Introduction
Dilated cardiomyopathy (DCM) is a progressive
myocardial disease of unknown etiology characterized by left
ventricular dilation and contractile dysfunction with normal
ventricular wall thickness (1).
It is the most common type of primary cardiomyopathy, with an
estimated prevalence of 1 in 2,500 individuals (1). DCM may lead to ventricular systolic
dysfunction and progressive heart failure, ventricular and
supraventricular arrhythmias, conduction system anomalies,
thromboembolic events and sudden cardiac death (1–5).
Indeed, DCM is the third most common cause of heart failure and the
most frequent reason for heart transplantation (1). A broad range of acquired causes have
been implicated in the pathogenesis of DCM, including viral
infections that often induce myocarditis, cardiac toxins or toxic
chemotherapeutic agents, the chronic excessive consumption of
alcohol, autoimmune abnormality and mitochondrial, metabolic,
endocrinal or nutritional disorders (1,6–9).
However, following a thorough review of secondary causes, a
substantial portion of DCM cases remain unexplained and such DCM is
defined as idiopathic DCM, among which 25–50% of DCM cases occur in
at least two closely related family members, hence termed familial
DCM (10). Aggregating evidence
has demonstrated that genetic defects play a crucial role in the
development of DCM, and mutations in >60 genes have been
associated with DCM (10–22). Among the DCM-associated genes, the
majority encode sarcomeric, Z-disc and cytoskeletal proteines that
are responsible for the generation and transmission of contractile
force, and are inherited predominantly in an autosomal dominant
mode, with the autosomal recessive, X-linked, or mitochondrial mode
of inheritance less frequent (10). Nevertheless, these established
DCM-associated genes only account for less than a third of the
examined cases and each gene has a low mutational frequency, with
the majority occurring in <1% of patients with DCM (23). Therefore, the genetic components
underpinning DCM in an overwhelming majority of patients remain to
be identified.
Previous studies have underscored the pivotal roles
of the cardiac transcription factors in cardiac morphogenesis and
the proliferation, specification and differentiation of
cardiomyocytes, including the GATA zinc finger-containing
transcription factor and the NK homeodomain transcription factor
families (24–29); a great number of mutations in GATA
binding protein (GATA)4, GATA5, GATA6 and NK2 transcription factor
related, locus 5 (NKX2.5) have shown to be been involved in various
congenital cardiovascular malformations and arrhythmias in humans,
including atrial septal defect, ventricular septal defect,
tetralogy of Fallot, double outlets of the right ventricle,
endocardial cushion defect, atrial fibrillation and cardiac
conduction block (30–64). Furthermore, GATA4, GATA6 and
NKX2.5 have also been causally linked to human DCM (16–20). Given that the expression profile
and functional characteristics of GATA5 overlap at least in part
with those of GATA4, GATA6 and NKX2.5, and that GATA5 physically
interacts with NKX2.5, collaborating to synergistically activate
several key cardiac target genes, such as the sarcomeric genes
encoding α-myosin heavy chain, β-myosin heavy chain, cardiac
troponin C and troponin I, and other genes coding for atrial
natriuretic factor (ANF), brain natriuretic peptide and connexin40
(16,24,27,64–68), it is justifiable to make a
hypothesis that genetically defective GATA5 can contribute to the
development of DCM.
Materials and methods
Ethics statement
This study conforms to the principles outlined in
the Declaration of Helsinki. The study protocol was reviewed and
approved by the local institutional ethics committee of Shanghai
Tenth People’s Hospital, Tongji University, Shanghai, China.
Written informed consent was obtained from all participants or
their guardians prior to enrollment in the study.
Study population
Patients affected with idiopathic DCM were recruited
from the Han Chinese population. Prior to recruitment, all patients
were evaluated by detailed history, physical examination, chest
radiography, electrocardiogram, echocardiography and exercise
performance testing. Cardiac catheterization, ventricular
angiography and myocardial biopsy were performed only if there was
a strong clinical indication. Idiopathic DCM was diagnosed
according to the criteria established by the World Health
Organization/International Society and Federation of Cardiology
Task Force on the Classification of Cardiomyopathy: a left
ventricular end-diastolic diameter >27 mm/m2 and an
ejection fraction <40% or fractional shortening <25% in the
absence of abnormal loading conditions, coronary artery disease,
congenital heart malformations and other systemic disorders
(17–19,69). Patients with concomitant disease
that may contribute to heart failure, such as coronary artery
disease, congenital heart disease, valvular heart disease, viral
myocarditis and essential hypertension, were excluded. Familial DCM
was defined when DCM occurred in 2 or more first-degree family
relatives. The available family members of the index patients were
also included in this study and for the deceased or unavailable
relatives, the medical records were reviewed. The control
population consisted of ethnically-matched healthy individuals.
Peripheral venous blood samples were obtained from all
participants.
Mutational scan of GATA5
Genomic DNA was isolated from the peripheral blood
leucocytes of each study subject using the Wizard Genomic DNA
Purification kit (Promega, Madison, WI, USA). The coding exons and
exon-intron boundaries of the GATA5 gene were sequenced in
130 unrelated patients with idiopathic DCM. When a mutation was
identified in an index patient, the available relatives of the
mutation carrier and 200 unrelated healthy controls were
subsequently genotyped for GATA5. The primer pairs used to
amplify the coding regions and splice junction sites of
GATA5 by polymerase chain reaction (PCR) were designed as
previously described (46). PCR
was conducted using HotStar TaqDNA polymerase (Qiagen, Hilden,
Germany) on a Veriti Thermal Cycler (Applied Biosystems, Foster,
CA, USA) with standard conditions and concentrations of reagents.
The amplified product was fractionated on a 1% agarose gel and then
extracted using the QIAquick Gel Extraction kit (Qiagen), as
described in a previous study of ours (19). Both strands of each PCR product
were sequenced using the BigDye® Terminator version 3.1
Cycle Sequencing kit under an ABI PRISM 3130 XL DNA Analyzer (both
from Applied Biosystems). DNA sequences were analyzed using the DNA
Sequencing Analysis Software version 5.1 (Applied Biosystems). The
identified sequence variation was verifieded by re-sequencing of an
independent PCR-generated amplicon from the same subject.
Additionally, for an identified variant, the single nucleotide
polymorphism (SNP; http://www.ncbi.nlm.nih.gov/SNP) and human gene
mutation (HGM; http://www.hgmd.org) databases were
queried to confirm its novelty.
Alignment of multiple amino acid
sequences of GATA5 across species
The amino acid sequence of human GATA5 was aligned
with that of the chimpanzee, rhesus monkey, dog, cattle, mouse,
rat, fowl, zebrafish and frog using the online Muscle program
(http://www.ebi.ac.uk/Tools/msa/muscle/).
Functional analysis in silico
The disease-causing potential of a GATA5
sequence variation was predicted by MutationTaster (an online
program at http://www.mutationtaster.org), which automatically
yields a probability for the variation to be either a pathogenic
mutation or a benign polymorphism. Notably, the P-value used here
is the probability of the correct prediction (i.e., a value close
to 1 indicates a high accuracy of the prediction). Additionally,
another online program, PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), was also used
to evaluate the possible effect of an amino acid substitution on
the function of GATA5.
Expression plasmids and site-directed
mutagenesis
The recombinant expression plasmid, GATA5-pcDNA3.1,
was constructed as previously described (46). The reporter plasmid,
ANF-luciferase (ANF-luc), which contains the 2,600-bp 5′-fanking
region of the ANF gene, was a kind gift from Dr Ichiro
Shiojima at Chiba University School of Medicine, Chiba, Japan. The
identified mutation was introduced into the wild-type GATA5
expression plasmid using a QuickChange II XL Site-Directed
Mutagenesis kit (Stratagene, La Jolla, CA, USA) with a
complementary pair of primers. The full-length cDNA of mutant GATA5
was sequenced to confirm the desired mutation and to exclude any
other non-synonymous variations.
Reporter gene assays
HEK-293 cells (from our cell bank) were seeded in
12-well plates and cultured in Dulbecco’s modified Eagle’s medium
supplemented with 10% fetal calf serum for 24 h before being
transfected with the empty pcDNA3.1 vector, wild-type or mutant
GATA5-pcDNA3.1 expression construct along with the Firefly
luciferase reporter plasmid. In addition, the Renilla
luciferase vector pGL4.75 (hRluc/CMV; Promega) was co-transfected
into the cells as an internal control for transfection efficiency.
The cells were transfected with 0.4 μg of pcDNA3.1,
wild-type or mutant GATA5-pcDNA3.1, 1.0 μg of ANF-luc, and
0.04 μg of pGL4.75 using Lipofectomine 2000 transfection
reagent (Invitrogen, Carlsbad, CA, USA). For co-transfection
experiments, 0.2 μg of wild-type GATA5-pcDNA3.1, 0.2
μg of mutant GATA5-pcDNA3.1, 1.0 μg of ANF-luc and
0.04 μg of pGL4.75 were used. The cells were incubated at
37°C and harvested 48 h after transfection. The luciferase activity
of the lysates was measured using the Dual-Luciferase Reporter
assay system (Promega) according to the manufacturer’s
instructions. The activity of the ANF promoter was presented as the
fold activation of Firefly luciferase relative to Renilla
luciferase. For wild-type or mutant GATA5, a minimum of 3
independent experiments were performed in triplicate.
Statistical analysis
Continuous variables are expressed as the means ±
standard deviation (SD). The Student’s unpaired t-test was used to
compare the continuous variables between 2 groups. A comparison of
the categorical variables between 2 groups was carried out using
Pearson’s χ2 test. A two-tailed P-value <0.05 was
considered to indicate a statistically significant difference.
Results
Baseline clinical characteristics of the
study population
A cohort of 130 genetically unrelated patients with
idiopathic DCM was clinically investigated in contrast to 200
unrelated control (healthy) individuals. They had no apparent
traditional risk factors predisposing to DCM. All the patients
presented with the typical DCM phenotype as previously described
(1,17–19,69). The control individuals had normal
echocardiographic parameters without evidence of structural cardiac
diseases. The baseline clinical characteristics of the study
population are summarized in Table
I.
 | Table IBaseline clinical characteristics of
the study participants. |
Table I
Baseline clinical characteristics of
the study participants.
|
Characteristics | Patients
(n=130) | Controls
(n=200) | P-value |
|---|
| Age (years) | 56.8±12.4 | 57.3±11.5 | 0.7085 |
| Male (%) | 72 (55) | 110 (55) | 0.9453 |
| Family history of
DCM (%) | 35 (27) | 0 (0) | <0.0001 |
| SBP (mmHg) | 114.2±13.6 | 125.4±10.9 | <0.0001 |
| DBP (mmHg) | 76.5±8.8 | 84.7±7.1 | <0.0001 |
| HR (bpm) | 92.4±11.3 | 75.8±9.5 | <0.0001 |
| LVEDD (mm) | 68.2± 8.2 | 48.1±6.4 | <0.0001 |
| LVESD (mm) | 56.5±7.3 | 36.2±6.9 | <0.0001 |
| LVEF (%) | 37.2±10.4 | 62.7±8.0 | <0.0001 |
| NYHA function class
(%) | | | |
| I | 26 (20) | NA | NA |
| II | 52 (40) | NA | NA |
| III | 40 (31) | NA | NA |
| IV | 12 (9) | NA | NA |
Novel GATA5 mutation identified
By sequence analysis of the GATA5 gene in the
130 unrelated patients with idiopathic DCM, a heterozygous missense
mutation was identified in a patient, with a mutational prevalence
of approximately 0.77%. Specifically, a substitution of adenine for
guanine in the second nucleotide of codon 240 (c.719G>A),
predicting the transition of glycine (G) into aspartic acid (D) at
amino acid position 240 (p.G240D) was identified in the index
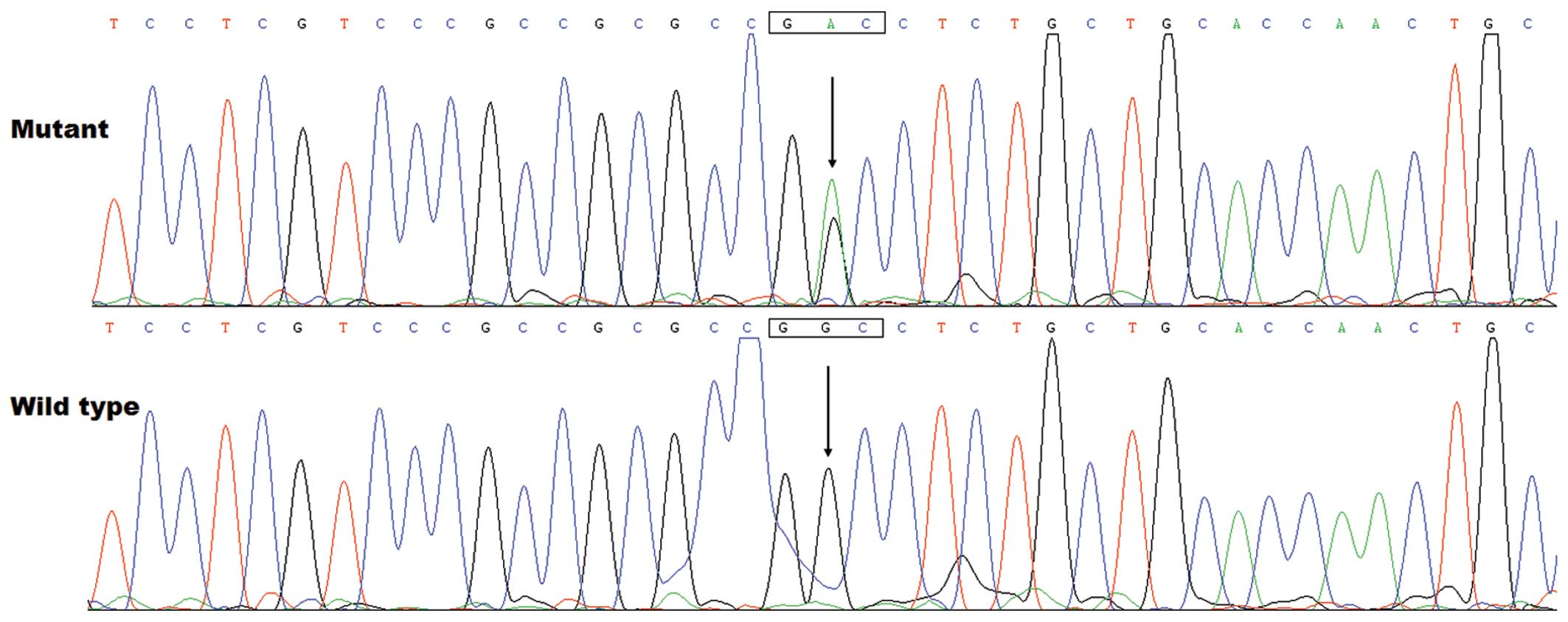
patient from family 1. The sequence chromatograms showing the
detected heterozygous GATA5 mutation of c.719G>A compared
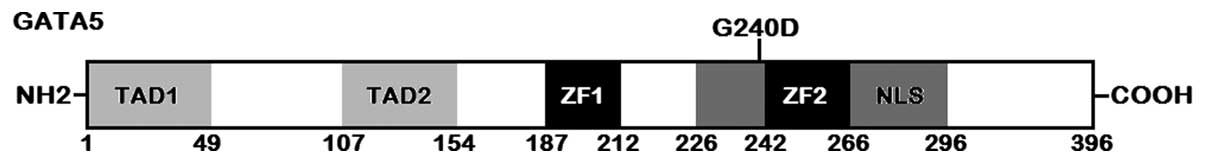
with its control sequence are shown in Fig. 1. A schematic diagram of GATA5
protein with the identified mutation marked above the structural
domains is displayed in Fig. 2.
The mutation was neither detected in the 200 control subjects nor
reported in the public databases for human sequence variations,
including the SNP and HGM databases, suggesting that it was a novel
mutation. The genetic screening of the family of the proband
revealed that the mutation was present in all affected family
members alive, but absent in unaffected family members examined.
Analysis of the pedigree demonstrated that the mutation
co-segregated with DCM transmitted as an autosomal dominant trait
in the family with complete penetrance. Besides, the father of the
proband (I-1), younger sister (II-6) and younger brother (II-7)
also had ventricular septal defect together with second-degree
atrioventricular block, paroxismal atrial fibrillation and
ventricular septal defect, respectively. The pedigree structure of
the family is exhibited in Fig.
3. The phenotypic characteristics and status of GATA5 mutation
of the affected family members are provided in Table II.
| Table IIPhenotypic characteristics of the
affected pedigree members. |
Table II
Phenotypic characteristics of the
affected pedigree members.
| Individual | Gender | Age (years) | Cardiac
phenotype | LVEDD (mm) | LVESD (mm) | LVEF (%) | LVFS (%) | ECG findings | GATA5 mutation |
|---|
| I-1 | M | 65a | DCM, VSD | 78 | 66 | 29 | 15 | AVB | NA |
| II-3 | M | 50 | DCM | 75 | 60 | 41 | 20 | | +/− |
| II-6 | F | 47 | DCM | 64 | 50 | 32 | 21 | PAF | +/− |
| II-7 | M | 43 | DCM, VSD | 60 | 47 | 35 | 23 | | +/− |
| III-3 | M | 26 | DCM | 57 | 46 | 19 | 19 | | +/− |
Alignment of multiple GATA5 protein
sequences across species
A cross-species alignment of GATA5 protein sequences
revealed that the altered amino acid, p.G240, was completely
conserved evolutionarily in various species (Fig. 4).
Disease-causing potential analyzed in
silico
The GATA5 sequence variation of c.719G>A
was predicted by MutationTaster to be a causative mutation with a
P-value of almost 1.0. The corresponding amino acid substitution of
p.G240D was also predicted by PolyPhen-2 to be possibly damaging,
with a score of 0.968 (sensitivity, 0.61; specificity, 0.93).
Diminished transcriptional activity of
the GATA5 mutant
The same amount (0.4 μg) of wild-type and
G240D-mutant GATA5 activated the ANF promoter by ~12- and
~2-fold, respectively (Fig. 5).
When the same amount of wild-type GATA5 (0.2 μg) was
co-transfected with G240D-mutant GATA5 (0.2 μg), the induced
activation of the ANF promoter was ~4-fold. These results
demonstrate that the G240D-mutant GATA5 has a significantly
decreased transcriptional activity compared with its wild-type
counterpart.
Discussion
In the present study, a novel heterozygous GATA5
mutation, p.G240D, was identified in a family with DCM transmitted
in an autosomal dominant manner. In the family, the missense
mutation co-segregated with DCM with complete penetrance. This
sequence variation altered the amino acid that was completely
conserved evolutionarily, and was predicted to be a pathogenic
mutation by MutationTaster and PolyPhen-2. Functional assays
unveiled that the mutant GATA5 was associated with significantly
reduced transcriptional activity. These findings demonstrate that
the GATA5 loss-of-function mutation can contribute to the
development of DCM.
To date, 6 members of the GATA transcription factor
family have been identified in vertebrates and parsed into 2
subfamilies based on their expression patterns. GATA1-3
genes are prominently expressed in hematopoietic cell lineages
where they regulate differentiation-specific gene expression in
T-lymphocytes, erythroid cells and megakaryocytes; GATA4-6
genes are expressed in various mesoderm- and endoderm-derived
tissues such as heart, liver, lung, gonad and gut tissues, where
they play critical roles in regulating tissue-specific gene
expression (70). In humans, the
GATA5 gene was mapped on chromosome 20q13.33 by fuorescence
in situ hybridization, which encodes a protein of 396 amino
acids (71). The structural
domains of GATA5 comprise 2 transcriptional activation domains
(TAD1, amino acids 1–49; TAD2, amino acids 107–154), 2 adjacent
zinc fingers (ZF1, amino acids 187–212; ZF2, amino acids 242–266)
and 1 nuclear localization signal (NLS, amino acids 226–296). The 2
TADs are crucial for the proper transcriptional activity of GATA5.
The C-terminal ZF2 is essential for DNA sequence recognition and
binding to the consensus motif (T/A)GATA(A/G), within the promoters
of target genes; the N-terminal ZF1 is responsible for sequence
specificity and stability of protein-DNA binding, and both ZFs can
also interact directly with other regulatory proteins. The NLS is
required for the sub-cellular trafficking and nuclear distribution
of GATA5 (44). The GATA5
mutation of p.G240D identified in this study is located in the NLS,
and thus may be expected to influence the transcriptional activity
of GATA5 by interfering with the nuclear localization of GATA5.
It has been validated that GATA5 is an upstream
mediator of multiple genes expressed during cardiogenesis,
including the ANF gene (72). Hence, the functional effect of the
GATA5 mutation may be ascertained at least in part by analyzing the
transcriptional activation of the ANF promoter in
appropriate cells. In the present study, the functional
characteristics of the novel GATA5 mutation (p.G240D) identified in
the patients with familial DCM were explored by transactivational
analysis in HEK-293 cells and the results unveiled that the
G240D-mutant GATA5 had a significantly reduced transcriptional
activity. These data indicate that haploinsufficiency or
dominant-negative effect resulted from GATA5 mutation is likely to
be an alternative molecular mechanism underlying DCM.
The findings that genetically defective GATA5
confers enhanced susceptibility to DCM may be partially ascribed to
the abnormal development of the myocardium. In zebrafish, the
depletion of the GATA5 transcription factor has been shown to lead
to cardiac morphogenetic defects and the loss of myocardial tissue.
In addition, in zebrafish, the Gata5 and Gata6 genes
are redundant for the specification of cardiomyocytes. Embryos
depleted of these two gene products were heartless and restoring
either gene product was sufficient to rescue cardiomyocyte
specification. By contrast, embryos depleted of Gata4 and
Gata6, or Gata4 and Gata5, developed defective
heart tubes, suggesting that a specific pair of GATA transcription
factors is essential for cardiomyocyte specification (68). Moreover, as a negative regulator
of myocardial proliferative growth in zebrafish embryos, the GRL
transcription factor diminishes cardiomyocyte volume and inhibits
cell proliferation, resulting in a marked reduction in heart size.
These GRL-induced cardiac effects were counterbalanced by the
transcriptional activator Gata5 but not Gata4, which promotes
cardiomyocyte expansion in the embryo, suggesting that GRL
negatively regulates embryonic heart growth by opposing Gata5
(73). In mice, the targeted
deletion of Gata5 has been shown to result in hypoplastic
hearts and partially penetrant bicuspid aortic valves (74). Furthermore, GATA5 interacts with
GATA4 and GATA6 and cooperatively regulates cardiac myocyte
proliferation (65,66). In a new mouse model with
Gata5-mutant allele lacking exons 2 and 3, although
Gata5(−/−) mice were viable,
Gata4(+/−)5(−/−) mutants died at
mid-gestation and exhibited profound cardiovascular defects,
including abnormalities of cardiomyocyte proliferation and cardiac
chamber maturation (66). In
double-gene deletion mouse models, compound Gata4/Gata5 and
Gata5/Gata6 mutants died embryonically or perinatally due to
severe congenital heart defects. Almost all
Gata4(+/−)Gata5(+/−) mutant embryos had double outlet right
ventricles and large ventricular septal defects. The compound loss
of a Gata5 and a Gata6 allele also led to double
outlet right ventricles associated with subaortic ventricular
septal defects (65). Taken
together, these results highlight the existence of important
genetic interactions between GATA5 and the two other cardiac GATA
factors in cardiovascular development.
In humans, a great number of GATA5 mutations have
been previously associated with various congenital cardiovascular
deformities, including atrial septal defect, ventricular septal
defect, tetralogy of Fallot, double outlet right ventricle, aortic
stenosis, patent ductus arteriosus and bicuspid aortic valve
(40–44). In the present study, all the
mutation carriers had DCM, but only 2 mutation carriers also had
ventricular septal defect. The pronunced discrepancy in the
GATA5-related phenotypes may be explained by one or more of the
following reasons. Firstly, since some developmental anomalies in
cardiac structure may be restored spontaneously, we cannot rule out
the possibility that some mutation carriers had minor cardiac
septal defects that healed on their own soon after birth. In
addition, distinct genetic backgrounds may partially account for
the phenotypic heterogeneity. Finally, mutations such as p.G240D
may be merely a genetic modifier that confers vulnerability to the
diseases, rather than a direct cause, other genetic determinants
and environmental risk factors may be required for the onset of
congenital heart diseases.
In conclusion, to the best of our knowledge, this is
the first study on the association of GATA5 loss-of-function
mutation with increased susceptibility to human DCM, which provides
novel insight into the molecular mechanisms responsible for DCM,
suggesting a potential molecular target for the antenatal
prevention and gene-specific treatment of DCM.
Acknowledgments
The authors are extremely thankful to the
participants for their dedication to the study. This study was
supported in part by grants from the National Natural Science Fund
of China (81270161, 81271927 and 81370301), the Natural Science
Fund of Shanghai, China (13ZR1438400 and 14ZR1438000), the
Experimental Animal Fund of Shanghai, China (12140902800), and the
Key Program of Basic Research of Shanghai, China (14JC1405500).
References
|
1
|
Maron BJ, Towbin JA, Thiene G,
Antzelevitch C, Corrado D, Arnett D, Moss AJ, Seidman CE and Young
JB: Contemporary definitions and classification of the
cardiomyopathies: an American Heart Association Scientific
Statement from the Council on Clinical Cardiology, Heart Failure
and Transplantation Committee; Quality of Care and Outcomes
Research and Functional Genomics and Translational Biology
Interdisciplinary Working Groups; and Council on Epidemiology and
Prevention. Circulation. 113:1807–1816. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Abdo AS, Kemp R, Barham J and Geraci SA:
Dilated cardiomyopathy and role of antithrombotic therapy. Am J Med
Sci. 339:557–560. 2010.PubMed/NCBI
|
|
3
|
Aleksova A, Carriere C, Zecchin M, Barbati
G, Vitrella G, Di Lenarda A and Sinagra G: New-onset left bundle
branch block independently predicts long-term mortality in patients
with idiopathic dilated cardiomyopathy: data from the Trieste Heart
Muscle Disease Registry. Europace. 16:1450–1459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Disertori M, Quintarelli S, Mazzola S,
Favalli V, Narula N and Arbustini E: The need to modify patient
selection to improve the benefits of implantable
cardioverter-defibrillator for primary prevention of sudden death
in non-ischaemic dilated cardiomyopathy. Europace. 15:1693–1701.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Koutalas E, Kanoupakis E and Vardas P:
Sudden cardiac death in non-ischemic dilated cardiomyopathy: a
critical appraisal of existing and potential risk stratification
tools. Int J Cardiol. 167:335–341. 2013. View Article : Google Scholar
|
|
6
|
Gaaloul I, Riabi S, Harrath R, Hunter T,
Hamda KB, Ghzala AB, Huber S and Aouni M: Coxsackievirus B
detection in cases of myocarditis, myopericarditis, pericarditis
and dilated cardiomyopathy in hospitalized patients. Mol Med Rep.
10:2811–2818. 2014.PubMed/NCBI
|
|
7
|
Xu HF, Ding YJ, Zhang ZX, Wang ZF, Luo CL,
Li BX, Shen YW, Tao LY and Zhao ZQ: MicroRNA-21 regulation of the
progression of viral myocarditis to dilated cardiomyopathy. Mol Med
Rep. 10:161–168. 2014.PubMed/NCBI
|
|
8
|
Kong Q, Li X, Wu W, Yang F, Liu Y, Lai W,
Pan X, Gao M and Xue Y: Increased circulating T-helper 22 cells in
patients with dilated cardiomyopathy. Mol Med Rep. 10:359–364.
2014.PubMed/NCBI
|
|
9
|
Yoshikawa T: Contribution of acquired
factors to the pathogenesis of dilated cardiomyopathy. -The cause
of dilated cardiomyopathy: genetic or acquired? (Acquired-Side).
Circ J. 75:1766–1773. 2011. View Article : Google Scholar
|
|
10
|
McNally EM, Golbus JR and Puckelwartz MJ:
Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin
Invest. 123:19–26. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wahbi K, Béhin A, Bécane HM, Leturcq F,
Cossée M, Laforêt P, Stojkovic T, Carlier P, Toussaint M, Gaxotte
V, Cluzel P, Eymard B and Duboc D: Dilated cardiomyopathy in
patients with mutations in anoctamin 5. Int J Cardiol. 168:76–79.
2013. View Article : Google Scholar
|
|
12
|
Ruppert V, Meyer T, Richter A, Maisch B
and Pankuweit S: German Competence Network of Heart F:
Identification of a missense mutation in the melusin-encoding
ITGB1BP2 gene in a patient with dilated cardiomyopathy. Gene.
512:206–210. 2013. View Article : Google Scholar
|
|
13
|
Pankuweit S1, Ruppert V, Jónsdóttir T,
Müller HH and Meyer T: German Competence Network of Heart Failure:
The HLA class II allele DQB1 0309 is associated with dilated
cardiomyopathy. Gene. 531:180–183. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Agrawal PB, Pierson CR, Joshi M, Liu X,
Ravenscroft G, Moghadaszadeh B, Talabere T, Viola M, Swanson LC,
Haliloğlu G, Talim B, Yau KS, Allcock RJ, Laing NG, Perrella MA and
Beggs AH: SPEG interacts with myotubularin, and its deficiency
causes centronuclear myopathy with dilated cardiomyopathy. Am J Hum
Genet. 95:218–226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matsa LS, Sagurthi SR, Ananthapur V, Nalla
S and Nallari P: Endothelin 1 gene as a modifier in dilated
cardiomyopathy. Gene. 548:256–262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Costa MW, Guo G, Wolstein O, Vale M,
Castro ML, Wang L, Otway R, Riek P, Cochrane N, Furtado M,
Semsarian C, Weintraub RG, Yeoh T, Hayward C, Keogh A, Macdonald P,
Feneley M, Graham RM, Seidman JG, Seidman CE, Rosenthal N, Fatkin D
and Harvey RP: Functional characterization of a novel mutation in
NKX2-5 associated with congenital heart disease and adult-onset
cardiomyopathy. Circ Cardiovasc Genet. 6:238–247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Li RG, Li L, Qiu XB, Yuan F, Xu L, Li X,
Xu YJ, Jiang WF, Jiang JQ, Liu X, Fang WY, Zhang M, Peng LY, Qu XK
and Yang YQ: GATA4 loss-of-function mutation underlies familial
dilated cardiomyopathy. Biochem Biophys Res Commun. 439:591–596.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhao L, Xu JH, Xu WJ, Yu H, Wang Q, Zheng
HZ, Jiang WF, Jiang JF and Yang YQ: A novel GATA4 loss-of-function
mutation responsible for familial dilated cardiomyopathy. Int J Mol
Med. 33:654–660. 2014.
|
|
19
|
Li J, Liu WD, Yang ZL, Yuan F, Xu L, Li RG
and Yang YQ: Prevalence and spectrum of GATA4 mutations associated
with sporadic dilated cardiomyopathy. Gene. 548:174–181. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Xu L, Zhao L, Yuan F, Jiang WF, Liu H, Li
RG, Xu YJ, Zhang M, Fang WY, Qu XK, Yang YQ and Qiu XB: GATA6
loss-of-function mutations contribute to familial dilated
cardiomyopathy. Int J Mol Med. 34:1315–1322. 2014.PubMed/NCBI
|
|
21
|
Dhandapany PS, Razzaque MA, Muthusami U,
Kunnoth S, Edwards JJ, Mulero-Navarro S, Riess I, Pardo S, Sheng J,
Rani DS, Rani B, Govindaraj P, Flex E, Yokota T, Furutani M,
Nishizawa T, Nakanishi T, Robbins J, Limongelli G, Hajjar RJ,
Lebeche D, Bahl A, Khullar M, Rathinavel A, Sadler KC, Tartaglia M,
Matsuoka R, Thangaraj K and Gelb BD: RAF1 mutations in
childhood-onset dilated cardiomyopathy. Nat Genet. 46:635–639.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
22
|
Roh JI, Cheong C, Sung YH, Lee J, Oh J,
Lee BS, Lee JE, Gho YS, Kim DK, Park CB, Lee JH, Lee JW, Kang SM
and Lee HW: Perturbation of NCOA6 leads to dilated cardiomyopathy.
Cell Rep. 8:991–998. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Flack E and Kannankeril PJ: The genetics
of dilated cardiomyopathy. Heart Rhythm. 9:397–398. 2012.
View Article : Google Scholar
|
|
24
|
Pikkarainen S, Tokola H, Kerkelä R and
Ruskoaho H: GATA transcription factors in the developing and adult
heart. Cardiovasc Res. 63:196–207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Oka T, Xu J and Molkentin JD:
Re-employment of developmental transcription factors in adult heart
disease. Semin Cell Dev Biol. 18:117–131. 2007. View Article : Google Scholar
|
|
26
|
Kikuchi K, Holdway JE, Werdich AA,
Anderson RM, Fang Y, Egnaczyk GF, Evans T, Macrae CA, Stainier DY
and Poss KD: Primary contribution to zebrafish heart regeneration
by gata4(+) cardiomyocytes. Nature. 464:601–605. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Akazawa H and Komuro I: Cardiac
transcription factor Csx/Nkx2-5: Its role in cardiac development
and diseases. Pharmacol Ther. 107:252–268. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kasahara A, Cipolat S and Chen Y:
Mitochondrial fusion directs cardiomyocyte differentiation via
calcineurin and Notch signaling. Science. 342:734–737. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cai H, Katoh-Kurasawa M, Muramoto T,
Santhanam B, Long Y, Li L, Ueda M, Iglesias PA, Shaulsky G and
Devreotes PN: Nucleocytoplasmic shuttling of a GATA transcription
factor functions as a development timer. Science. 343:12495312014.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Garg V, Kathiriya IS, Barnes R,
Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS,
Hirayama-Yamada K, Joo K, Matsuoka R, Cohen JC and Srivastava D:
GATA4 mutations cause human congenital heart defects and reveal an
interaction with TBX5. Nature. 424:443–447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rajagopal SK, Ma Q, Obler D, Shen J,
Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V,
Srivastava D, Goldmuntz E, Broman KW, Benson DW, Smoot LB and Pu
WT: Spectrum of heart disease associated with murine and human
GATA4 mutation. J Mol Cell Cardiol. 43:677–685. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Yang YQ, Li L, Wang J, Liu XY, Chen XZ,
Zhang W, Wang XZ, Jiang JQ, Liu X and Fang WY: A novel GATA4
loss-of-function mutation associated with congenital ventricular
septal defect. Pediatr Cardiol. 33:539–546. 2012. View Article : Google Scholar
|
|
33
|
Wang J, Sun YM and Yang YQ: Mutation
spectrum of the GATA4 gene in patients with idiopathic atrial
fibrillation. Mol Biol Rep. 39:8127–8135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yang YQ, Wang J, Liu XY, Chen XZ, Zhang W
and Wang XZ: Mutation spectrum of GATA4 associated with congenital
atrial septal defects. Arch Med Sci. 9:976–983. 2013. View Article : Google Scholar
|
|
35
|
Yang YQ, Gharibeh L, Li RG, Xin YF, Wang
J, Liu ZM, Qiu XB, Xu YJ, Xu L, Qu XK, Liu X, Fang WY, Huang RT,
Xue S and Nemer G: GATA4 loss-of-function mutations underlie
familial tetralogy of fallot. Hum Mutat. 34:1662–1671. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang E, Sun S, Qiao B, Duan W, Huang G, An
Y, Xu S, Zheng Y, Su Z, Gu X, Jin L and Wang H: Identification of
functional mutations in GATA4 in patients with congenital heart
disease. PLoS One. 8:e621382013. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xiang R, Fan LL, Huang H, Cao BB, Li XP,
Peng DQ and Xia K: A novel mutation of GATA4 (K319E) is responsible
for familial atrial septal defect and pulmonary valve stenosis.
Gene. 534:320–323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Posch MG, Boldt LH, Polotzki M, Richter S,
Rolf S, Perrot A, Dietz R, Ozcelik C and Haverkamp W: Mutations in
the cardiac transcription factor GATA4 in patients with lone atrial
fibrillation. Eur J Med Genet. 53:201–203. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Jiang JQ, Shen FF, Fang WY, Liu X and Yang
YQ: Novel GATA4 mutations in lone atrial fibrillation. Int J Mol
Med. 28:1025–1032. 2011.PubMed/NCBI
|
|
40
|
Jiang JQ, Li RG, Wang J, Liu XY, Xu YJ,
Fang WY, Chen XZ, Zhang W, Wang XZ and Yang YQ: Prevalence and
spectrum of GATA5 mutations associated with congenital heart
disease. Int J Cardiol. 165:570–573. 2013. View Article : Google Scholar
|
|
41
|
Wei D, Bao H, Zhou N, Zheng GF, Liu XY and
Yang YQ: GATA5 loss-of-function mutation responsible for the
congenital ventriculoseptal defect. Pediatr Cardiol. 34:504–511.
2013. View Article : Google Scholar
|
|
42
|
Wei D, Bao H, Liu XY, Zhou N, Wang Q, Li
RG, Xu YJ and Yang YQ: GATA5 loss-of-function mutations underlie
tetralogy of fallot. Int J Med Sci. 10:34–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Huang RT, Xue S, Xu YJ, Zhou M and Yang
YQ: Somatic GATA5 mutations in sporadic tetralogy of Fallot. Int J
Mol Med. 33:1227–1235. 2014.PubMed/NCBI
|
|
44
|
Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu
L, Liu H, Li RG, Xu YJ, Wang Q, Zheng HZ, Li X, Wang XZ, Zhang M,
Qu XK and Yang YQ: GATA5 loss-of-function mutations associated with
congenital bicuspid aortic valve. Int J Mol Med. 33:1219–1226.
2014.PubMed/NCBI
|
|
45
|
Yang YQ, Wang J, Wang XH, Wang Q, Tan HW,
Zhang M, Shen FF, Jiang JQ, Fang WY and Liu X: Mutational spectrum
of the GATA5 gene associated with familial atrial fibrillation. Int
J Cardiol. 157:305–307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang XH, Huang CX, Wang Q, Li RG, Xu YJ,
Liu X, Fang WY and Yang YQ: A novel GATA5 loss-of-function mutation
underlies lone atrial fibrillation. Int J Mol Med. 31:43–50.
2013.
|
|
47
|
Kodo K, Nishizawa T, Furutani M, Arai S,
Yamamura E, Joo K, Takahashi T, Matsuoka R and Yamagishi H: GATA6
mutations cause human cardiac outflow tract defects by disrupting
semaphorin-plexin signaling. Proc Natl Acad Sci USA.
106:13933–13938. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Lin X, Huo Z, Liu X, Zhang Y, Li L, Zhao
H, Yan B, Liu Y, Yang Y and Chen YH: A novel GATA6 mutation in
patients with tetralogy of Fallot or atrial septal defect. J Hum
Genet. 55:662–667. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Zheng GF, Wei D, Zhao H, Zhou N, Yang YQ
and Liu XY: A novel GATA6 mutation associated with congenital
ventricular septal defect. Int J Mol Med. 29:1065–1071.
2012.PubMed/NCBI
|
|
50
|
Wang J, Luo XJ, Xin YF, Liu Y, Liu ZM,
Wang Q, Li RG, Fang WY, Wang XZ and Yang YQ: Novel GATA6 mutations
associated with congenital ventricular septal defect or tetralogy
of fallot. DNA Cell Biol. 31:1610–1617. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Huang RT, Xue S, Xu YJ and Yang YQ:
Somatic mutations in the GATA6 gene underlie sporadic tetralogy of
Fallot. Int J Mol Med. 31:51–58. 2013.
|
|
52
|
Wang X, Ji W, Wang J, Zhao P, Guo Y, Xu R,
Chen S and Sun K: Identification of two novel GATA6 mutations in
patients with nonsyndromic conotruncal heart defects. Mol Med Rep.
10:743–748. 2014.PubMed/NCBI
|
|
53
|
Yang YQ, Wang XH, Tan HW, Jiang WF, Fang
WY and Liu X: Prevalence and spectrum of GATA6 mutations associated
with familial atrial fibrillation. Int J Cardiol. 155:494–496.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Yang YQ, Li L, Wang J, Zhang XL, Li RG, Xu
YJ, Tan HW, Wang XH, Jiang JQ, Fang WY and Liu X: GATA6
loss-of-function mutation in atrial fibrillation. Eur J Med Genet.
55:520–526. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Li J, Liu WD, Yang ZL and Yang YQ: Novel
GATA6 loss-of-function mutation responsible for familial atrial
fibrillation. Int J Mol Med. 30:783–790. 2012.PubMed/NCBI
|
|
56
|
Moak JP, Maron BJ, Seidman CE and Seidman
JG: Congenital heart disease caused by mutations in the
transcription factor NKX2-5. Science. 281:108–111. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Guntheroth W, Chun L, Patton KK,
Matsushita MM, Page RL and Raskind WH: Wenckebach periodicity at
rest that normalizes with tachycardia in a family with a NKX2.5
mutation. Am J Cardiol. 110:1646–1650. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Perera JL, Johnson NM, Judge DP and
Crosson JE: Novel and highly lethal NKX2.5 missense mutation in a
family with sudden death and ventricular arrhythmia. Pediatr
Cardiol. 35:1206–1212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Izumi K, Noon S, Wilkens A and Krantz ID:
NKX2.5 mutation identification on exome sequencing in a patient
with heterotaxy. Eur J Med Genet. 57:558–561. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Qu XK, Qiu XB, Yuan F, Wang J, Zhao CM,
Liu XY, Zhang XL, Li RG, Xu YJ, Hou XM, Fang WY, Liu X and Yang YQ:
A novel NKX2.5 loss-of-function mutation associated with congenital
bicuspid aortic valve. Am J Cardiol. 114:1891–1895. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Xie WH, Chang C, Xu YJ, Li RG, Qu XK, Fang
WY, Liu X and Yang YQ: Prevalence and spectrum of Nkx2.5 mutations
associated with idiopathic atrial fibrillation. Clinics (Sao
Paulo). 68:777–784. 2013. View Article : Google Scholar
|
|
62
|
Huang RT, Xue S, Xu YJ, Zhou M and Yang
YQ: A novel NKX2.5 loss-of-function mutation responsible for
familial atrial fibrillation. Int J Mol Med. 31:1119–1126.
2013.PubMed/NCBI
|
|
63
|
Yu H, Xu JH, Song HM, Zhao L, Xu WJ, Wang
J, Li RG, Xu L, Jiang WF, Qiu XB, Jiang JQ, Qu XK, Liu X, Fang WY,
Jiang JF and Yang YQ: Mutational spectrum of the NKX2-5 gene in
patients with lone atrial fibrillation. Int J Med Sci. 11:554–563.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
McCulley DJ and Black BL: Transcription
factor pathways and congenital heart disease. Curr Top Dev Biol.
100:253–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Laforest B and Nemer M: GATA5 interacts
with GATA4 and GATA6 in outflow tract development. Dev Biol.
358:368–378. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Singh MK, Li Y, Li S, Cobb RM, Zhou D, Lu
MM, Epstein JA, Morrisey EE and Gruber PJ: Gata4 and Gata5
cooperatively regulate cardiac myocyte proliferation in mice. J
Biol Chem. 285:1765–1772. 2010. View Article : Google Scholar :
|
|
67
|
Haworth KE, Kotecha S, Mohun TJ and
Latinkic BV: GATA4 and GATA5 are essential for heart and liver
development in Xenopus embryos. BMC Dev Biol. 8:742008. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Holtzinger A and Evans T: Gata5 and Gata6
are functionally redundant in zebrafish for specification of
cardiomyocytes. Dev Biol. 312:613–622. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Elliott P, O’Mahony C, Syrris P, Evans A,
Rivera Sorensen C, Sheppard MN, Carr-White G, Pantazis A and
McKenna WJ: Prevalence of desmosomal protein gene mutations in
patients with dilated cardiomyopathy. Circ Cardiovasc Genet.
3:314–322. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Molkentin JD: The zinc finger-containing
transcription factors GATA-4, -5, and -6. Ubiquitously expressed
regulators of tissue-specific gene expression. J Biol Chem.
275:38949–38952. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
71
|
Nemer G, Qureshi ST, Malo D and Nemer M:
Functional analysis and chromosomal mapping of Gata5, a gene
encoding a zinc finger DNA-binding protein. Mamm Genome.
10:993–999. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
McBride K and Nemer M: Regulation of the
ANF and BNP promoters by GATA factors: lessons learned for cardiac
transcription. Can J Physiol Pharmacol. 79:673–681. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Jia H, King IN, Chopra SS, Wan H, Ni TT,
Jiang C, Guan X, Wells S, Srivastava D and Zhong TP: Vertebrate
heart growth is regulated by functional antagonism between Gridlock
and Gata5. Proc Natl Acad Sci USA. 104:14008–14013. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Laforest B, Andelfinger G and Nemer M:
Loss of Gata5 in mice leads to bicuspid aortic valve. J Clin
Invest. 121:2876–2887. 2011. View Article : Google Scholar : PubMed/NCBI
|