Introduction
Cardiovascular disease, a leading cause of mortality
worldwide, commonly presents as myocardial infarction (MI)
(1). The ischemia-induced
formation of coronary collateral circulation serves as a
compensatory mechanism that provides blood perfusion to ischemic
myocardial tissue, thus playing a significant role in the prognosis
of MI (2). Coronary artery
disease, including MI, is also a common complication contributing
to morbidity and mortality in diabetic patients (3). In diabetes, angiogenesis is impaired
(4), which may underlie the poor
prognosis of diabetic patients with cardiovascular complications
(5). Therefore, therapeutic
angiogenesis has been proposed as an exciting strategy to enhance
perfusion to the ischemic myocardium of diabetic patients with
MI.
Vascular endothelial growth factor (VEGF) plays a
pivotal role in blood vessel formation during the developmental
stage and in the regulation of hypoxia-induced angiogenesis
(6). VEGF expression is regulated
by hypoxia, oxidative stress and nitric oxide (NO), and elevated
VEGF levels have been observed in the retina and glomeruli of
diabetic rats (7,8). However, the levels of both VEGF and
its receptors are decreased in myocardial tissue in diabetic
animals (9). This preclinical
observation suggests a potential molecular explanation for
inadequate collateral vascular formation in response to myocardial
ischemia in diabetic patients.
Protein kinase C (PKC) is a family of cytoplasmic
serine/threonine kinases with a wide variety of functions in
intracellular signal transduction (10). The activation of PKC is associated
with vascular complications under diabetic conditions (11). The β isoforms of PKC (PKCβ1 and
PKCβ2) are expressed in cardiovascular tissue and can be activated
by free fatty acids and under hyperglycemic conditions (12,13). In diabetic rats, the activation of
PKCβ is elevated in vascular tissue and is involved in the
dysfunction of the cardiac microvascular barrier (14). Furthermore, in endothelial cells,
PKCβ has been shown to inhibit the activation of Akt by insulin or
VEGF, and to interrupt the regulation of Akt-dependent endothelial
NO synthase (eNOS) by insulin in obesity-associated insulin
resistance (15). In a previous
study, a PKCβ-selective inhibitor, LY333531, was shown to prevent
diabetic vascular complications both in vitro and in
vivo (16). However, the
effects of the inhibition of PKCβ on angiogenesis in the ischemic
myocardium under diabetic conditions remain unclear.
Statins are inhibitors of the
3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase, the
rate-limiting enzyme in cholesterol biosynthesis. This class of
drugs can also induce lipid-independent benefits, including the
improvement of endothelial function, the inhibition of
inflammation, the facilitation of angiogenesis and, consequently,
the reduction of the myocardial or cerebral infarct size (17). The promoting effect of statins on
angiogenesis is likely mediated through the VEGF-dependent Akt/eNOS
pathway. Accordingly, statins have been shown to promote
angiogenesis in response to ischemia, which is mediated by VEGF
(18,19). In the present study, we therefore
aimed to investigate the effects of combined treatment with
rosuvastatin and LY333531 on angiogenesis in myocardial tissue in
diabetic rats with MI.
Materials and methods
Cell culture and treatments
Human umbilical vein endothelial cells (HUVECs) were
purchased from the American Type Culture Collection (ATCC,
Manassas, VA, USA). The cells were cultured in Dulbecco’s modified
Eagle’s medium (DMEM) supplemented with 10% bovine serum albumin
(BSA; Gibco-Invitrogen Inc., Carlsbad, CA, USA), 0.05 mg/ml
endothelial growth factor (Sigma, St. Louis, MO, USA), 0.1 mg/ml
heparin, 100 units/ml penicillin and 100 mg/ml streptomycin at 37°C
under 5% CO2. Cells between the second and fourth
passages were used for the experiments. The cells were divided into
5 treatment groups as follows: i) cells cultured in normal glucose
medium (N-G, 5.6 mmol/l); ii) cells cultured in high glucose medium
(H-G, 33.3 mmol/l); iii) cells cultured in high glucose medium and
treated with rosuvastatin (RSV, 0.1 μmol/l); iv) cells
cultured in high glucose medium and treated with LY333531 (LY, 10
nmol/l); and v) cells cultued in high glucose medium and treated
with rosuvastatin plus LY333531 (RSV + LY, 0.1 μmol/l+10
nmol/l). Rosuvastatin (obtained from AstraZeneca, Cheshire, UK) and
LY333531 (from Eli Lilly & Co., Indianapolis, IN, USA) were
dissolved in dimethyl sulfoxide (DMSO, 0.2%; Sigma).
Tube formation assay
The formation of tube-like structures by HUVECs on
Matrigel (BD Biosciences, Oxford, UK) was assessed as previously
described (20). The cells were
seeded in gel-precoated wells and cultured in the absence or
presence of either rosuvastatin alone or in combination with
LY333531 in H-G medium at 37°C for 24 h with 5% CO2.
Cells cultured in N-G medium served as the controls. Tube formation
was observed under a microscope (Olympus Corp., Tokyo, Japan).
Images were analyzed using Image-Pro Plus software (Media
Cybernetics, Inc., Rockville, MD, USA). The degree of tube
formation was assessed by measuring the length of tubes in 3 random
fields (magnification, ×40) from each well. Tube formation under
different treatment conditions was normalized to that under normal
glucose conditions.
Migration assay
A wound healing assay was performed to measure the
unidirectional migration of HUVECs, as previously described
(21). The cells were seeded in a
6-well plate at a density of 5×104 cells/well, and
incubated at 37°C with 5% CO2 for 24 h. After being
washed with PBS twice, the cells were incubated in N-G medium
containing 1% FBS for 12 h. The wells were then scraped with a
200-μl pipette tip to mechanically injure the cells. The
wells were rinsed with PBS twice to remove cellular debris and
dislodge the cells. After injury, the cells were cultured in the
presence of the different reagents for 24 h. Images were captured
24 h later using an Olympus microscope (Olympus Corp.) at ×40
magnification. The migration ability of the cells was calculated
using the following formula: 100% (width at 24 h/width at 0 h)
×100%, as previously described (22).
Cell proliferation assay
Cell proliferation was assessed using the
3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay (Sigma). The HUVECs were seeded into 96-well plates at a
density of 2,000 cells/well and were incubated in N-G medium
containing 1% FBS at 37°C with 5% CO2 for 24 h. The
cells were then transferred to fresh medium in the absence or
presence of the different reagents. Each group had 3 replicate
wells. Following incubation for different periods of time, 10
μl of MTT were added to each well and the cells were further
incubated at 37°C for 4 h with 5% CO2. The supernatant
fluid was then removed and 100 μl of DMSO were added to each
well. The absorbance (OD value) at a wavelength of 490 nm was
measured using a microplate reader (Bio-Tek ELx808; BioTek
Instruments, Inc., Winooski, VT, USA), as previously described
(23).
Animal model
Male Sprague-Dawley (SD) rats, (weighing 200–250 g,
from the Experimental Animal Center, Fudan University, Shanghai,
China) were used in the experiment. All procedures were approved by
the Animal Care and Use Committee of the Sixth People’s Hospital
affiliated to Shanghai Jiaotong University, Shanghai, China and
were in compliance with the regulations of the Animal Welfare Act
of the National Institutes of Health Guide for the Care and Use of
Laboratory Animals. The animals were allowed to acclimatize for 2
weeks prior to the experiment.
After an overnight fast, the animals were
administered streptozotocin [50 mg/kg, intraperitonealy (i.p.)
daily] (Sigma) or the vehicle (citrate saline, i.p.) (14). One week after the streptozotocin
injection, blood glucose levels were measured. Animals with a
glucose level ≤300 mg/dl were excluded from the study.
After 1 week of the administration of the
streptozotocin injection, the model of MI was created as described
in our previous study (24). In
brief, a total of 55 diabetic rats were anesthetized by an
intraperitoneal injection of pentobarbital (50 mg/kg) and were
ventilated using an animal respirator (DW-200; Alcott Biotech,
Shanghai, China) with room air following endotracheal intubation.
The electrical activity of the mouse hearts was continuously
monitored during the experiment using an lead II electrocardiogram
(ECG). Heart exposure was performed by a thoracotomy at the left
fourth intercostal space. The left anterior descending coronary
artery (LAD) was ligated using a 6-0 prolene suture, 1–2 mm below
the tip of the left atrial appendage. LAD ligation was confirmed by
ST segment elevation. Another 10 diabetic rats, as a control group,
underwent thoracotomy and incision of the pericardial sac, but not
LAD ligation.
Animal protocols
After establishing the animal model, all surviving
animals were randomly assigned into the following groups: i) the MI
group: rats with MI were treated with the vehicle (normal saline
orally) for 4 weeks starting 1 day after LAD ligation; ii) the RSV
group: rats with MI were administered rosuvastatin orally (5 mg/kg
daily); iii) the LY333531 group: rats with MI were administered
LY333531 orally (10 mg/kg daily); iv) the RSV + LY333531 group:
rats with MI were orally administered rosuvastatin (5 mg/kg daily)
plus LY333531 (10 mg/kg daily); and v) another sham-operated and
non-diabetic group of animals was administered the vehicle (normal
saline). All surviving animals were sacrificed on day 28. Before
the rats were sacrificed, an echocardiographic examination was
performed to evaluate the remodeling process.
After the rats were euthanized, whole blood was
collected from the inferior vena cava and the hearts were
harvested. One cross-section of left ventricular (LV) myocardial
tissue at the level of the papillary muscles, approximately 2 mm,
was collected and fixed in 10% formalin followed by embedding in
paraffin for histological examination. The remaining LV tissue was
quickly separated into the peri-infarct zone and remote zone, and
the tissue was snap-frozen and mechanically homogenized in liquid
nitrogen, and resuspended in lysis buffer to which phosphatase was
added. Subsequently, the tissue was further lysed in a tightly
fitting cylinder homogenizer. The sample was then stored at −80°C
for later analysis.
Echocardiography
An echocardiographic examination was performed on
day 28 following the induction of MI using a standard Acuson
Sequoia 512 ultrasound system equipped with a 15-MHz probe
(Siemens, Erlangen, Germany). Both two-dimensional and M-mode
echocardiography were performed. The LV end-diastolic diameter
(LVEDD) and the LV end-systolic diameter (LVESD) were measured. In
addition, the LV ejection fraction (LVEF) and fractional shortening
(FS) were calculated, as previously described (25). All measurements from 5 consecutive
cardiac cycles were averaged and analyzed by a single observer
blinded to the treatment protocol.
Histological analysis\
Under deep anesthesia with pentobarbital sodium (60
mg/kg, i.p.), the heart of a rat was excised and cut into
transverse slices (2-mm-thick), which were then fixed in 10%
formalin solution for 24 h. The samples were then embedded in
paraffin and sectioned at 4 μm. Immunohistochemical analysis
was performed as previously described (26). In brief, deparaffinized tissue
sections (4-μm-thick) were blocked by 10% goat serum for 30
min at 37°C. Rabbit polyclonal antibodies against CD31 (ab28364;
capillary density maker; diluted at 1:500) and VEGF (ab46154;
diluted at 1:250; Abcam, Cambridge, MA, USA) were employed as
primary antibodies. Following overnight incubation with the primary
antibodies at 4°C, the sections were treated with goat polyclonal
secondary antibody to rabbit IgG (goat anti-rabbit IgG H&L,
ab97047; diluted at 1:1,000; Abcam). All sections were
counterstained with hematoxylin. Immunoreactivity for CD31 and VEGF
was measured using the Image-Pro Plus 4.0 analysis system (Media
Cybernetics, Inc., Bethesda, MD, USA). Capillaries were identified
by positive staining for CD31. The results were expressed as an
average amount of capillaries per ×10 field, as previously
described (27).
Tissue staining with Masson’s trichrome stain was
performed in the border area to evaluate collagen deposition.
Sirius Red Staining (a collagen-specific dye) was also used to
clearly discriminate between the cardiomyocytes and collagen.
Collagen volume fraction was analyzed using ImageJ software and
expressed as the average percentage of collagen staining of 20
randomized high power fields.
Enzyme-linked immunosorbent assay
(ELISA)
The plasma levels of transforming growth factor
(TGF)-β1, VEGF and hypoxia-inducible factor (HIF)-1α were
determined using solid-phase sandwich ELISA kits (USCN Life Science
Inc., Wuhan, China), according to the manufacturer’s
instructions.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was prepared from 150 mg LV free wall
using TRIzol reagent (Invitrogen, Life Technologies), followed by
chloroform extraction and isopropanol precipitation. Genomic DNA
was eliminated by incubating with DNase I (0.1
μl−1, 37°C) for 30 min followed by acid
phenol-chloroform extraction. RNA was quantified by
spectrophotometric absorbance at 260 nm (A260), with its purity
confirmed by measuring the A260/A280 ratio.
The integrity of RNA was evaluated by ethidium bromide staining on
a denaturing agarose gel. Total RNA (2 μg) was then reverse
transcribed using oligo(dT) primer and SuperScript II RT
(Invitrogen-Life Technologies, Carlsbad, CA, USA).
Quantitative (real-time) PCR was performed as
described in a previous study of ours (28) starting with 12.5 ng cDNA and both
sense and antisense primers at a concentration of 900 nM
(Invitrogen-Life Technologies) in a final volume of 25 μl,
using SYBR-Green Master Mix (Applied Biosystems, Denver, CO, USA).
Fluorescence was monitored and analyzed using a GeneAmp 7000
detection system instrument (Applied Biosystems). The PCR reactions
were performed 42 times by a three-step cycle procedure
(denaturation 95°C, 15 sec; annealing 60°C, 30 sec; extension 72°C,
30 sec) following an initial stage (95°C, 10 min). A ∆Ct value was
obtained to quantify the mRNA levels and was normalized to an
endogenous control (GAPDH mRNA) for each sample. A relative
quantification ΔΔCt method was used for the comparison between
groups. Oligonucleotide primers were designed using Primer Express
software (Applied Biosystems). The primers used in this study were
as follows: VEGF forward, 5′-CGAGACGCAGCGACAAGGCA-3′ and reverse,
5′-ACCTTCTCCAAACCGTTGGCA-3′; HIF-1α forward,
5′-GCCCAGTGAGAAAGGGGAAA-3′ and reverse, 5′-CGGCTGGTTACTGCTGGTAT-3′;
and GAPDH sense, 5′-TGATGGGTGTGAACCACGAG-3′ and antisense,
3′-CCCTTCCACGATGCCAAAGT-5′.
Western blot analysis
The infarcted heart tissue from the LV free wall or
the non-infarcted heart tissue at the same site was used for
western blot analysis. Equal amounts of protein (30 μg) were
separated on a 8–12% Tris-glycine gel (Novex; Invitrogen-Life
Technologies) and transferred onto polyvinylidene difluoride (PVDF)
membranes (Roche Applied Science, Penzberg, Germany). After
blocking with 5% skim milk, the membranes were incubated with
primary antibodies against phospho-Akt Ser473 (sc-7985-R; 1:1,000
dilution; Santa Cruz Biotechnology, Santa Cruz, CA, USA) and
phospho-eNOS Thr1117 (sc-21871-R; 1:500 dilution; Santa Cruz
Biotechnology) overnight at 4°C, followed by incubation with
horseradish peroxidase (HRP)-conjugated secondary antibody.
Immunoreactions were visualized using an ECL detection kit
(Amersham Biosciences, Piscataway, NJ, USA). All protein expression
levels were adjusted against GAPDH intensity (cat. no. 2118; Cell
Signaling Technology, Danvers, MA, USA). Bands were quantified by
densitometry using ImageJ software (version 1.41; National
Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data are expressed as the means ± SD. All data
analyses were performed using SPSS 13.0 statistical software (SPSS,
Inc., Chicago, IL. USA). One-way ANOVA followed by the
Student-Newman-Keuls test was used to compare the effects of the
different treatments on various parameters. A P-value <0.05 was
considered to indicate a statistically significant difference.
Results
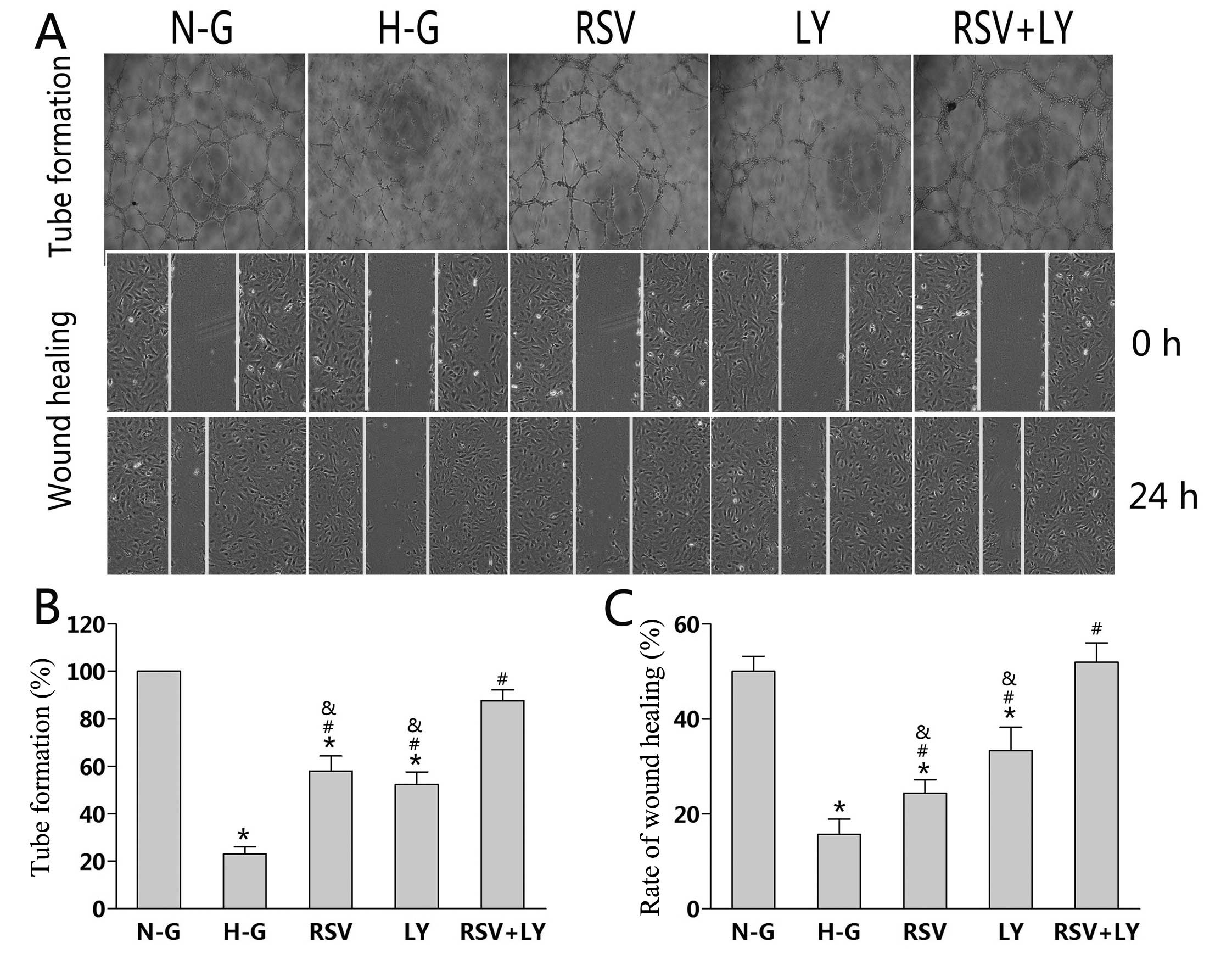
Effect of combination treatment with
rosuvastatin and LY333531 on tube formation and migration of
HUVECs
Ten hours after plating, the HUVECs cultured in N-G
and H-G medium treated with rosuvastatin + LY333531 had an
elongated shape and had invaded the collagen gel to form tubes. By
contrast, the HUVECs cultured in H-G medium remained in individual
clusters. Twenty-four hours after plating, the HUVECs cultured in
N-G medium formed a more extensive tube network than the HUVECs
cultured in H-G medium (P<0.01; Fig. 1A). Single treatment with either
rosuvastatin or LY333531 significantly promoted tube formation by
HUVECs compared to the H-G goup (P<0.05; Fig. 1B). The tube-forming ability of the
HUVECs was more enhanced following treatment with rosuvastatin +
LY333531 than with treatment with either agent alone (P<0.05;
Fig. 1B).
Wound healing was assessed following culture for 24
h under different conditions (Fig.
1A). Compared to the N-G group, culture in H-G medium inhibited
the wound healing ability of the HUVECs (P<0.05; Fig. 1C). Treatment with rosuvastatin or
LY333531 alone increased the wound healing ability of the HUVECs by
10 and 18%, respectively, as compared to the H-G group (P<0.05).
Treatment with rosuvastatin + LY333531 further facilitated wound
closure; an increase of 32% was observed (Fig. 1C) compared to the cells treated
with either agent alone (P<0.05).
Effect of combination treatment with
rosuvastatin and LY333531 on cardiac function in rats
Twenty-eight days following surgery, the rats with
MI demonstrated LV dilation, a greater LVESD and LVEDD, and
decreased FS in comparison to the sham-operated rats (P<0.05;
Fig. 2). Treatment with
rosuvastatin + LY333531 improved these parameters (P<0.05;
Fig. 2B–D), while treatment with
either rosuvastatin or LY333531 alone numerically decreased LVESD
and LVEDD althgouth no statistically significant differences were
observed (P>0.05; Fig. 2B and
C).
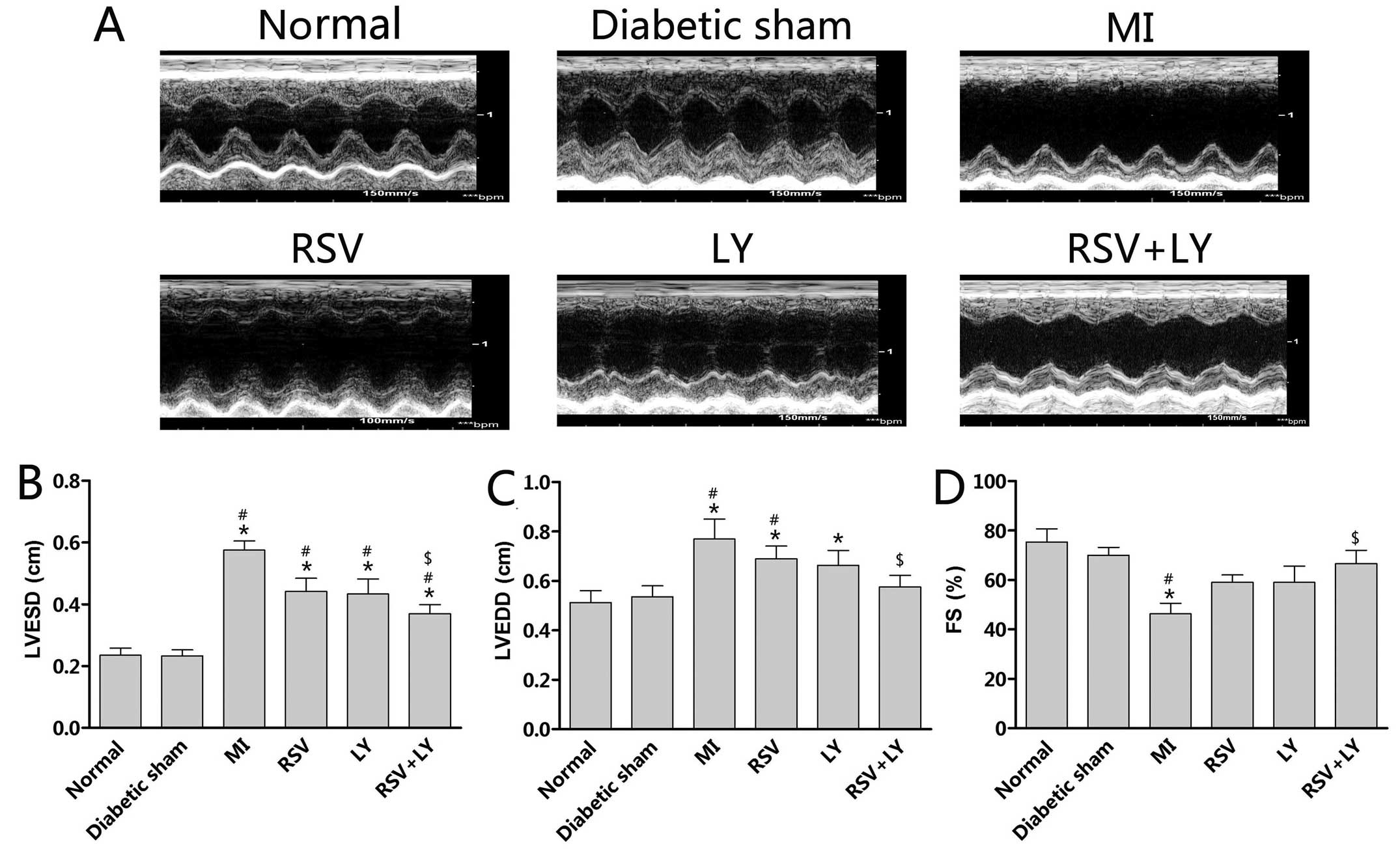 | Figure 2(A) Representative M-mode
echocardiograms obtained from the normal (non-diabetic), diabetic
sham-operated, RSV, LY and RSV + LY group rats. Summary data of
echocardiographic measurements in each group of animals: (B) LVESD,
(C) LVEDD and (D) FS. Data are expressed as the means ± SD; n=8–10
rats per group. *P<0.05 vs. normal group;
#P<0.05 vs. diabetic sham-operated group;
$P<0.05 vs. MI group. Sham, sham-operated; RSV,
rosuvastatin; LY, LY333531; MI, myocardial infarction; LVESD, left
ventricular end-systolic diameter; LVEDD, left ventricular
end-diastolic diameter; FS, fractional shortening. |
Effect of combination treatment with
rosuvastatin and LY333531 on myocardial fibrosis in rats
The rats with MI had increased serum TGF-β1 levels
and myocardial interstitial fibrosis compared to the diabetic rats
in the sham-operated group (P<0.05; Fig. 3A and B). Treatment with
rosuvastatin, LY333531, or rosuvastatin + LY333531 for 28 days
reduced serum TGF-β1 levels (P<0.05; Fig. 3A). Treatment with rosuvastatin +
LY333531, as compared to treatment with either agent alone, induced
a greater reduction in interstitial fibrosis (P<0.05; Fig. 3B) as indicated by Masson’s
thrichrome staining (Fig. 3C) and
Sirius Red Staining (Fig.
3D).
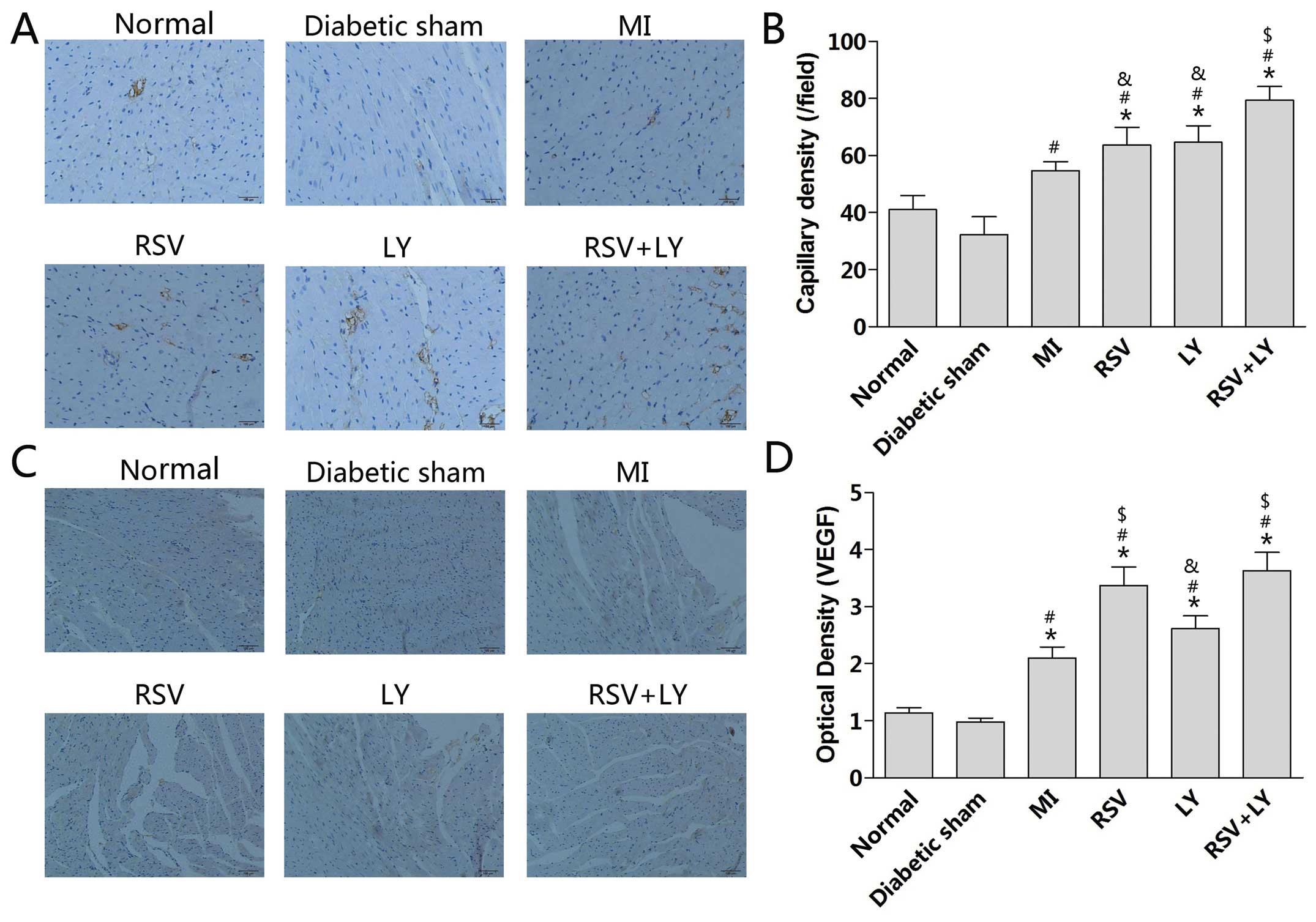
Effect of combination treatment with
rosuvastatin and LY333531 on capillary density in rats
The induction of MI in the rats increased capillary
density in the peri-infarct zone as compared to rats in the
sham-operated group (P<0.05; Fig.
4A and B). Treatment with either rosuvastatin or LY333531 alone
did not change the capillary density, while treatment with
rosuvastatin + LY333531 increased capillary density (P<0.05;
Fig. 4B). The expression levels
of VEGF were increased after the induction of MI (P<0.05;
Fig. 4C and D). Treatment with
rosuvastatin alone or rosuvastatin + LY333531 further increased the
VEGF expression levels (P<0.05; Fig. 4D).
Effect of combination treatment with
rosuvastatin and LY333531 on VEGF and HIF-1α expression in
rats
The rats with MI had increased serum VEGF levels
compared with the rats in the sham-operated group (79±6.1 vs.
31±4.9 pg/ml; P<0.05; Fig.
5A). Treatment with rosuvastatin alone (132±7.0 vs. 79±6.1
pg/ml, P<0.05), but not with LY333531 alone (108±8.7 pg/ml),
further elevated the serum VEGF levels. Treatment with rosuvastatin
+ LY333531 induced a significant increase in VEGF levels compared
to treatment with rosuvastatin alone (167±7.2 vs. 132±7.0 pg/ml,
P<0.05; Fig. 5A). MI increased
myocardial VEGF mRNA levels (Fig.
5C). Treatment with rosuvastatin alone (1.53±0.10-fold
increase, P<0.05; Fig. 5C) or
in combination with LY333531 further elevated the VEGF mRNA levels
(1.56±0.13-fold increase, P<0.05; Fig. 5C) compared with the MI group.
There were no significant differences observed between single and
co-treatment groups.
MI did not significantly alter the myocardial VEGF
protein levels (Fig. 5E).
Treatment with either rosuvastatin or LY333531 alone did not affect
the VEGF protein levels, while higher VEGF protein levels were
observed following combination treatment (P<0.05; Fig. 5E).
MI also increased the serum HIF-1α protein levels
and the myocardial HIF-1α protein and mRNA levels (P<0.05;
Fig. 5B–F). The HIF-1α levels
were not significantly affected by single or combination
treatment.
Effect of combination treatment with
rosuvastatin and LY333531 on Akt and eNOS phosphorylation in
rats
MI on its own did not induce Akt or eNOS
phosphorylation (Fig. 6).
Although there was a numerical increase in Akt phosphorylation
following treatment with either rosuvastatin or LY333531 alone, the
change did not reach statistical significance (P>0.05; Fig. 6A), while treatment with
rosuvastatin + LY333531 significantly increased Akt phosphorylation
(P<0.05). Monotherapy with either rosuvastatin or LY333531
significantly increased eNOS phosphorylation (P<0.05; Fig. 6B), and this increase was even more
pronounced following treatment with rosuvastatin + LY333531
(P<0.05).
Discussion
The present study demonstrated that treatment with
rosuvastatin or LY333531 reversed the high glucose-induced
impairment of HUVEC tube formation and migration. These effects on
HUVECs were further enhanced when the cells were treated with both
rosuvastatin and LY333531. Using a rat diabetic model, we found
that co-treatment with rosuvastatin and LY3333531, but not
treatment with either agent alone, improved the cardiac function of
the rats with MI. Oure results further revealed that co-treatment
with rosuvastatin and LY333531 had a greater effect on capillary
density in the peri-infarct zone than treatment with either agent
alone. This effect was mediated by the upregulation of the
VEGF-dependent Akt/eNOS signaling pathway. Taken together, and to
the best of our knoweledge, the results of the present study
provide the first evidence of improved cardiac function following
combination treatment with rosuvastatin and LY333531, as compared
to treatment with either agent alone in a rat model of MI under
diabetic conditions. In addition combination treatment had a
pro-angiogenesis effect.
The coronary collateral circulation is a critical
mechanism of adaptation of the heart to prevent cardiac dysfunction
caused by ischemic insults (29,30). The mechanisms underlying the
development of coronary collaterals are largely unknown, and the
role of myocardial ischemia as an inhibitory factor is unclear
(31,32). The degree of collateral coronary
development varies greatly among patients with myocardial ischemia
(33). A growing body of evidence
suggests that diabetic patients experience accelerated
atherosclerosis and, as shown angiographically and in autopsy
studies, they also have an impaired ability to form coronary
collaterals in response to myocardial ischemia (33,34). In addition, in vitro
studies have demonstrated decreased tree-like tubular network
formation by HUVECs cultured under high glucose conditions
(35). Different concepts have
been proposed to explain the impaired angiogenic response under
diabetic conditions (4). One of
the commonly accepted mechanisms includes the decreased cardiac
expression of VEGF and its receptors in diabetic states (9).
Statins have been suggested to exert
cardioprotective benefits through mechanisms independent of
lipid-lowering effects, including anti-inflammatory effects,
improvement of endothelial function and NO synthesis. The
angiogenic effects of statins remain controversial (36). Zaitone and Abo-Gresha (37) demonstrated that rosuvastatin
promoted angiogenesis in diabetic rats with MI. The present study
demonstrated that rosuvastatin significantly increased tube-like
network formation and the migration of HUVECs cultured in
high-glucose medium, whereas the formation of coronary collaterals
was not further enhanced by rosuvastatin following the induction of
MI in rats with diabetes. However, an increased VEGF expression was
observed in the rosuvastatintreated rats, indicating that factors
other than decreased VEGF levels in the diabetic myocardium are
involved in the impaired development of coronary collaterals.
Hyperglycemia has been reported to induce the activation of PKCβ2,
which has been associated with diabetic complications, as treatment
with the PKCβ2-specific inhibitor, LY333531, has been shown to be
effective in diabetic myocardial hypertrophy, retinopathy and
nephropathy (21,38,39). Furthermore, it has been
established that the hyperglycemia-induced activation of PKCβ2
inhibits the insulin-mediated Akt-dependent regulation of eNOS in
obesity-associated insulin resistance (15). Consistently, our previous study
confirmed that the inhibition of PKCβ2 by LY333531 not only
increased the phosphorylation levels of Akt and eNOS, but also
improved coronary collateral development in response to MI in
diabetic rats, indicating that the activation of PKCβ2 may be
involved in the downregulation of the Akt/eNOS angiogenic pathway
under diabetic conditions (40).
Indeed, in the present study, we observed that treatment with
LY333531 alone increased the Akt-dependent eNOS activation without
increasing VEGF expression. However, it has been demonstrated that
the LY333531-associated inhibition of PKCβ2 enhances the myocardial
ischemia-triggered increase in VEGF expression levels, leading to
the improvement of impaired angiogenesis in diabetic rats (41).
Most importantly, the present study suggests that
combination treatment with rosuvastatin and LY333531 is more
effective in promoting the revovery of cardiac function than
treatment with rosuvastatin or LY333531 alone. The extent of
angiogenesis measured by CD31 staining also indicated that the
combination treatment exerted more pronounced angiogenic effects
compared to the other treatment groups. This is consistent with the
findings regarding the changes in VEGF protein levels in the
combination treatment group. Additionally, the extent of Akt and
eNOS phosphorylation was more pronounced in the combination group
than in the groups treated with rosuvastatin or LY333531 alone.
Therefore, enhanced neovascularization in the infarcted myocardium
induced by combination treatment with rosuvastatin and LY333531
seems to ameliorate dysfunctional ventricular remodeling. However,
it has also been shown that treatment with both statins and
LY333531 alone decreases the expression of TGF-β1 with a
concomitant decrease in myocardial fibrosis (42,43). In the present study, we cannot
exclude the possibility that the decreased extent of myocardial
fibrosis by statins and LY333531 also contributed to the
improvement of cardiac function. In particular, we did not observe
a more pronounced decrease in serum TGF-β1 protein levels in the
group treated with the combination of both agents as compared to
the groups treated with either agent alone.
In conclusion, to the best of our knowledge, the
results of the present study suggest, for the first time, that
treatment with rosuvastatin in combination with LY333531 is more
beneficial compared to treatment with either statins or LY333531
alone during an MI event under diabetic conditions. The beneficial
effects are likely attributed to the increased numbers of coronary
collaterals.
Acknowledgments
The present study was supported a grant from the
Science and Technology Commission of Shanghai Municipality to D.H.
(no. 14ZR1432000).
References
|
1
|
Nordlie MA, Wold LE and Kloner RA: Genetic
contributors toward increased risk for ischemic heart disease. J
Mol Cell Cardiol. 39:667–679. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Habib GB, Heibig J, Forman SA, Brown BG,
Roberts R, Terrin ML, et al: Influence of coronary collateral
vessels on myocardial infarct size in humans: results of phase I
Thrombolysis in Myocardial Infarction (TIMI) trial: the TIMI
Investigators. Circulation. 83:739–746. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Samuel SM, Thirunavukkarasu M, Penumathsa
SV, Koneru S, Zhan L, Maulik G, et al: Thioredoxin-1 gene therapy
enhances angiogenic signaling and reduces ventricular remodeling in
infarcted myocardium of diabetic rats. Circulation. 121:1244–1255.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Boodhwani M, Sodha NR, Mieno S, Xu SH,
Feng J, Ramlawi B, et al: Functional, cellular, and molecular
characterization of the angiogenic response to chronic myocardial
ischemia in diabetes. Circulation. 116(11 Suppl): 131–137. 2007.
View Article : Google Scholar
|
|
5
|
Martin A, Komada MR and Sane DC: Abnormal
angiogenesis in diabetes mellitus. Med Res Rev. 23:117–145. 2003.
View Article : Google Scholar
|
|
6
|
Shweiki D, Itin A, Soffer D and Keshet E:
Vascular endothelial growth factor induced by hypoxia may mediate
hypoxia-initiated angiogenesis. Nature. 359:843–845. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hammes HP, Lin J, Bretzel RG, Brownlee M
and Breier G: Upregulation of the vascular endothelial growth
factor/vascular endothelial growth factor receptor system in
experimental background diabetic retinopathy of the rat. Diabetes.
47:401–406. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cooper ME, Vranes D, Youssef S, Stacker
SA, Cox AJ, Rizkalla B, et al: Increased renal expression of
vascular endothelial growth factor (VEGF) and its receptor VEGFR-2
in experimental diabetes. Diabetes. 48:2229–2239. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chou E, Suzuma I, Way KJ, Opland D,
Clermont AC, Naruse K, et al: Decreased cardiac expression of
vascular endothelial growth factor and its receptors in
insulin-resistant and diabetic States: a possible explanation for
impaired collateral formation in cardiac tissue. Circulation.
105:373–379. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mellor H and Parker PJ: The extended
protein kinase C super-family. Biochem J. 332:281–292. 1998.
|
|
11
|
Geraldes P and King GL: Activation of
protein kinase C isoforms and its impact on diabetic complications.
Circ Res. 106:1319–1331. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Inoguchi T, Battan R, Handler E, Sportsman
JR, Heath W and King GL: Preferential elevation of protein kinase C
isoform II and diacylglycerol levels in the aorta and heart of
diabetic rats: differential reversibility to glycemic control by
islet cell transplantation. Proc Natl Acad Sci USA. 89:11059–11063.
1992. View Article : Google Scholar
|
|
13
|
Sheetz MJ and King GL: Molecular
understanding of hyperglycemia’s adverse effects for diabetic
complications. JAMA. 288:2579–2588. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wei L, Yin Z, Yuan Y, Hwang A, Lee A, Sun
D, et al: A PKC-beta inhibitor treatment reverses cardiac
microvascular barrier dysfunction in diabetic rats. Microvasc Res.
80:158–165. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Naruse K, Rask-Madsen C, Takahara N, Ha
SW, Suzuma K, Way KJ, et al: Activation of vascular protein kinase
C-beta inhibits Akt-dependent endothelial nitric oxide synthase
function in obesity-associated insulin resistance. Diabetes.
55:691–698. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
He Z and King GL: Protein kinase C beta
isoform inhibitors: a new treatment for diabetic cardiovascular
diseases. Circulation. 110:7–9. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sata M, Nishimatsu H, Osuga J, Tanaka K,
Ishizaka N, Ishibashi S, et al: Statins augment collateral growth
in response to ischemia but they do not promote cancer and
atherosclerosis. Hypertension. 43:1214–1220. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Erbs S, Beck EB, Linke A, Adams V, Gielen
S, Kränkel N, et al: High-dose rosuvastatin in chronic heart
failure promotes vasculogenesis, corrects endothelial function, and
improves cardiac remodeling–results from a randomized,
double-blind, and placebo-controlled study. Int J Cardiol.
146:56–63. 2011. View Article : Google Scholar
|
|
19
|
Siddiqui AJ, Gustafsson T, Fischer H,
Widegren U, Hao X, Mansson-Broberg A, et al: Simvastatin enhances
myocardial angiogenesis induced by vascular endothelial growth
factor gene transfer. J Mol Cell Cardiol. 37:1235–1244.
2004.PubMed/NCBI
|
|
20
|
Miura S, Matsuo Y and Saku K:
Transactivation of KDR/Flk-1 by the B2 receptor induces tube
formation in human coronary endothelial cells. Hypertension.
41:1118–1123. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakamura S, Chikaraishi Y, Tsuruma K,
Shimazawa M, Hara H, et al: Ruboxistaurin, a PKCbeta inhibitor,
inhibits retinal neovascularization via suppression of
phosphorylation of ERK1/2 and Akt. Exp Eye Res. 90:137–145. 2010.
View Article : Google Scholar
|
|
22
|
Shi F, Wang YC, Zhao TZ, Zhang S, Du TY,
Yang CB, et al: Effects of simulated microgravity on human
umbilical vein endothelial cell angiogenesis and role of the
PI3K-Akt-eNOS signal pathway. PLoS One. 7:e403652012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Huang FY, Mei WL, Li YN, Tan GH, Dai HF,
Guo JL, et al: Toxicarioside A inhibits tumor growth and
angiogenesis: involvement of TGF-β/endoglin signaling. PLoS One.
7:e503512012. View Article : Google Scholar
|
|
24
|
Xin P, Zhu W, Li J, Ma S, Wang L, Liu M,
et al: Combined local ischemic postconditioning and remote
perconditioning recapitulate cardioprotective effects of local
ischemic preconditioning. Am J Physiol Heart Circ Physiol.
298:H1819–H1831. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Derumeaux G, Mulder P, Richard V,
Chagraoui A, Nafeh C, Bauer F, et al: Tissue Doppler imaging
differentiates physiological from pathological pressure-overload
left ventricular hypertrophy in rats. Circulation. 105:1602–1608.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Tang JM, Wang JN, Zhang L, Zheng F, Yang
JY, Kong X, et al: VEGF/SDF-1 promotes cardiac stem cell
mobilization and myocardial repair in the infarcted heart.
Cardiovasc Res. 91:402–411. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Xie J, Lu W, Gu R, Dai Q, Zong B, Ling L,
et al: The impairment of ILK related angiogenesis involved in
cardiac maladaptation after infarction. PLoS One. 6:e241152011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yu T, Zhu W, Gu B, Li S, Wang F, Liu M, et
al: Simvastatin attenuates sympathetic hyperinnervation to prevent
atrialfibrillation during the postmyocardial infarction remodeling
process. J Appl Physiol. 113:1937–1944. 2012. View Article : Google Scholar
|
|
29
|
Chilian WM, Penn MS, Pung YF, Dong F,
Mayorga M, Ohanyan V, et al: Coronary collateral growth - back to
the future. J Mol Cell Cardiol. 52:905–911. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hoole SP, White PA, Read PA, Heck PM, West
NE, O’Sullivan M, et al: Coronary collaterals provide a constant
scaffold effect on the left ventricle and limit ischemic left
ventricular dysfunction in humans. J Appl Physiol. 112:1403–1409.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fujita M, Ikemoto M, Kishishita M, Otani
H, Nohara R, Tanaka T, et al: Elevated basic fibroblast growth
factor in pericardial fluid of patients with unstable angina.
Circulation. 94:610–613. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Chilian WM, Mass HJ, Williams SE, Layne
SM, Smith EE, Scheel KW, et al: Microvascular occlusions promote
coronary collateral growth. Am J Physiol. 258:H1103–H1111.
1990.PubMed/NCBI
|
|
33
|
Abaci A, Oguzhan A, Kahraman S, Eryol NK,
Unal S, Arinc H, et al: Effect of diabetes mellitus on formation of
coronary collateral vessels. Circulation. 99:2239–2242. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yarom R, Zirkin H, Stammler G and Rose AG:
Human coronary microvessels in diabetes and ischaemia. Morphometric
study of autopsy material. J Pathol. 166:265–270. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Dubois S, Madec AM, Mesnier A, Armanet M,
Chikh K, Berney T, et al: Glucose inhibits angiogenesis of isolated
human pancreatic islets. J Mol Endocrinol. 45:99–105. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Elewa HF, El-Remessy AB, Somanath PR and
Fagan SC: Diverse effects of statins on angiogenesis: new
therapeutic avenues. Pharmacotherapy. 30:169–176. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Zaitone SA and Abo-Gresha NM: Rosuvastatin
promotes angiogenesis and reverses isoproterenol-induced acute
myocardial infarction in rats: role of iNOS and VEGF. Eur J
Pharmacol. 691:134–142. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Liu Y, Lei S, Gao X, Mao X, Wang T, Wong
GT, et al: PKCβ inhibition with ruboxistaurin reduces oxidative
stress and attenuates left ventricular hypertrophy and dysfunction
in rats with streptozotocin-induced diabetes. Clin Sci (Lond).
122:161–173. 2012. View Article : Google Scholar
|
|
39
|
Kelly DJ, Zhang Y, Hepper C, Gow RM,
Jaworski K, Kemp BE, et al: Protein kinase C beta inhibition
attenuates the progression of experimental diabetic nephropathy in
the presence of continued hypertension. Diabetes. 52:512–518. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Wang F, Huang D, Zhu W, Li S, Yan M, Wei
M, et al: Selective inhibition of PKCβ2 preserves cardiac function
after myocardial infarction and is associated with improved
angiogenesis of ischemic myocardium in diabetic rats. Int J Mol
Med. 32:1037–1046. 2013.PubMed/NCBI
|
|
41
|
Ikeda A, Matsushita S and Sakakibara Y:
Inhibition of protein kinase Cβ ameliorates impaired angiogenesis
in type I diabetic mice complicating myocardial infarction. Circ J.
76:943–949. 2012. View Article : Google Scholar
|
|
42
|
Ma YX, Li WH and Xie Q: Rosuvastatin
inhibits TGF-beta1 expression and alleviates myocardial fibrosis in
diabetic rats. Pharmazie. 68:355–358. 2013.PubMed/NCBI
|
|
43
|
Palaniyandi SS, Ferreira JC, Brum PC and
Mochly-Rosen D: PKCβII inhibition attenuates myocardial infarction
induced heart failure and is associated with a reduction of
fibrosis and pro-inflammatory responses. J Cell Mol Med.
15:1769–1777. 2011. View Article : Google Scholar :
|