Introduction
It is well recognized that Parkinson’s disease (PD),
one of the most common neurodegenerative movement disorders, is
characterized by the selective degeneration of dopaminergic neurons
in the nigrostriatal system (1).
Recent statistics show that PD affects approximately 2% of the
population over the age of 60 and the incidence is expected to rise
dramatically in the next 25 years with the extension of life
expectancy by improved health care (2). Although the detailed mechanisms
responsible for PD are still under investigation (3), extensive research over the last
several decades has indicated that mitochondrial dysfunction and
oxidative stress resulting from the excessive production of
reactive oxygen species (ROS) play crucial roles in the
pathogenesis of PD (4,5). Oxidative stress induced by ROS
induces the opening of the mitochondrial permeability transition
pore and the dissipation of mitochondrial membrane potential (MMP)
(6) and, subsequently, apoptotic
activators are released from the mitochondria (7), finally resulting in the apoptosis of
dopaminergic neurons observed in PD (8).
Advances in our understanding of dopaminergic
neuronal apoptosis in PD have been achieved by studies on
Parkinsonism induced by
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) and
1-methyl-4-phenylpyridinium ion (MPP+) (9,10).
Moreover, pheochromocytoma cells (PC12 cells), derived from a
clonal rat pheochromocytoma cell line, have been widely used as
cellular models of PD, as these cells share characteristics with
midbrain dopaminergic neurons (11,12). In addition, previous studies have
demonstrated that MPP+-induced cytotoxicity in PC12
cells, which induces apoptotic characteristics accompanied by
mitochondrial dysfunction and oxidative stress, is a classic
cellular model of PD (13,14).
Therefore, in this study, we investigated the effects of three
plant flavonoids on cytotoxicity in MPP+-treated PC12
cells to determine whether they may be of preventive or potential
therapeutic value in PD.
Naturally occurring plant-derived flavonoids have
been suggested to play a role in protecting the central nervous
system against oxidative and excitotoxic stress, although the
mechanisms of action require further study (15). In this study, using
MPP+ as the oxidative insult in PC12 cells, we
investigated the mechanisms responsible for neurotoxicity and
attempted to identify the possible sites of action of three of the
most potent protective flavonoids, apigenin (AP), galangin (GA) and
genkwanin (GE) (the chemical structures are shown in Fig. 1). AP (4′,5,7-trihydroxyflavone),
GA (3,5,7-trihydroxyflavone) and GE
(4′,5-dihydroxy-7-methoxyflavone), which are very similar in
structure, have been reported to possess a number of similar
biological activities, including antioxidant/free radical
scavenging activity, antitumor effects and anti-inflammatory
activity (16–19). Furthermore, AP has been shown to
inhibit Aβ-mediated oxidative damage in nerve cells and in animal
models of Alzheimer’s disease (20,21). Additionally, several antioxidants
and free radical scavengers, including luteolin (22), morin (23) and myricetin (24), which also have a similar chemical
structure to that of these three flavonoid compounds, have been
reported to attenuate the oxidative toxicity induced by
MPP+. However, to the best of our knowlege, no study has
been published to date on the protective effects of these three
flavonoid compounds against MPP+-induced toxicity in
vivo or in vitro. Therefore, in the present study, we
aimed to investigate whether these compounds exert protective
effects against MPP+-induced neurotoxicity in PC12 cells
and to explore the underlying molecular mechanisms of these
neuroprotective effects.
Materials and methods
Chemicals and reagents
AP, GA and GE, purchased from Sigma-Aldrich (St.
Louis, MO, USA), were dissolved in dimethyl sulfoxide (DMSO)
(<0.1%) and diluted with serum-free medium prior to each
experiment. MPP+ and methylthiazolyldiphenyl-tetrazolium
bromide (MTT) were also purchased from Sigma-Aldrich.
2′,7′-Dichlorofluorescin diacetate (DCFH-DA) was obtained from
Invitrogen Life Technologies (Carlsbad, CA, USA). Dulbecco’s
modified Eagle’s medium (DMEM), fetal bovine serum, penicillin and
streptomycin were purchased from Gibco (Grand Island, NY, USA). All
other reagents and chemicals used in the study were of analytical
grade.
Cell culture and drug treatment
The PC12 cells, obtained from the American Type
Culture Collection (ATCC, Rockville, MD, USA), were routinely
cultured in DMEM supplemented with heat-inactivated horse serum
(5%, v/v), heat-inactivated fetal calf serum (5%, v/v), penicillin
(100 IU/ml) and streptomycin (100 μg/ml) at 37°C in a
humidified atmosphere of 5% CO2/95% air. The culture
medium was changed every 2 days. In all the experiments, apart from
the assessment of cell viability, the cells were incubated for 24 h
and were then treated for 4 h with or without various
concentrations of AP, GA and GE (3, 6 and 12 μM), prior to
the addition of MPP+ (final concentration, 1,000
μM) for an additional 48 h. The control cells were treated
in the same manner without the addition of the test compounds and
MPP+ to the free serum culture medium. All experiments
were repeated at least 3 times for each treatment condition in each
experiment.
MTT assay
Cell survival was quantified by the colorimetric MTT
assay using a previously described protocol (14). Briefly, the PC12 cells were seeded
in 96-well culture plates at a density of 2×104
cells/well; following treatment with the drugs, MTT solution was
added to the cell cultures at a final concentration of 1 mg/ml
followed by incubation for a further 4 h at 37°C. The supernatant
was carefully aspirated and 150 μl of DMSO were added to
dissolve the formazan crystals. The 96-well microplate was then
transferred to a microplate reader (BMG Labtech, Offenbury,
Germany) and the absorbance was read at 570 nm. Cell viability was
expressed as the percentage of the untreated controls.
Lactate dehydrogenase (LDH) activity
assay
Cytotoxicity was quantitatively assessed by
measuring the activity of LDH released from the damaged cells into
the culture medium (25). The
PC12 cells were seeded in 96-well culture plates at a density of
2×104 cells/well and treated according to the procedures
as described above. At the end of the treatments, the medium was
collected for the measurement of the extracellular LDH level; the
cells were then treated with 0.5% Triton X-100, after being
centrifuged at 10,000 × g and the supernatant was used for the
measurement of the intracellular LDH level by spectrophotometrical
determination at 440 nm following the procedures provided in the
assay kits (Nanjing Jiancheng Bioengineering Institute, Nanjing,
China). LDH release was expressed as the percentage of the total
LDH activity (LDH in the medium + LDH in the cells), according to
the following equation: LDH release (%) = (LDH activity in the
medium/total LDH activity) x100. Cultures under normal conditions
(control group) represent the basal LDH release.
Measurement of intracellular ROS
production
Intracellular ROS production was measured using the
intracellular peroxide-sensitive fluorescent probe, DCFH-DA, as
previously described (25).
Briefly, the PC12 cells were seeded in 96-well black culture plates
at a density of 2×104 cells/well and treated according
to the procedures as described above. Following treatment with
MPP+ and the drugs, the cells were washed twice with
D-Hank’s solution and incubated with DCFH-DA at a final
concentration of 10 μM for 30 min at 37°C in dark. After the
cells were washed twice with D-Hank’s solution to remove the
extracellular DCFH-DA, the fluorescence intensity was measured
using a fluorescence microplate reader (Tecan, Groedig, Austria) at
an excitation wavelength of 485 nm and an emission wavelength of
538 nm. The measured fluorescence values were expressed as the fold
changes relative to the control group.
Additionally, the PC12 cells were seeded in 6-well
culture plates at a density of 1×106 cells/well. At the
end of the drug treatment, the cells were washed twice with
D-Hank’s solution and incubated with DCFH-DA (final concentration,
10 μM) for 30 min at 37°C in the dark. Changes in ROS
production were then assessed using a fluorescence microscope
(Olympus, Tokyo, Japan).
Measurement of MMP
A JC-1 kit (Beyotime, Haimen, China) was used to
measure the mitochondrial depolarization in the PC12 cells
according to the manufacturer’s instructions.
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl benzimidazolyl
carbocyanine iodide (JC-1) is a cationic dye for detecting MMP
changes, where mitochondrial depolarization is indicated by an
increase in the green/red fluorescence intensity ratio. In the
mitochondria of healthy cells, JC-1 forms aggregates and fluoresces
red. When the MMP collapses, the cationic dye remains in the
cytoplasm as green fluorescence, its monomeric form. Briefly, on
the one hand, PC12 cells were seeded in 96-well black culture
plates at a density of 2×104 cells/well. At the end of
the drug treatment, the cells were washed twice with D-Hank’s
solution and incubated with JC-1 reagent (final concentration, 10
μg/ml) for 20 min at 37°C in the dark. Subsequently, the
green (excitation, 490; emission, 530 nm) and red (excitation, 525;
emission, 590 nm) fluorescence were measured using a fluorescence
plate reader (Tecan). The measured green/red fluorescence ratios
were expressed as fold changes relative to the control group.
On the other hand, the PC12 cells were seeded in
6-well culture plates at a density of 1×106 cells/well.
At the end of the drug treatment, the cells were washed twice with
D-Hank’s solution and incubated with JC-1 (final concentration, 10
μg/ml) for 20 min at 37°C in dark. Changes in MMP were
assessed using a fluorescence microscope (Olympus).
Flow cytometric analysis of
apoptosis
The apoptotic rate was measured by flow cytometry
according to the protocol provided with the Annexin V-FITC/PI kit
(Sigma-Aldrich). Briefly, the PC12 cells were seeded in 6-well
culture plates at a density of 1×106 cells/well. At the
end of the drug treatment, the cells were harvested by
centrifugation at 1,000 × g, washed twice with ice-cold
phosphate-buffered saline (PBS) and resuspended in binding buffer
at a concentration of 1×106 cells/ml. A total of 5
μl of 20 μg/ml Annexin V-FITC and 50 μg/ml
propidium iodide (PI) were added and the tube was incubated for 30
min in the dark. The quantitative analysis of apoptosis was carried
out using a flow cytometer (BD Biosciences, San Jose, CA, USA).
Quadrants were positioned on Annexin V-FITC/PI dot plots, allowing
living cells (Annexin Ⅴ-FITC−/PI−),
early/primary apoptotic cells (Annexin
V-FITC+/PI−), late/secondary apoptotic cells
(Annexin Ⅴ-FITC+/PI+) and necrotic cells
(Annexin V-FITC−/PI+) to be distinguished
(26). Data were analyzed using
CellQuest™ software (BD Biosciences).
Western blot analysis
Western blo analysis was performed to investigate
the changes in the protein levels of Bcl-2 and Bax. The PC12 cells
were seeded onto 100-mm dishes at 5×106 cells/dish and
allowed to grow until confluent. The cells were then washed twice
with ice-cold D-Hank’s solution after drug treatment and lysed
using protein lysis buffer. The lysates were collected by scraping
from the plates and then centrifugation at 13,000 × g at 4°C for 15
min. The supernatant was separated and stored at −80°C until
use.
Western blot analysis was performed according to a
procedure described previously (27). Briefly, protein samples were
electrophoresed by SDS-PAGE for 2 h at 80 V and then transferred
onto polyvinylidene fluoride membranes for 40 min at 200 mA. The
blots were blocked for 2 h at room temperature in fresh blocking
buffer (0.1% Tween-20 in Tris-buffered saline, pH 7.4, containing
5% non-fat dried milk) and subsequently incubated at 4°C overnight
with primary antibodies against Bcl-2 (1:300; Santa Cruz
Biotechnology, Inc., Santa Cruz, CA, USA), Bax (1:200; Santa Cruz
Biotechnology, Inc.) or GAPDH (1:500; Santa Cruz Biotechnology,
Inc.) in blocking solution. Subsequently, the membranes were washed
with TBS-T (Tris-buffer saline containing 0.1% Tween-20) 3 times
and incubated with horseradish peroxidase-conjugated secondary
antibody at 1:5,000 in PBS with 5% non-fat dry milk at room
temperature for 1 h. To verify the equal loading of samples, the
membranes were incubated with monoclonal antibody GAPDH, followed
by a horseradish peroxidase-conjugated goat anti-mouse IgG. The
membrane again was washed with TBS-T 3 times and finally, the
protein bands were visualized using ECL western blotting detection
reagents (Amersham Biosciences, Buckinghamshire, UK). The intensity
of each band was analyzed using Image J software (NIH Image;
National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
All quantitative data are presented as the means ±
standard error of the mean (SEM). The changes in variable
parameters between the treated groups and the control group were
analyzed by one-way ANOVA followed by Dunnett’s test as a post hoc
comparison. A value of p<0.05 was considered to indicate a
statistically significant difference in all cases.
Results
Effects of the test compounds on
MPP+-induced cytotoxicity in PC12 cells
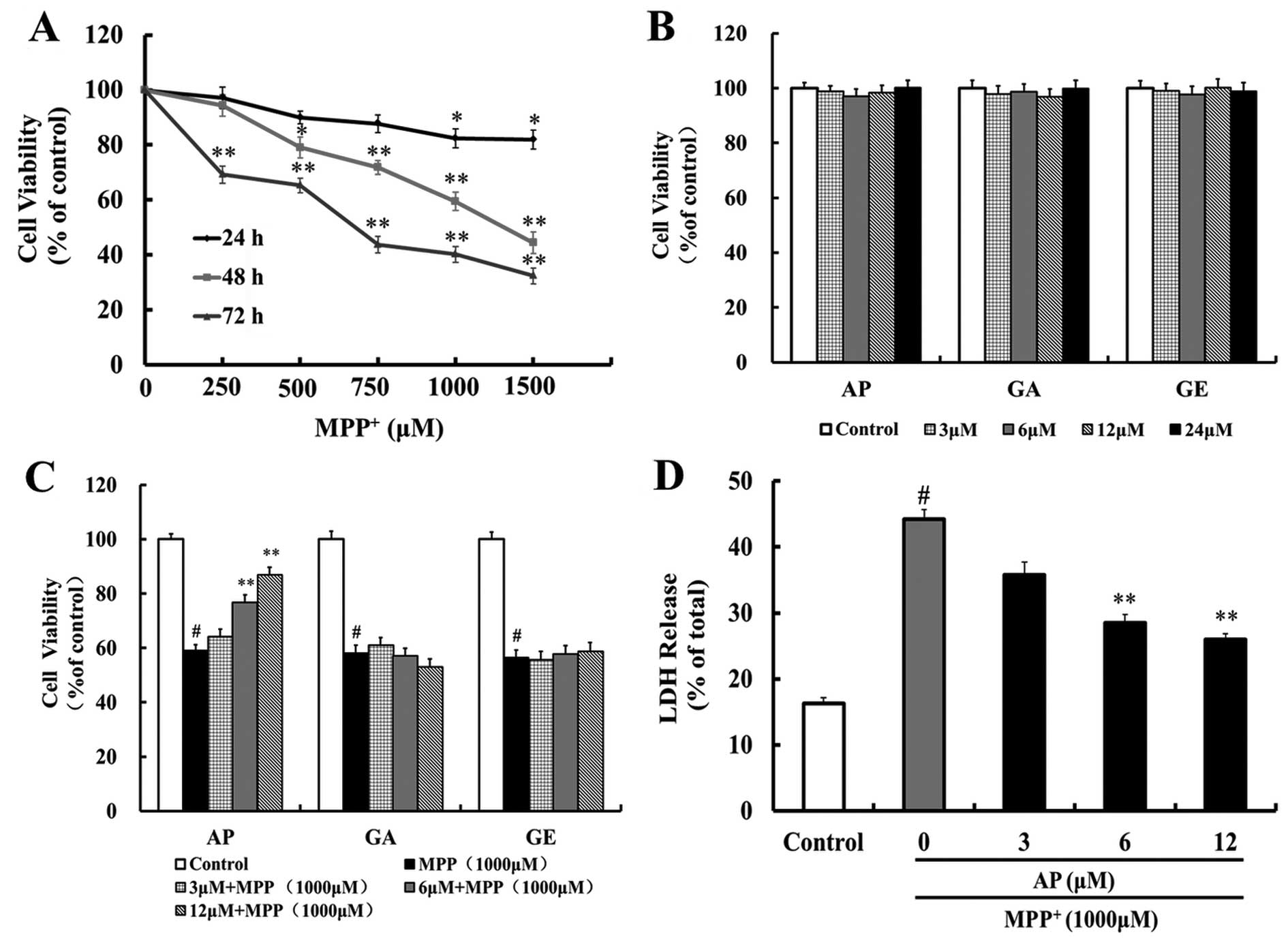
To investigate the effects of MPP+ on
PC12 cells, we exposed the cells to a range of concentrations of
MPP+ (250–1,500 μM) for various periods of time
(24, 48 and 72 h). There was a concentration- and time-dependent
decrease in cell viability following exposure to MPP+
(Fig. 2A). The cells exposed to
1,000 μM MPP+ for 48 h exhibited 59.5% of the
cell viability observed in the control cells. Therefore, we used
1,000 μM MPP+ treatent for 48 h as the optimal
standard concentration and time point for the induction of
apoptosis in the subsequent experiments.
We then investigated the neuroprotective effects of
the 3 flavonoid compounds (AP, GA and GE). These 3 compounds alone
did not have any cytotoxic effects at concentrations ranging
between 3–24 μM (Fig. 2B).
The PC12 cells were pretreated with these test compounds (3, 6 and
12 μM) for 4 h, and then exposed to 1,000 μM
MPP+ for 48 h. MTT assays indicated that pre-treatment
with AP (6 and 12 μM) markedly increased the cell viability
(p<0.01 and p<0.01, respectively) as compared with the
MPP+ group and the survival rate was 76.8 and 86.9% of
the controls, respectively (Fig.
2C). However, pre-treatment with GA and GE (3–12 μM) did
not increase the cell viability when compared with the
MPP+ group (Fig. 2C).
Therefore, in the subsequent experiments, we focused on the
protective effects of AP against MPP+ neurotoxicity.
Effect of AP on the
MPP+-induced release of LDH
To further investigate the protective effects of AP,
LDH assay, another indicator of cell toxicity, was performed. LDH
is a stable cytoplasmic enzyme present in all cells and is rapidly
released into the cell culture supernatant upon damage to he plasma
membrane. As shown in Fig. 2D,
when the PC12 cells were incubated with 1,000 μM
MPP+ for 48 h, the percentage of LDH being released
increased from 16.2 (controls) to 44.1%. Pre-treatment with AP (6
and 12 μM) significantly attenuated the
MPP+-induced release of LDH to 28.4 and 25.9% (p<0.01
and p<0.01, respectively), as compared to the
MPP+-treated control group.
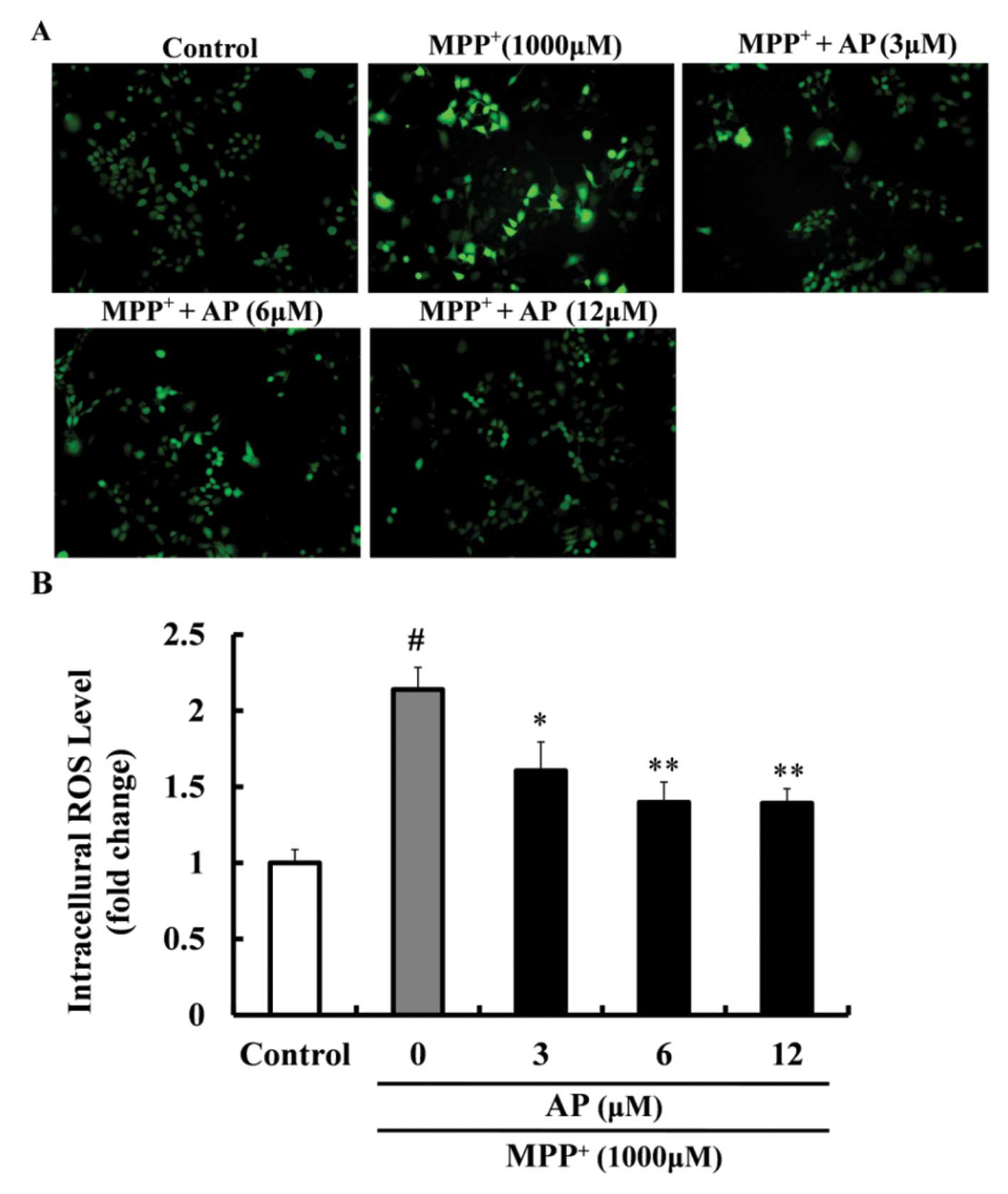
Effect of AP on the intracellular ROS
level in MPP+-treated PC12 cells
As shown in Fig.
3, exposure of the PC12 cells to 1,000 μM
MPP+ for 48 h led to a significant increase in the
levels of ROS (2.14-fold increase relative to the control value).
However, the overproduction of the intracellular ROS level was
markedly inhibited by pre-treatment with AP at concentrations of 3,
6 and 12 μM (p<0.05, p<0.01 and p<0.01,
respectively), when compared with the cells cultured with
MPP+ only.
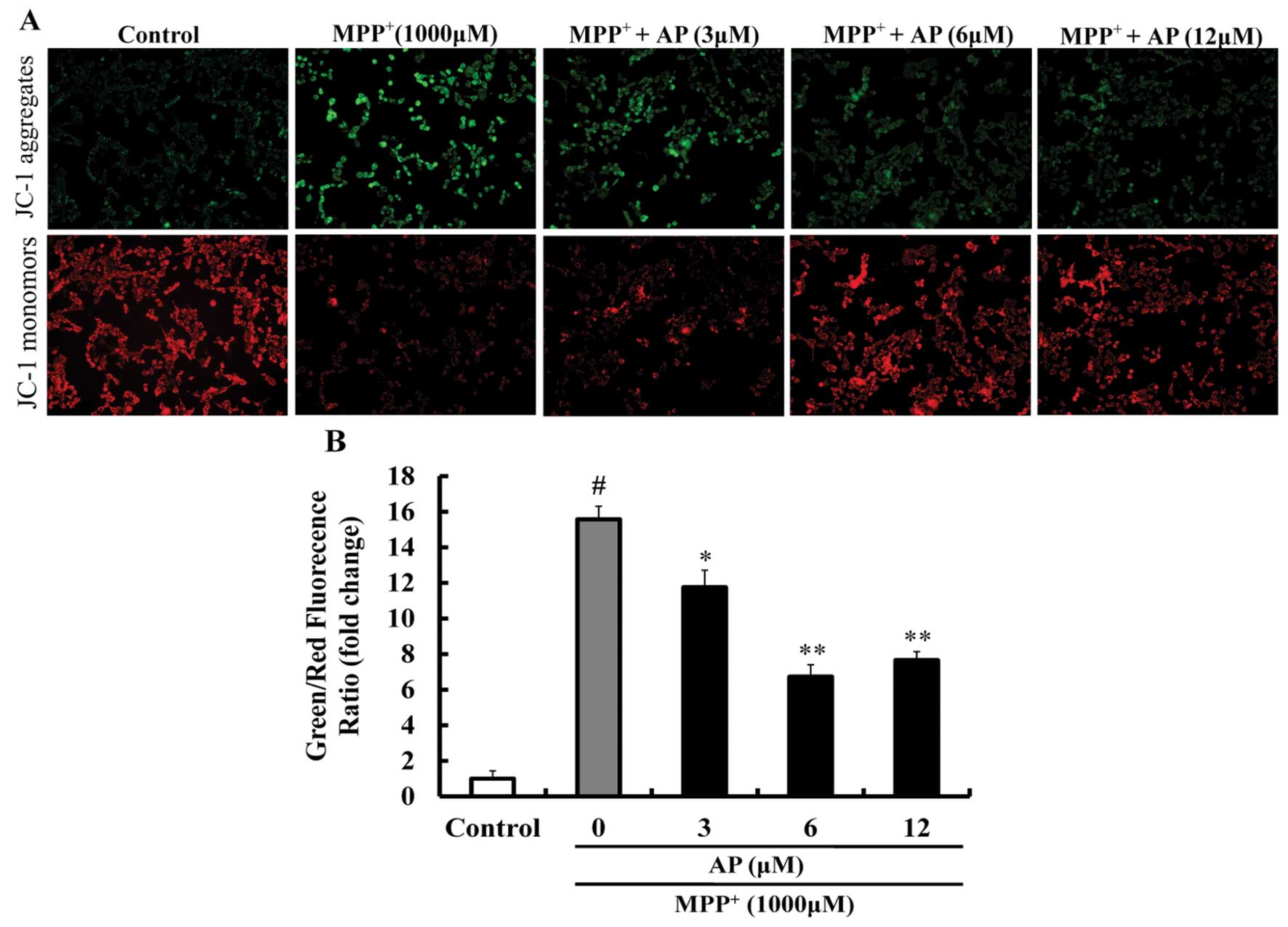
Effect of AP on MMP in
MPP+-treated PC12 cells
As shown in Fig.
4, following exposure to 1,000 μM MPP+ for 48
h, JC-1 aggregates within the normal mitochondria were dispersed to
the monomeric form (green fluorescence) and the ratio of green/red
fluorescence intensity was significantly increased (p<0.01 vs.
the control group), suggesting that MPP+ induced a
significant decrease in MMP. However, pre-treatment with AP (3, 6
and 12 μM) markedly prevented the decrease in MMP induced by
MPP+ (p<0.05, p<0.01 and p<0.01, respectively),
as compared to the group treated with MPP+ alone.
Effect of AP on MPP+-induced
apoptosis in PC12 cells
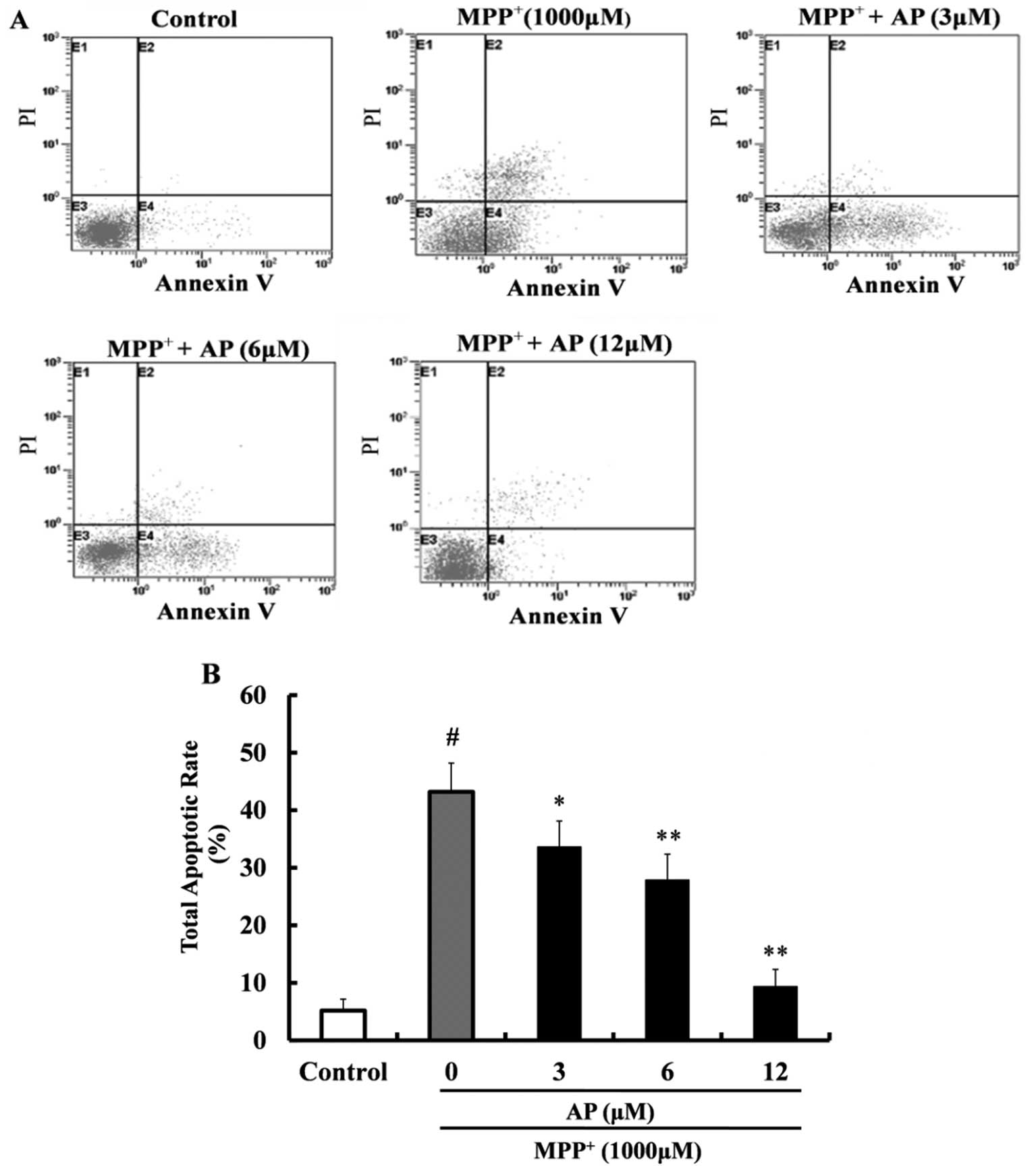
Annexin V-FITC and PI double staining was used to
detect apoptosis. As shown in Fig.
5, following exposure to 1,000 μM MPP+ for 48
h, the percentage of apoptotic cells was significantly increased
(43.2%) in comparison with the control group (5.2%). However,
pre-treatment with AP (3, 6 and 12 μM) markedly reduced the
cell apoptotic rate (p<0.05, p<0.01 and p<0.01,
respectively) as compared with the MPP+ group and the
cell apoptotic rate was 33.6, 27.9 and 9.4%, respectively.
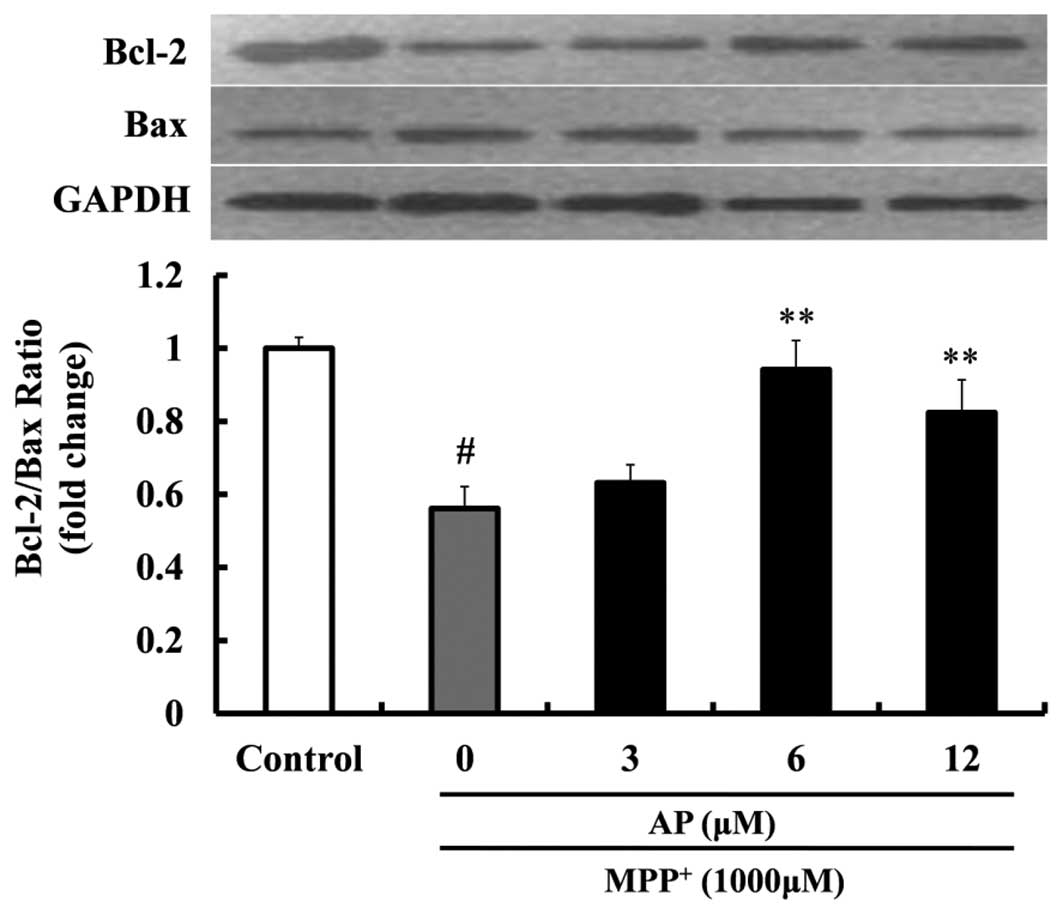
Effect of AP on the expression of Bcl-2
and Bax in MPP+-treated PC12 cells
To elucidate the molecular mechanisms responsible
for the protective effects exerted by AP against PC12 cell
apoptosis induced by MPP+, we measured the ratio of
Bcl-2/Bax protein expression. Following exposure to 1,000 μM
MPP+ for 48 h, as shown in Fig. 6, the ratio of Bcl-2/Bax was
sharply decreased (1.79-fold decrease relative to control,
p<0.01). However, when the cells were pre-treated with AP at the
concentrations of 6 and 12 μM, the above changes induced by
MPP+ were significantly mitigated. The ratio of
Bcl-2/Bax was significantly increased (p<0.01 and p<0.01,
respectively), compared with the cells treated with MPP+
alone.
Discussion
Various pharmacological and surgical treatments have
been used in patients with PD; however, some of these have
significant adverse effects and most do not halt or retard the
degeneration of dopaminergic neurons (28). Thus, in recent years, considerable
attention has been paid to the development of neuroprotective drugs
from natural origins as a therapeutic strategy for PD (29). Since naturally occurring
plant-derived flavonoids have been shown to play a useful role in
protecting the central nervous system (15), in this study, we investigated the
neuroprotective effects of the three most potent protective natural
flavonoids, including AP, GA and GE, using the model of
MPP+-induced neurotoxicity in PC12 cells. Our data
suggested that the neuroprotective effects of these natural
flavonoids varied dramatically, although they are structurally
similar. Significant protective effects of AP were observed against
MPP+-induced cell death, while GA and GE exerted no
protective effects. We conclude that this effect may well be due to
the position and number of free phenolic hydroxyl groups (OH
groups) in their structures. Free OH group is a typical reactive
functional group, particularly when it is attached to a C-7 or C-4′
of flavonoid structure (16). A
comparison of the AP structure (4′,5,7-trihydroxyflavone) with that
of GA (3,5,7-trihydroxyflavone) (4′,5-dihydroxy-7-methoxyflavone)
and GE shows that the 5-, 7- and 4′-OH substitutions are important.
For GE, one OH group (at the C-7 position) is methoxylated; for GA,
a free 4′-OH group is lacking (Fig.
1). Therefore, AP, with three free OH groups at C-5, C-7 and
C-4′ positions, exerts significant protective effect against
MPP+-induced neurotoxicity in PC12 cells. Our data
further suggest that AP ameliorates the MPP+-induced
production of ROS, increases the number of viable cells, attenuates
the release of LDH, prevents the loss of MMP, reduces the total
number of apoptotic cells and elevates the ratio of Bcl-2/Bax. Even
in the long-term procedure of MPP+-induced cytotoxicity
in PC12 cells, significant neuroprotective effects of AP can be
observed.
There is convincing evidence that the overproduction
of ROS, which leads to a pro-oxidant state known as oxidative
stress, plays an important role in the etiology and/or progression
of a number of neurological diseases and is responsible for
neurodegeneration (5,30). The excessive production of ROS can
cause severe impairment of cellular functions, such as peroxidize
membrane lipids, oxidize protein and attack cytoplasmic RNA and
mitochondrial DNA (31).
Moreover, previous data have demonstrated that ROS are involved in
the apoptotic mechanism of MPP+-mediated neurotoxicity
and may contribute to the apoptotic processes that are associated
with the development of PD (32).
In addition, ROS generated by MPP+ may be at least
partly responsible for the opening of mitochondrial permeability
transition pores and the collapse of MMP (33). As mentioned above, our experiment
data suggested that treatment with MPP+ resulted in a
significant increase in ROS production which was consistent with
previous research papers (14,22) and pre-treatment with AP markedly
reduced the generation of intracellular ROS. Based on these
findings, it can be concluded that AP ameliorates oxidative damage
induced by MPP+ in PC12 cells at least partly through
the scavenging of ROS.
Mitochondria are well known to be the principal
source of intracellular ROS production (5). The production of ROS in the
mitochondria is accelerated by ROS themselves, termed ROS-induced
ROS release and ROS generation in only small numbers of
mitochondria can affect neighboring mitochondria, eventually
propagating the ROS surge to the whole cell through this positive
feedback loop (6). The
overproduction of ROS rapidly causes a decrease in MMP and then
this depolarization of MMP results in the release of apoptogenic
factors, such as cytochrome c from the intermembrane space
to the cytoplasm. Subsequently, cytochrome c and other
apoptogenic factors trigger the caspase family and induce cells
apoptosis (5,34). In this study, the involvement of
the mitochondria in MPP+-induced apoptosis was
investigated by evaluating the loss of MMP. Our data indicated that
the treatment of the PC12 cells with MPP+ markedly
reduced MMP, which was detected by JC-1 staining, and the reduction
in MMP induced by MPP+ was reversed by pre-treatment
with AP. These results strongly suggest that AP stabilizes the
MPP+-induced MMP disruption and that this may be
attributed to the oxidative stress-reducing action of AP.
In addition, the mitochondrial apoptotic pathway is
the best known intrinsic apoptotic pathway (35). Although the precise mechanisms
through which the Bcl-2 family acts remain unclear, it has been
established that the Bcl-2 family does indeed play a pivotal role
in the mitochondrial apoptotic pathway (14). The Bcl-2 family proteins consist
of two subgroups according to structural homology: the
anti-apoptotic proteins, such as Bcl-2 and the pro-apoptotic
proteins, including Bax (36). It
is now evident that Bcl-2 or Bax control the mitochondrial
permeability transition pores or influence other early
mitochondrial perturbations (37). Therefore, Bcl-2 or Bax may
facilitate the passage of certain important proteins, such as
cytochrome c or other apoptosis-inducing factors that may
trigger the activation of a caspase cascade and result in
apoptosis. Cell survival in the early phases of the apoptotic
cascade depends mostly on the balance between the anti-apoptotic
and pro-apoptotic proteins of the Bcl-2 family (38). In this regard, the Bcl-2/Bax ratio
may predict the apoptotic fate of the cell better than the absolute
concentrations of either molecule alone (39). In the present study, we
investigated the effects of AP on the expression levels of Bcl-2
and Bax in MPP+-treated cells by western blot analysis.
Our data indicated that MPP+ significantly decreased the
ratio of Bcl-2/Bax, a result that was consistent with that of
previous studies (40). However,
pre-treatment with AP increased the expression of Bcl-2, while
significantly decreasing the expression of Bax, thus ameliorating
the MPP+-induced reduction in the Bcl-2/Bax ratio in
PC12 cells. Therefore, the effects of AP on MPP+-induced
apoptosis may be at least be partially mediated through the
regulation of the expression of Bcl-2 and Bax. These results
suggest that the neuroprotective effects of AP are associated with
the inhibition of apoptosis through the mitochondrial pathway.
In conclusion, the results from our study
demonstrate that AP exerts a protective effect against
MPP+-induced neurotoxicity in PC12 cells and that the
neuroprotective effects of AP are, at least partially mediated
through the inhibition of oxidative stress, the stabilization of
mitochondrial function and the reduction of neuronal apoptosis via
the mitochondrial pathway. To the best of our knowledge, this is
the first study to demonstrate that AP rescues neuronal cells from
MPP+-induced apoptosis in vitro. The present
results suggest that treatment with AP may prove to be an effecive
therapeutic approach for neuroprotection in PD. The neuroprotective
effects of this compound in PD require further investigation in
primary neuronal cultures, as well as in animal models of PD.
Acknowledgments
This study was supported by grant no. 2011BAI01B01
from the Key Technologies Research and Development Program of
China, grant no. 06CXTD004 from the Program for Innovative Research
Team of Higher Education of Guangdong Province of China, grant no.
2012A080202002 from the Science and Technology Planning Project of
Guangdong Province of China and educational finance grant no. 276
(2014) from the Special Funds from Central Finance of China in
Support of the Development of Local Colleges and University.
Abbreviations:
|
PD
|
Parkinson’s disease
|
|
ROS
|
reactive oxygen species
|
|
MMP
|
mitochondrial membrane potential
|
|
MPP+
|
1-methyl-4-phenylpyridinium ion
|
|
PI
|
propidium iodide
|
|
AP
|
apigenin
|
|
GA
|
galangin
|
|
GE
|
genkwanin
|
|
DCFH-DA
|
2′,7′-dichlorofluorescin diacetate
|
|
LDH
|
lactate dehydrogenase
|
|
JC-1
|
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethyl benzimidazolyl
carbocyanine iodide
|
References
|
1
|
de Lau LM and Breteler MM: Epidemiology of
Parkinson’s disease. Lancet Neurol. 5:525–535. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Henchcliffe C and Beal MF: Mitochondrial
biology and oxidative stress in Parkinson disease pathogenesis. Nat
Clin Pract Neurol. 4:600–609. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kanthasamy A, Jin H, Mehrotra S, Mishra R,
Kanthasamy A and Rana A: Novel cell death signaling pathways in
neurotoxicity models of dopaminergic degeneration: relevance to
oxidative stress and neuroinflammation in Parkinson’s disease.
Neurotoxicology. 31:555–561. 2010. View Article : Google Scholar :
|
|
4
|
Winklhofer KF and Haass C: Mitochondrial
dysfunction in Parkinson’s disease. Biochim Biophys Acta.
1802:29–44. 2010. View Article : Google Scholar
|
|
5
|
Yan MH, Wang X and Zhu X: Mitochondrial
defects and oxidative stress in Alzheimer disease and Parkinson
disease. Free Radic Biol Med. 62:90–101. 2013. View Article : Google Scholar :
|
|
6
|
Zorov DB, Filburn CR, Klotz LO, Zweier JL
and Sollott SJ: Reactive oxygen species (ROS)-induced ROS release:
a new phenomenon accompanying induction of the mitochondrial
permeability transition in cardiac myocytes. J Exp Med.
192:1001–1014. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Roucou X and Martinou JC: Conformational
change of Bax: a question of life or death. Cell Death Differ.
8:875–877. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tipton KF and Singer TP: Advances in our
understanding of the mechanisms of the neurotoxicity of MPTP and
related compounds. J Neurochem. 61:1191–1206. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Langston JW and Ballard PA Jr: Parkinson’s
disease in a chemist working with
1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N Engl J Med.
309:3101983. View Article : Google Scholar
|
|
10
|
Tetrud JW, Langston JW, Garbe PL and
Ruttenber AJ: Mild parkinsonism in persons exposed to
1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP). Neurology.
39:1483–1487. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Greene LA and Tischler AS: Establishment
of a noradrenergic clonal line of rat adrenal pheochromocytoma
cells which respond to nerve growth factor. Proc Natl Acad Sci USA.
73:2424–2428. 1976. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mao QQ, Ip SP, Ko KM, Tsai SH, Zhao M and
Che CT: Peony glycosides protect against corticosterone-induced
neurotoxicity in PC12 cells. Cell Mol Neurobiol. 29:643–647. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Jiang B, Zhang H, Bi J and Zhang XL:
Neuroprotective activities of catalpol on
MPP+/MPTP-induced neurotoxicity. Neurol Res. 30:639–644.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou J, Sun Y, Zhao X, Deng Z and Pu X:
3-O-demethylswertipunicoside inhibits MPP+-induced
oxidative stress and apoptosis in PC12 cells. Brain Res.
1508:53–62. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schroeter H, Spencer JP, Rice-Evans C and
Williams RJ: Flavonoids protect neurons from oxidized
low-density-lipoprotein-induced apoptosis involving c-Jun
N-terminal kinase (JNK), c-Jun and caspase-3. Biochem J.
358:547–557. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li S, Chou G, Hseu Y, Yang H, Kwan H and
Yu Z: Isolation of anticancer constituents from flos genkwa (Daphne
genkwa Sieb et Zucc) through bioassay-guided procedures. Chem Cent
J. 7:1592013. View Article : Google Scholar
|
|
17
|
Jacobson KA, Moro S, Manthey JA, West PL
and Ji XD: Interactions of flavones and other phytochemicals with
adenosine receptors. Adv Exp Med Biol. 505:163–171. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jaganathan SK and Mandal MJ:
Antiproliferative effects of honey and of its polyphenols: a
review. J Biomed Biotechnol. 2009:8306162009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shukla S and Gupta S: Apigenin: a
promising molecule for cancer prevention. Pharm Res. 27:962–978.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu R, Zhang T, Yang H, Lan X, Ying J and
Du G: The flavonoid apigenin protects brain neurovascular coupling
against amyloid-β25–35-induced toxicity in mice. J
Alzheimers Dis. 24:85–100. 2011.
|
|
21
|
Zhao L, Wang JL, Wang YR and Fa XZ:
Apigenin attenuates copper-mediated β-amyloid neurotoxicity through
antioxidation, mitochondrion protection and MAPK signal
inactivation in an AD cell model. Brain Res. 1492:33–45. 2013.
View Article : Google Scholar
|
|
22
|
Wruck CJ, Claussen M, Fuhrmann G, Römer L,
Schulz A, Pufe T, Waetzig V, Peipp M, Herdegen T and Götz ME:
Luteolin protects rat PC12 and C6 cells against MPP+
induced toxicity via an ERK dependent Keap1-Nrf2-ARE pathway. J
Neural Transm. (Suppl): 57–67. 2007. View Article : Google Scholar
|
|
23
|
Zhang ZT, Cao XB, Xiong N, Wang HC, Huang
JS, Sun SG and Wang T: Morin exerts neuroprotective actions in
Parkinson disease models in vitro and in vivo. Acta Pharmacol Sin.
31:900–906. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Zhang K, Ma Z, Wang J, Xie A and Xie J:
Myricetin attenuated MPP+-induced cytotoxicity by
anti-oxidation and inhibition of MKK4 and JNK activation in MES23.5
cells. Neuropharmacology. 61:329–335. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kong SZ, Xian YF, Ip SP, Lai XP, Shi XG,
Lin ZX and Su ZR: Protective effects of hydroxysafflor yellow A on
β-amyloid-induced neurotoxicity in PC12 cells. Neurochem Res.
38:951–960. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang D, Wong HK, Feng YB and Zhang ZJ:
Paeoniflorin, a natural neuroprotective agent, modulates multiple
anti-apoptotic and pro-apoptotic pathways in differentiated PC12
cells. Cell Mol Neurobiol. 33:521–529. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu XH, Pan LL, Chen PF and Zhu YZ:
Leonurine improves ischemia-induced myocardial injury through
antioxidative activity. Phytomedicine. 17:753–759. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Toulouse A and Sullivan AM: Progress in
Parkinson’s disease-where do we stand? Prog Neurobiol. 85:376–392.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kwon IH, Choi HS, Shin KS, Lee BK, Lee CK,
Hwang BY, Lim SC and Lee MK: Effects of berberine on
6-hydroxydo-pamine-induced neurotoxicity in PC12 cells and a rat
model of Parkinson’s disease. Neurosci Lett. 486:29–33. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Federico A, Cardaioli E, Da Pozzo P,
Formichi P, Gallus GN and Radi E: Mitochondria, oxidative stress
and neurodegeneration. J Neurol Sci. 322:254–262. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Uttara B, Singh AV, Zamboni P and Mahajan
RT: Oxidative stress and neurodegenerative diseases: a review of
upstream and downstream antioxidant therapeutic options. Curr
Neuropharmacol. 7:65–74. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lu XL, Yao XL, Liu Z, Zhang H, Li W, Li Z,
Wang GL, Pang J, Lin Y, Xu Z, et al: Protective effects of
xyloketal B against MPP+-induced neurotoxicity in
Caenorhabditis elegans and PC12 cells. Brain Res. 1332:110–119.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cassarino DS, Parks JK, Parker WD Jr and
Bennett JP Jr: The parkinsonian neurotoxin MPP+ opens
the mitochondrial permeability transition pore and releases
cytochrome c in isolated mitochondria via an oxidative mechanism.
Biochim Biophys Acta. 1453:49–62. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ling YH, Liebes L, Zou Y and Perez-Soler
R: Reactive oxygen species generation and mitochondrial dysfunction
in the apoptotic response to bortezomib, a novel proteasome
inhibitor, in human H460 non-small cell lung cancer cells. J Biol
Chem. 278:33714–33723. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Muñoz-Pinedo C: Signaling pathways that
regulate life and cell death: evolution of apoptosis in the context
of self-defense. Adv Exp Med Biol. 738:124–143. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ghibelli L and Diederich M: Multistep and
multitask Bax activation. Mitochondrion. 10:604–613. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Martinou JC and Youle RJ: Mitochondria in
apoptosis: Bcl-2 family members and mitochondrial dynamics. Dev
Cell. 21:92–101. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chen Q and Lesnefsky EJ: Blockade of
electron transport during ischemia preserves bcl-2 and inhibits
opening of the mitochondrial permeability transition pore. FEBS
Lett. 585:921–926. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schelman WR, Andres RD, Sipe KJ, Kang E
and Weyhenmeyer JA: Glutamate mediates cell death and increases the
Bax to Bcl-2 ratio in a differentiated neuronal cell line. Brain
Res Mol Brain Res. 128:160–169. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Selvaraj S, Watt JA and Singh BB: TRPC1
inhibits apoptotic cell degeneration induced by dopaminergic
neurotoxin MPTP/MPP+. Cell Calcium. 46:209–218. 2009.
View Article : Google Scholar : PubMed/NCBI
|