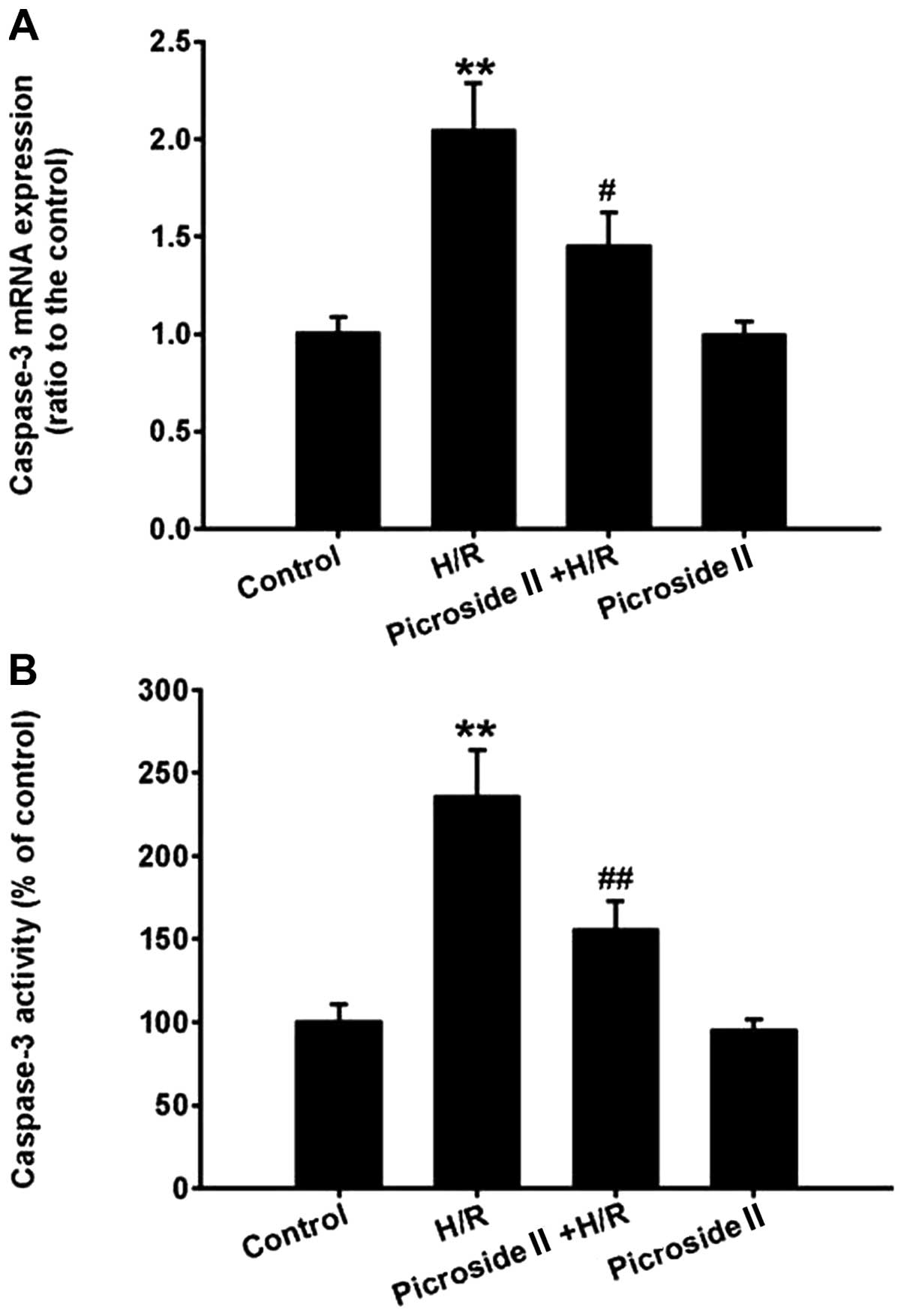
The Hoechst 33342 staining assay showed that the
percentage of apoptotic cells was significantly increased after
hypoxia for 3 h and reoxygenation for 12 h compared to the control
group. Consistent with the results of Hoechst 33342 staining, the
mRNA expression and activity of caspase-3, the effector caspase of
the mitochondrial apoptosis pathway, were also markedly increased
in the H/R group. These effects induced by H/R were significantly
inhibited by pretreatment of picroside II (Figs. 2 and 3). However, picroside II alone had no
effect on cardiomyocyte apoptosis and the mRNA expression and
activity of caspase-3.
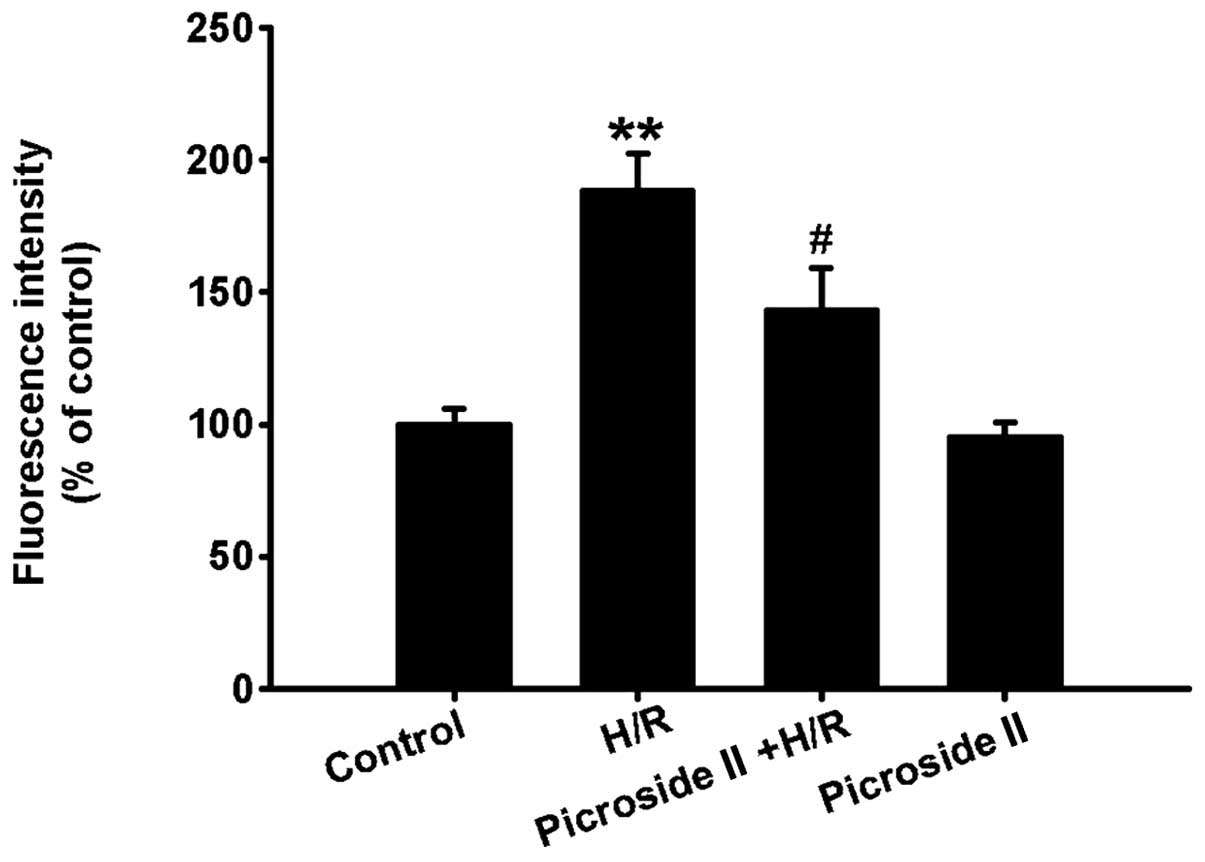
Effect of picroside II on H/R-induced
intracellular ROS production
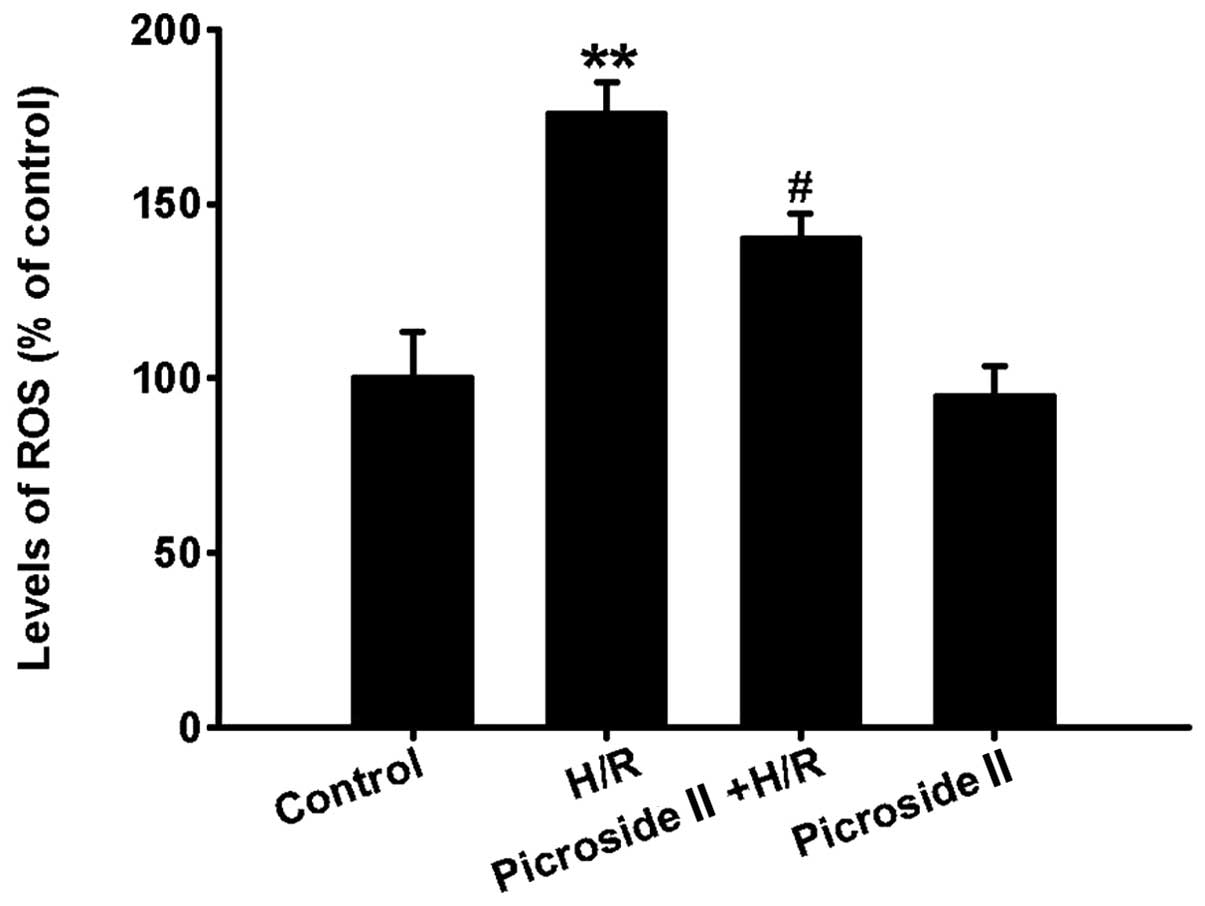
Compared to the control group, the levels of
intracellular ROS were clearly increased after hypoxia for 3 h and
reoxygenation for 12 h, which was indicated by the increased
fluorescence intensity. However, pretreatment of picroside II
significantly inhibited the production of intracellular ROS induced
by H/R (Fig. 4). Picroside II
alone had no effect on the production of intracellular ROS.
Effect of picroside II on H/R-induced
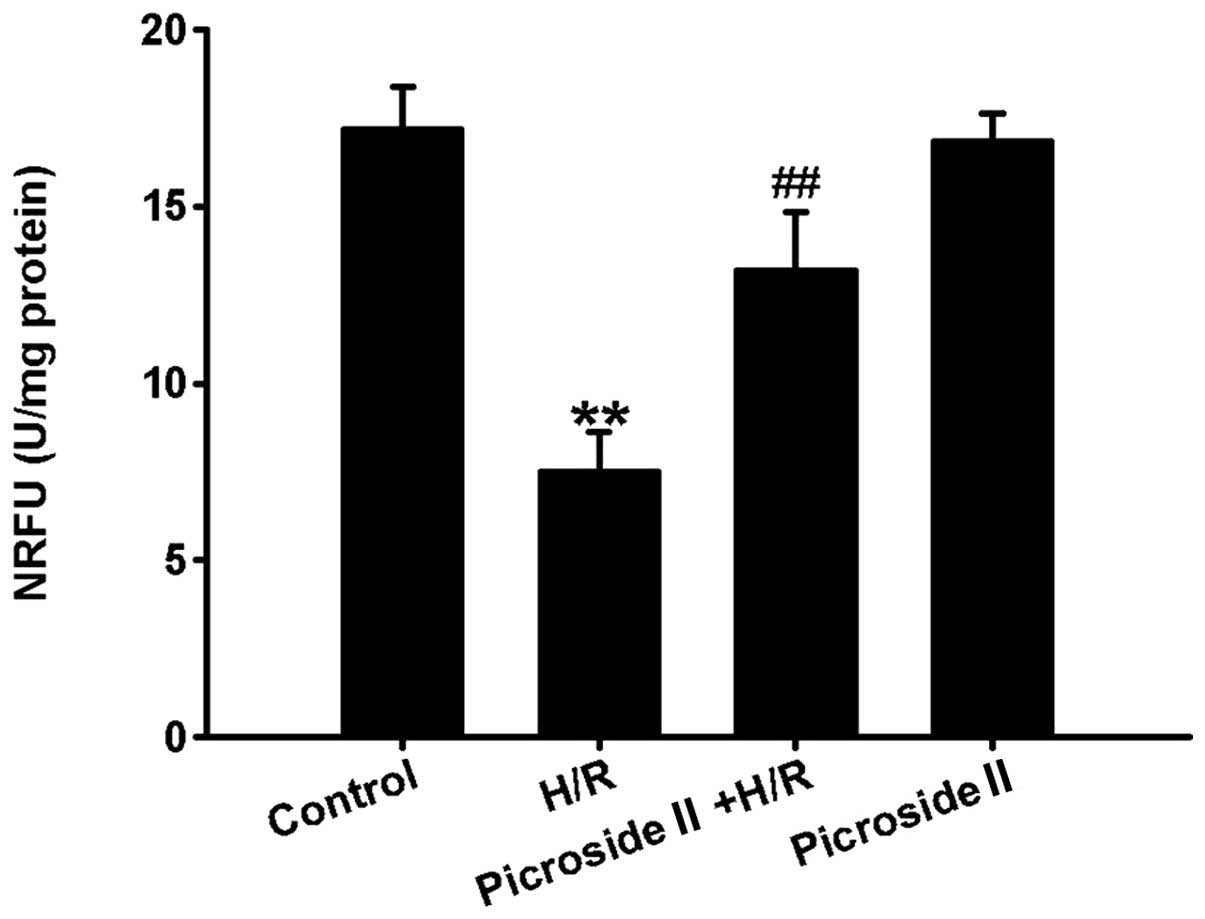
mPTP opening
The opening of mPTP was determined with calcein-AM
in the presence of cobalt chloride. Calcein-AM fluorescent dye is
able to move freely between mitochondrial and cytosol via the
opening of mPTP. Cobalt chloride can quench calcein-AM
fluorescence, but it cannot eliminate calcein-AM fluorescence in
mitochondria when the mPTP closed. Due to this property, the
fluorescence intensity of calcein-AM following treatment with
cobalt chloride can reflect the extent of mPTP opening. The results
showed that the fluorescence intensity was significantly decreased
in the H/R group, indicating that the extent of mPTP opening is
enhanced following H/R. However, pretreatment of picroside II was
able to markedly increase the fluorescence intensity compared to
the H/R group (Fig. 5). Picroside
II alone had no effect on the fluorescence intensity. These results
indicated that picroside II decreased the degree of mPTP opening in
response of H/R-induced injury.
Effect of picroside II on mitochondrial
membrane potential after H/R
The results showed that mitochondrial membrane
potential was depolarized after hypoxia for 3 h and reoxygenation
for 12 h, which was indicated by the increased fluorescence
intensity of TMRE. Pretreatment of picroside II was able to
markedly decrease the fluorescence intensity of TMRE compared to
the H/R group (Fig. 6). However,
picroside II alone had no effect on the fluorescence intensity.
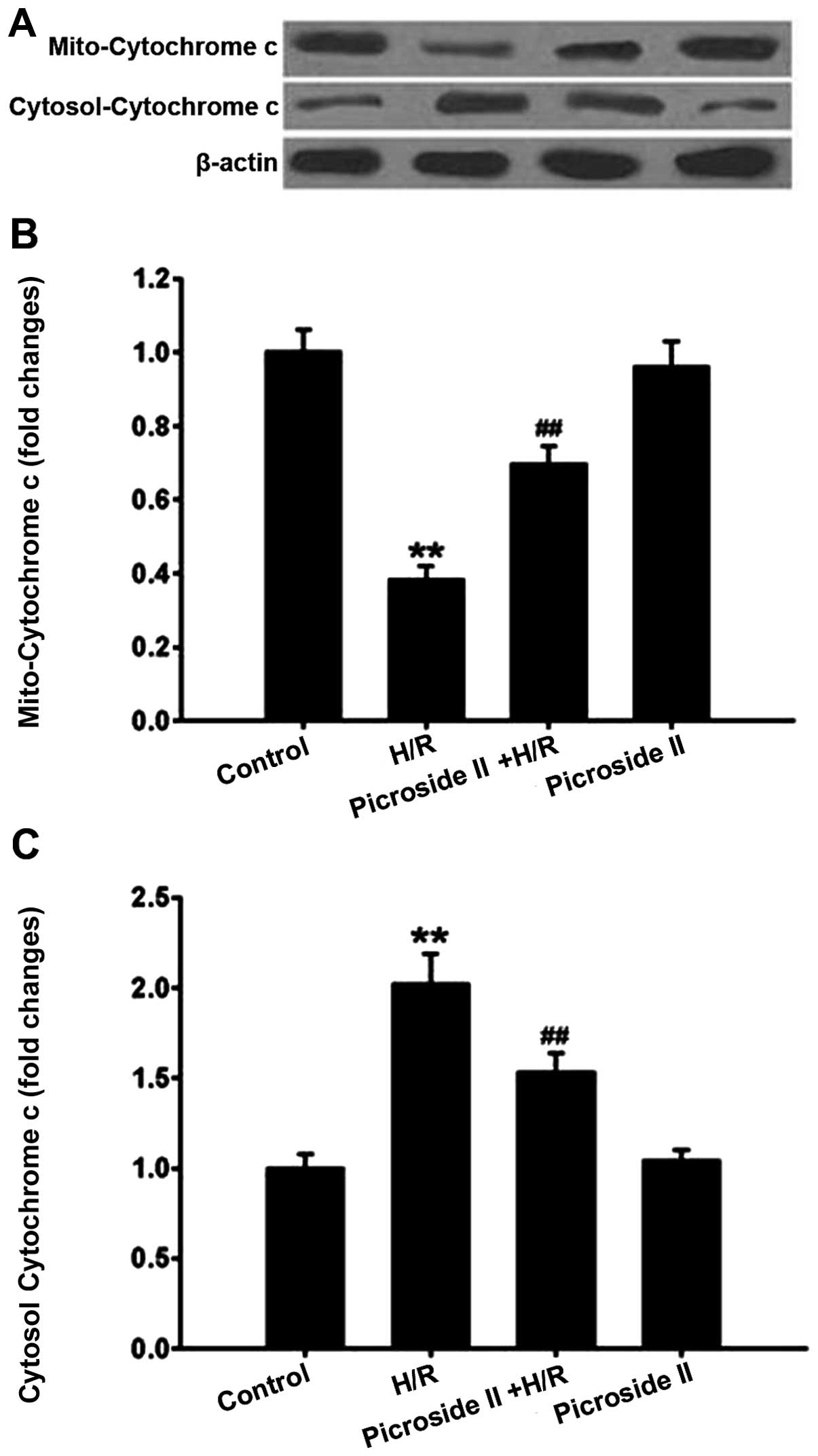
Effect of picroside II on H/R-induced
mitochondrial cytochrome c release
In mitochondria, there was a high level of
cytochrome c expression in the control group. However, the
protein expression of cytochrome c in mitochondria was
notably decreased following H/R. This H/R-induced effect was
significantly inhibited by pretreatment of picroside II. By
contrast, the cytochrome c expression in the cytosol was
lower in the control group compared to the H/R group. Pretreatment
of picroside II was able to significantly reduce the increase in
cytosolic cytochrome c expression induced by H/R (Fig. 7). Picroside II alone had no effect
on cytochrome c expression in the mitochondria and cytosol.
Taken together, these results suggested that picroside II has the
ability to inhibit cytochrome c release from the
mitochondria to cytosol.
Discussion
In the present study, the protective effect of
picroside II on H/R-induced cardiomyocyte apoptosis and the
underlying mechanisms in the H9c2 rat cardiomyocyte cell line were
investigated. The results indicated that pretreatment with
picroside II was able to protect cardiomyocytes from H/R-induced
apoptosis, which was associated with decreasing the opening degree
of mPTP, increasing mitochondrial membrane potential, inhibiting
cytochrome c release from the mitochondria to the cytosol
and downregulating caspase-3 expression and activity through a
mechanism involving a decrease in ROS production.
Myocardial I/R injury is a common clinical problem
and may result in serious consequences, including heart failure and
mortality. Although the exact pathophysiological mechanism leading
to myocardial I/R injury is not fully understood, oxidative damage
has long been considered a key factor in the initiation of I/R
injury (21). Preclinical studies
also consistently show that inhibition of ROS production can
significantly alleviate I/R-induced injury (22,23). Cardiomyocyte apoptosis is
well-recognized to play a crucial role in I/R injury (5,6).
Therefore, inhibition of cardiomyocyte apoptosis may be a potential
strategy to prevent I/R-induced injury. Although the causes of
cardiomyocyte apoptosis induced by I/R are complicated, it is
widely accepted that excessive production of ROS during I/R play an
important role in cardiomyocyte apoptosis (7,8).
The mitochondrial compartment is the main target of
intracellular ROS as they are particularly rich in polyunsaturated
fatty acids (24,25). Increased ROS can result in serious
dysfunction of mitochondrion, which is the main cause of
I/R-induced cardiomyocyte apoptosis (6). A pivotal factor mediating
mitochondrial dysfunction is the long-lasting opening of mPTP, a
large non-selective conductance pore located in the inner membrane
of mitochondria (15,26). Under physiological conditions, the
transient mPTP opening may occur in normal cells (27). However, under pathological
conditions the long-lasting mPTP opening allows protons into
mitochondria, and subsequently results in the uncoupling of the
electron respiratory chain and the decrease in mitochondrial
membrane potential (26). The
decrease in mitochondrial membrane potential is one of the earliest
events in cell apoptosis (28).
The long-lasting mPTP opening increases mitochondrial permeability
and allows ions and solutes with molecular weights of <1.5 kDa
in cytoplasm freely entering the mitochondria, leading to
mitochondrial matrix swelling and loss of critical electrochemical
gradients (29). Another adverse
consequence of the mPTP opening is the cytochrome c release
from the mitochondria to the cytosol. Cytochrome c is a
well-known pro-apoptotic factor, and the translocation plays an
important role in apoptosis by activating caspase cascade
reactions. In the cytosol, cytochrome c binds the apoptotic
factor, Apaf-1, and induces Apaf-1 oligomerization, resulting in
recruitment and activation of procaspase-9 (30). In turn, activated caspase-9
cleaves and activates caspase-3 (a classical executioner caspase),
which terminally leads to cardiomyocyte apoptosis (31). Inhibition of the mPTP opening by
cyclosporine A (an inhibitor of mPTP) or genetic deletion of the
gene encoding CypD can protect against I/R-induced myocardial
injury (27,32). The above studies indicated that
the sustained opening of the mPTP plays an important role in cell
apoptosis. Strong evidence indicates that overproduction of ROS in
the I/R period is the primary triggers of the mPTP opening
(15). Decrease in ROS production
is able to markedly inhibit the opening of mPTP (26). Therefore, inhibition of ROS
overgeneration may be a promising strategy for prevention of
I/R-induced cardiomyocyte apoptosis through suppressing the mPTP
opening.
Picroside II, an iridoid glucoside extracted from
Picrorhiza scrophulariiflora Pennell, has been reported to
have multiple pharmacological actions, such as anti-inflammation
(33) and neuroprotective effect
(34). Most recently, it has been
reported that picroside II has the protective effect on H/R-induced
cardiomyocyte apoptosis (20),
but the underlying mechanisms are not fully understood. As
mentioned previously, picroside II scavenged oxygen free radical,
protected normal constructions of mitochondria membrane (19,35) and increased ROS-induced
mitochondrial dysfunction during I/R, which plays an important role
in cardiomyocyte apoptosis. Therefore, we hypothesized that the
protective mechanism of picroside II on H/R-induced cardiomyocyte
apoptosis may be associated with ameliorating mitochondrial
function by a decrease in ROS production. In the present study,
pretreatment of picroside II markedly inhibited H/R-induced
cardiomyocyte apoptosis concomitantly with a decrease in ROS
production. The further study showed that mitochondrial function
was also significantly ameliorated, which was indicated by the
decreased mPTP opening, increased mitochondrial membrane potential
and decreased cytochrome c release from mitochondria to
cytosol.
In conclusion, the present study suggests that
picroside II is able to inhibit H/R-induced cardiomyocyte apoptosis
through ameliorating mitochondrial function involving a mechanism
of decrease in ROS production.
Acknowledgments
The present study was supported by the Guangxi
Nature Science Foundation of China (grant no. 2013GXNSFBA019126 to
J. Z. Li), the Bureau of Public Health of Guangxi Province (grant
no. Z2013206) and the National Nature Science Foundation of China
(grant no. 81101476 to S. Y. Yu).
References
|
1
|
Dianat M, Esmaeilizadeh M, Badavi M,
Samarbaf-Zadeh AR and Naghizadeh B: Protective effects of crocin on
ischemia-reperfusion induced oxidative stress in comparison with
vitamin E in isolated rat hearts. Jundishapur J Nat Pharm Prod.
9:e171872014.PubMed/NCBI
|
|
2
|
Rodrigo R, Libuy M, Feliú F and Hasson D:
Molecular basis of cardioprotective effect of antioxidant vitamins
in myocardial infarction. Biomed Res Int. 2013:4376132013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wongcharoen W, Jai-Aue S, Phrommintikul A,
Nawarawong W, Woragidpoonpol S, Tepsuwan T, Sukonthasarn A, Apaijai
N and Chattipakorn N: Effects of curcuminoids on frequency of acute
myocardial infarction after coronary artery bypass grafting. Am J
Cardiol. 110:40–44. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Syrjälä SO, Tuuminen R, Nykänen AI,
Raissadati A, Dashkevich A, Keränen MA, Arnaudova R, Krebs R, Leow
CC, Saharinen P, Alitalo K and Lemström KB: Angiopoietin-2
inhibition prevents transplant ischemia-reperfusion injury and
chronic rejection in rat cardiac allografts. Am J Transplant.
14:1096–1108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Liu Y, Yang H, Song L, Li N, Han QY, Tian
C, Gao E, Du J, Xia YL and Li HH: AGGF1 protects from myocardial
ischemia/reperfusion injury by regulating myocardialapoptosis and
angiogenesis. Apoptosis. 19:1254–1268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Wang Y, Li X, Wang X, Lau W, Wang Y, Xing
Y, Zhang X, Ma X and Gao F: Ginsenoside Rd attenuates myocardial
ischemia/reperfusion injury via Akt/GSK-3β signaling and inhibition
of the mitochondria-dependent apoptotic pathway. PLoS One.
8:e709562013. View Article : Google Scholar
|
|
7
|
Wu B, Meng K, Ji Q, Yu K, Zhao X, Tony H,
Liu Y, Zhou Y, Chang C, Zhong Y, Zhu Z, Zhang W, Mao X and Zeng Q:
Interleukin-37 ameliorates myocardial ischaemia/reperfusion injury
in mice. Clin Exp Immunol. 176:438–451. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Peng YW, Buller CL and Charpie JR: Impact
of N-acetylcysteine on neonatal cardiomyocyte ischemia-reperfusion
injury. Pediatr Res. 70:61–66. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li JZ, Yu SY, Wu JH, Shao QR and Dong XM:
Paeoniflorin protects myocardial cell from doxorubicin-induced
apoptosis through inhibition of NADPH oxidase. Can J Physiol
Pharmacol. 90:1569–1575. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Teshima Y, Takahashi N, Nishio S, Saito S,
Kondo H, Fukui A, Aoki K, Yufu K, Nakagawa M and Saikawa T:
Production of reactive oxygen species in the diabetic heart. Roles
of mitochondria and NADPH oxidase. Circ J. 78:300–306. 2014.
View Article : Google Scholar
|
|
11
|
Matsushima S, Tsutsui H and Sadoshima J:
Physiological and pathological functions of NADPH oxidases during
myocardial ischemia-reperfusion. Trends Cardiovasc Med. 24:202–205.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Dröse S, Brandt U and Wittig I:
Mitochondrial respiratory chain complexes as sources and targets of
thiol-based redox-regulation. Biochim Biophys Acta. 1844:1344–1354.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Loor G, Kondapalli J, Iwase H, Chandel NS,
Waypa GB, Guzy RD, Vanden Hoek TL and Schumacker PT: Mitochondrial
oxidant stress triggers cell death in simulated
ischemia-reperfusion. Biochim Biophys Acta. 1813:1382–1394. 2011.
View Article : Google Scholar :
|
|
14
|
Ma WW, Hou CC, Zhou X, Yu HL, Xi YD, Ding
J, Zhao X and Xiao R: Genistein alleviates the
mitochondria-targeted DNA damage induced by β-amyloid peptides
25–35 in C6 glioma cells. Neurochem Res. 38:1315–1323. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li J, Umar S, Iorga A, Youn JY, Wang Y,
Regitz-Zagrosek V, Cai H and Eghbali M: Cardiac vulnerability to
ischemia/reperfusion injury drastically increases in late
pregnancy. Basic Res Cardiol. 107:2712012. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cui J, Li Z, Qian LB, Gao Q, Wang J, Xue
M, Lou XE, Bruce IC, Xia Q and Wang HP: Reducing the oxidative
stress mediates the cardioprotection of bicyclol against
ischemia-reperfusion injury inrats. J Zhejiang Univ Sci B.
14:487–495. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Meng FJ, Hou ZW, Li Y and Yu B: The
protective effect of picroside II against hypoxia/reoxygenation
injury in neonatal rat cardiomyocytes. Pharm Biol. 50:1226–1232.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li T, Liu JW, Zhang XD, Guo MC and Ji G:
The neuroprotective effect of picroside II from hu-huang-lian
against oxidative stress. Am J Chin Med. 35:681–691. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gao H and Zhou YW: Anti-lipid peroxidation
and protection of liver mitochondria against injuries by picroside
II. World J Gastroenterol. 11:3671–3674. 2005.PubMed/NCBI
|
|
20
|
Meng FJ, Jiao SM and Yu B: Picroside II
protects cardiomyocytes from hypoxia/reoxygenation-induced
apoptosis by activating the PI3K/Aktand CREB pathways. Int J Mol
Med. 30:263–270. 2012.PubMed/NCBI
|
|
21
|
de Vries DK, Kortekaas KA, Tsikas D,
Wijermars LG, van Noorden CJ, Suchy MT, Cobbaert CM, Klautz RJ,
Schaapherder AF and Lindeman JH: Oxidative damage in clinical
ischemia/reperfusion injury: a reappraisal. Antioxid Redox Signal.
19:535–545. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Pisarenko OI, Lankin VZ, Konovalova GG,
Serebryakova LI, Shulzhenko VS, Timoshin AA, Tskitishvili OV,
Pelogeykina YA and Studneva IM: Apelin-12 and its structural analog
enhance antioxidant defense in experimental myocardial ischemia and
reperfusion. Mol Cell Biochem. 391:241–250. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ma S, Zhang Z, Yi F, Wang Y, Zhang X, Li
X, Yuan Y and Cao F: Protective effects of low-frequency magnetic
fields on cardiomyocytes from ischemia reperfusion injury via ROS
and NO/ONOO−. Oxid Med Cell Longev. 2013:5291732013.
|
|
24
|
Gao C, Chen X, Li J, Li Y, Tang Y, Liu L,
Chen S, Yu H, Liu L and Yao P: Myocardial mitochondrial oxidative
stress and dysfunction in intense exercise: regulatory effects of
quercetin. Eur J Appl Physiol. 114:695–705. 2014. View Article : Google Scholar
|
|
25
|
Seleznev K, Zhao C, Zhang XH, Song K and
Ma ZA: Calcium-independent phospholipase A2 localizes in and
protects mitochondria during apoptotic induction by staurosporine.
J Biol Chem. 281:22275–22288. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yue R, Hu H, Yiu KH, Luo T, Zhou Z, Xu L,
Zhang S, Li K and Yu Z: Lycopene protects against
hypoxia/reoxygenation-induced apoptosis by preventing mitochondrial
dysfunction in primary neonatal mouse cardiomyocytes. PLoS One.
7:e507782012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Schriewer JM, Peek CB, Bass J and
Schumacker PT: ROS-mediated PARP activity undermines mitochondrial
function after permeability transition pore opening during
myocardial ischemia-reperfusion. J Am Heart Assoc. 2:e0001592013.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wadia JS, Chalmers-Redman RM, Ju WJ,
Carlile GW, Phillips JL, Fraser AD and Tatton WG: Mitochondrial
membrane potential and nuclear changes in apoptosis caused by serum
and nerve growth factor withdrawal: time course and modification by
(−)-deprenyl. J Neurosci. 18:932–947. 1998.PubMed/NCBI
|
|
29
|
Perrelli MG, Pagliaro P and Penna C:
Ischemia/reperfusion injury and cardioprotective mechanisms: Role
of mitochondria and reactive oxygen species. World J Cardiol.
3:186–200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Martin MC, Allan LA, Lickrish M, Sampson
C, Morrice N and Clarke PR: Protein kinase A regulates caspase-9
activation by Apaf-1 downstream of cytochrome c. J Biol Chem.
280:15449–15455. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Li P, Nijhawan D, Budihardjo I,
Srinivasula SM, Ahmad M, Alnemri ES and Wang X: Cytochrome c and
dATP-dependent formation of Apaf-1/caspase-9 complex initiates an
apoptotic protease cascade. Cell. 91:479–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Gedik N, Heusch G and Skyschally A:
Infarct size reduction by cyclosporine A at reperfusion involves
inhibition of the mitochondrial permeability transition pore but
does not improve mitochondrial respiration. Arch Med Sci.
9:968–975. 2013. View Article : Google Scholar
|
|
33
|
Guo Y, Xu X, Li Q, Li Z and Du F:
Anti-inflammation effects of picroside 2 in cerebral ischemic
injury rats. Behav Brain Funct. 6:432010. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhao L, Guo Y, Ji X and Zhang M: The
neuroprotective effect of picroside II via regulating the
expression of myelin basic protein after cerebral ischemia injury
in rats. BMC Neurosci. 15:252014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Cao Y, Liu JW, Yu YJ, Zheng PY, Zhang XD,
Li T and Guo MC: Synergistic protective effect of picroside II and
NGF on PC12 cells against oxidative stress induced by
H2O2. Pharmacol Rep. 59:573–579.
2007.PubMed/NCBI
|