Introduction
Bone is a dynamic tissue that constantly undergoes
remodeling in which a coupled process of bone formation by
osteoblasts and resorption by osteoclasts continues throughout life
(1). An imbalance between bone
formation and resorption results in excessive bone loss, a feature
of chronic inflammatory diseases such as rheumatoid arthritis,
osteomyelitis, bacterial arthritis and infection of orthopedic
implants (2).
Lipopolysaccharide (LPS), a component of the outer
membranes of gram-negative bacteria, was shown to be capable of
inducing bone resorption in vitro or in vivo
(3–8). LPS can also inhibit osteoblast
differentiation and function in cell cultures (9–12).
However, effective therapy, such as treatment with antibiotics and
surgery, against chronic inflammatory diseases such as
bacteria-induced bone destruction is limiting. Therefore, the study
and development of potential drugs that can restore osteoblast
function remains a fundamental objective in the prevention of bone
destruction in infective bone diseases.
C-Jun N-terminal kinases (JNKs) are protein kinases
involved in cell apoptosis and the inflammatory response (13,14). JNK1, JNK2 and JNK3 are genetic
loci encoded in the mammalian genome, with JNK1 and JNK2 being
ubiquitously expressed, while JNK3 is expressed predominantly in
the brain and to a lesser extent in the heart and testis (15). However, evidence suggesting that
JNK1, but not JNK2 or JNK3, is the key JNK family kinase
responsible for the phosphorylation of c-Jun and for the expression
of RNA polymerase III. In many cells, JNK1, but not JNK2, mediates
tumor necrosis factor-α (TNF-α)-induced cell apoptosis by
inhibiting cell differentiation and promoting the generation of
inflammatory cytokines such as interleukin-6 (IL-6) and LIF
(16–18). In a previous study, we found that
LPS induced osteoblast apoptosis and inhibited osteoblast
differentiation via activation of the JNK pathway (19). Therefore, the aim of the present
study was to evaluate the effect of JNK inhibition by SP600125 on
the apoptosis and osteoblast differentiation in MC3T3-E1 cells
stimulated with LPS.
Materials and methods
Reagents
Escherichia coli LPS (serotype 055:B5),
β-glycerophosphate (β-GP),
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), and
ascorbic acid were purchased from Sigma Chemical Co. (St. Louis,
MO, USA). The protease inhibitor cocktail, and the selective
mitogen-activated protein kinase (MAPK) inhibitors, SP600125 and
PD98059 were purchased from Calbiochem (San Diego, CA, USA).
Dulbecco’s modified Eagle’s medium (DMEM), fetal bovine serum (FBS)
and penicillin/streptomycin were purchased from Gibco (Rockville,
MD, USA). Antibodies to c-Jun N-terminal kinase 1 (JNK1),
extracellular signal-regulated kinase 1 (ERK1), alkaline
phosphatase (ALP) and caspase-3 were purchased from Cell Signaling
Technology (Beverly, MA, USA). Antibodies to Runx2, osterix (OSX),
osteocalcin (OCN), Bax, Bcl-2 and β-actin were purchased from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). MC3T3-E1 cells, an
osteoblast-like cell line was obtained from the American Type
Culture Collection (ATCC, Manassas, VA, USA). Other chemicals and
reagents used in this study were of analytical grade.
Cell culture
MC3T3-E1 cells were grown in DMEM supplemented with
10% (v/v) FBS, 1% (v/v) penicillin-streptomycin solution, 10 mM
HEPES solution and incubated at 37°C in 5% CO2
humidified air. To examine the effect of SP600125 on osteoblast
differentiation stimulated with LPS, MC3T3-E1 cells at a density of
5×104 cells/cm2 were cultured in osteogenic
differentiation medium (DMEM with 10% FBS, 10 mM HEPES, 50
μg/ml L-ascorbic acid and 5 mM β-GP) for 2 days. On
differentiation day 3, following pretreatment with or without
SP600125 for 2 h, the cells were treated with 100 and 1,000 ng/ml
LPS or without LPS at the indicated time points.
MTT assay
Cell metabolism was measured using MTT assay.
MC3T3-E1 cells at a density of 5×103
cells/cm2 were plated in 96-well culture plates and
cultured in 0.2 ml osteogenic differentiation medium. Following
pretreatment with 5, 10, 15, 25 and 50 μM SP600125 or
without SP600125 for 2 h, the cells were treated with 100 and 1,000
ng/ml LPS or without LPS for 1 and 3 days. At the end of treatment,
20 μl MTT (5 mg/ml) was added to each well. The cells were
cultured for an additional 4 h, the supernatant was removed, and
100 μl dimethyl sulfoxide (DMSO) was added to each well.
After agitation for 3 min, absorbance was measured at 490 nm using
a microplate reader (Thermo Fisher Scientific, Inc., Pittsburgh,
PA, USA). The experiment was performed in triplicate.
ALP activity determination
MC3T3-E1 cells at 5×105
cells/cm2 were seeded in 100-mm diameter culture dishes
and cultured in osteogenic differentiation medium for 2 days. On
differentiation day 3, following pretreatment with 25 and 50
μM SP600125 or without SP600125 for 2 h, the cells were
treated with 100 and 1,000 ng/ml LPS or without LPS in osteogenic
differentiation medium for 1 and 2 days. The cells were detached
aided by a cell scraper in phosphate-buffered saline (PBS), and
then centrifuged at 16,000 × g for 5 min. Cell pellets were placed
in 300 μl of lysis buffer (Cytobuster protein extraction
reagent; Novagen, Darmstadt, Germany) with 1X protease inhibitor
cocktail. The ALP activity in the lysates was determined by the
measurement of p-nitrophenyl phosphate (pNPP) using a commercial
assay kit (Beyotime Institute of Biotechnology, Shanghai, China)
according to the manufacturer’s instructions. Briefly, cell lysate
of each sample was added to the assay buffer (2 mM
MgCl2, 50 mM Tris-HCl/pH 9.8) containing 2.5 mM pNPP.
After 30-min incubation at 37°C, the reaction was terminated by the
addition of 0.2 M NaOH. Absorbance of the reaction mixture was
measured by spectrophotometry (Eppendorf BioSpectrometer; Eppendorf
AG, Hamburg, Germany) at 405 nm. A standard curve was also
generated using a series of diluted colored p-nitrophenol (pNP)
with known concentrations. The concentration was calculated by
projecting the optical densities on the standard curve. Total
protein concentrations were quantified by spectrophotometry
(Eppendorf BioSpectrometer). The relative ALP activity was defined
as micromoles of pNPP hydrolyzed per minute per 1 g of total
protein (18).
Measurement of caspase-3 activity
MC3T3-E1 cells at a density of 5×105
cells/cm2 were seeded in 100-mm diameter culture dishes
and cultured in osteogenic differentiation medium for 2 days. On
differentiation day 3, following pretreatment with 25 and 50
μM SP600125 or without SP600125 for 2 h, the cells were
treated with 100 and 1,000 ng/ml LPS or without LPS in osteogenic
differentiation medium for 1 and 2 days. The activity of caspase-3
was determined using the caspase-3 activity kit (Beyotime Institute
of Biotechnology) according to the manufacturer’s instructions.
Briefly, the cells were detached aided by a cell scraper in PBS
buffer, then centrifuged at 16,000 × g for 5 min and the cell
pellets were placed in 300 μl of lysis buffer (cytobuster
protein extraction reagent) with 1X protease inhibitor cocktail.
Total protein concentrations were quantified by spectrophotometry
(Eppendorf BioSpectrometer). The cell lysates of each sample were
then incubated with a mixture provided in the kit, containing
caspase-3 assay buffer, Ac-DEVD-pNA, the substrate of caspase-3.
After 2-h incubation at 37°C, the optical density (coloration)
resulting from the cleavage of the substrate and the release of
pNA, was detected and quantified by spectrophotometry (Eppendorf
BioSpectrometer) at 405 nm. A standard curve was also generated
using a series of diluted pNA with known concentrations. The
concentration was calculated by projecting the optical densities on
the standard curve. The relative caspase-3 activity was defined as
nanomoles of pNA per minute per 1 mg of total protein. All the
experiments were carried out in triplicate.
Quantitative PCR
Total RNA was isolated using TRIzol reagent
(Invitrogen, Grand Island, NY, USA) and quantified by
spectrophotometry. After isolation, 3 μg total RNA from each
sample was reverse transcribed (RT) utilizing the HiFi-MMLV cDNA
kit (Beijing CoWin Biotech, Beijing, China) according to the
manufacturer’s instructions. The primer sequences of
osteoblast-associated genes, caspase-3, Bax,
Bcl-2, JNK1 and ERK1 (Sangon Biotech Co.,
Ltd., Shanghai, China) and annealing temperatures used in this
study are listed in Table I.
Quantitative PCR (qPCR) was performed with a RealSYBR Mixture
(Beijing CoWin Biotech) according to the manufacturer’s
instructions. The qPCR reactions were performed in the ABI PRISM
7300 sequence detection system (Applied Biosystems, Grand Island,
NY, USA). In each reaction, 1 μl cDNA, 10 μl 2X
RealSYBR Mixture, and 0.25 μM forward and reverse primer in
a total volume of 20 μl were used. The reaction conditions
used were: 1 cycle of 95°C for 5 min followed by 40 cycles of 95°C
for 15 sec, annealing temperature for 30 sec and extension at 72°C
for 30 sec. qPCR for each sample was run in triplicate. β-actin was
used as an internal control, and the results were analyzed using
the standard 2−ΔΔCt method as previously described
(20).
 | Table IPrimer sequences used for
quantitative PCR. |
Table I
Primer sequences used for
quantitative PCR.
| Gene | Primer sequences
(5′→3′) | Accession no. | Annealing
temperature (°C) | Product size
(bp) |
|---|
| Runx2 | F:
TTCAACGATCTGAGATTTGTGGG | | | |
| R:
GGATGAGGAATGCGCCCTA | NM_001146038 | 62 | 221 |
| ALP | F:
GAGCGTCATCCCAGTGGAG | | | |
| R:
TAGCGGTTACTGTAGACACCC | NM_007433 | 62 | 158 |
| BSP | F:
CAGGGAGGCAGTGACTCTTC | | | |
| R:
AGTGTGGAAAGTGTGGCGTT | NM_008318 | 58 | 158 |
| OSX | F:
ATGGCGTCCTCTCTGCTTG | | | |
| R:
TGAAAGGTCAGCGTATGGCTT | NM_130458 | 62 | 156 |
| OCN | F:
GAGGGCAATAAGGTAGTGAA | | | |
| R:
CATAGATGCGTTTGTAGGC | NM_001037939 | 62 | 160 |
| Col1α1 | F:
CCCTGCCTGCTTCGTGTA | | | |
| R:
TTGAGTTTGGGTTGTTCGTC | BC003198 | 63 | 101 |
|
Caspase-3 | F:
TGGTGATGAAGGGGTCATTTATG | | | |
| R:
TTCGGCTTTCCAGTCAGACTC | BC038825 | 60 | 105 |
| Bax | F:
AGACAGGGGCCTTTTTGCTAC | | | |
| R:
AATTCGCCGGAGACACTCG | NM_007527 | 60 | 137 |
| Bcl-2 | F:
GTCGCTACCGTCGTGACTTC | | | |
| R:
GACCCCACCGAACTCAAAGAA | NM_177410 | 66 | 152 |
| β-actin | F:
GGCTGTATTCCCCTCCATCG | | | |
| R:
CCAGTTGGTAACAATGCCATGT | NM_007393 | 60 | 154 |
Western blot analysis
At the end of treatment, cell culture medium was
aspirated and the cells were detached in PBS by scraping. Detached
cells were centrifuged at 15,000 × g at 4°C for 15 min. Cell
pellets were then lysed in 300 μl lysis buffer (Cytobuster
protein extraction reagent; Novagen) with 25 mM NaF, 1 mM
Na3VO4, and 1X protease inhibitor cocktail.
Protein concentrations were quantified by spectrophotometry. For
western blotting, an equal amount of protein from each sample was
loaded on SDS-PAGE and electrotransferred onto PVDF membranes
(Millipore, Bedford, MA, USA). The membranes were then blocked with
5% (w/v) bovine serum albumin in TBST [10 mM Tris, 150 mM NaCl, and
0.1% (v/v) Tween-20, pH 7.5] for 1 h at room temperature, and
incubated with primary antibodies incubated with primary
antibodies: rabbit polyclonal anti-JNK, phosphorylated JNK,
β-actin, goat polyclonal anti-OSX at dilutions of 1:500 (Santa Cruz
Biotechnology, Inc.); rabbit polyclonal anti-ERK1/2 and
phosphorylated ERK1/2 at dilutions of 1:1,000 (Cell Signaling
Technology, Inc.). The secondary antibodies used for detection were
horseradish peroxidase (HRP)-conjugated goat anti-rabbit
immunoglobulin G (IgG) and HRP-conjugated rabbit anti-goat IgG
(both from Santa Cruz Biotechnology, Inc.). Chemiluminescence ECL
(Beyotime Institute of Biotechnology) was used to detect
immunoreactive protein signals. Protein signals were then
visualized on film and scanned and quantified using the ImageJ
software (National Institutes of Health Image, Bethesda, MA, USA).
For re-probing, PVDF membranes were stripped with 0.2 M NaOH for 10
min before blocking with another primary antibody. The expression
of molecules of interest was determined relative to β-actin.
Statistical analysis
Data are presented as the means ± standard deviation
(SD) for three or more independent experiments. Significant
differences were determined using factorial analysis of variance
(ANOVA). Statistical analysis was performed using SPSS13.0
software. P<0.05 was regarded significant and shown with an
asterisk in the bar diagrams.
Results
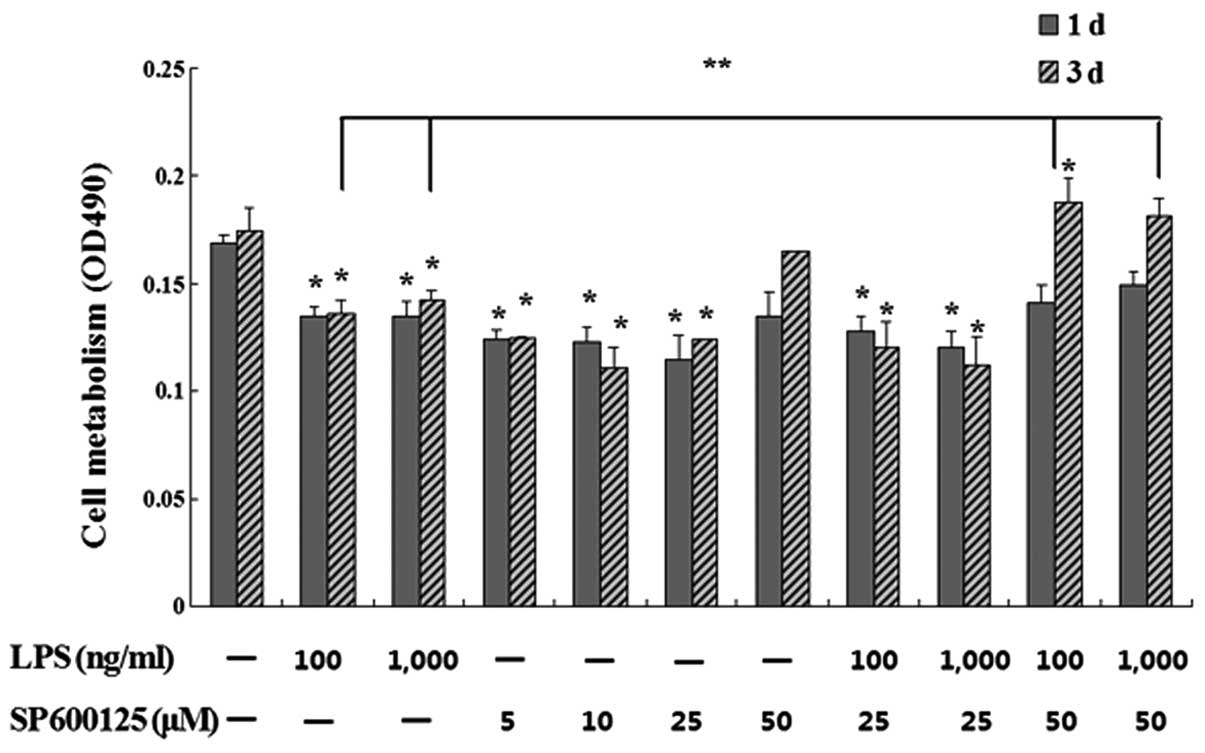
Effect of SP600125 on cell metabolism in
MC3T3-E1 cells suppressed by LPS
Following pretreatment with 5, 10, 15, 25 and 50
μM SP600125 or without SP600125 for 2 h, the cells were
treated with 100 and 1,000 ng/ml LPS or without LPS in the presence
of SP600125 for 1 and 3 days. MTT assays showed that SP600125 at 50
μM significantly restored cell metabolism downregulated by
LPS in MC3T3-E1 cells compared to the remainig treated cultures
(P<0.01) (Fig. 1).
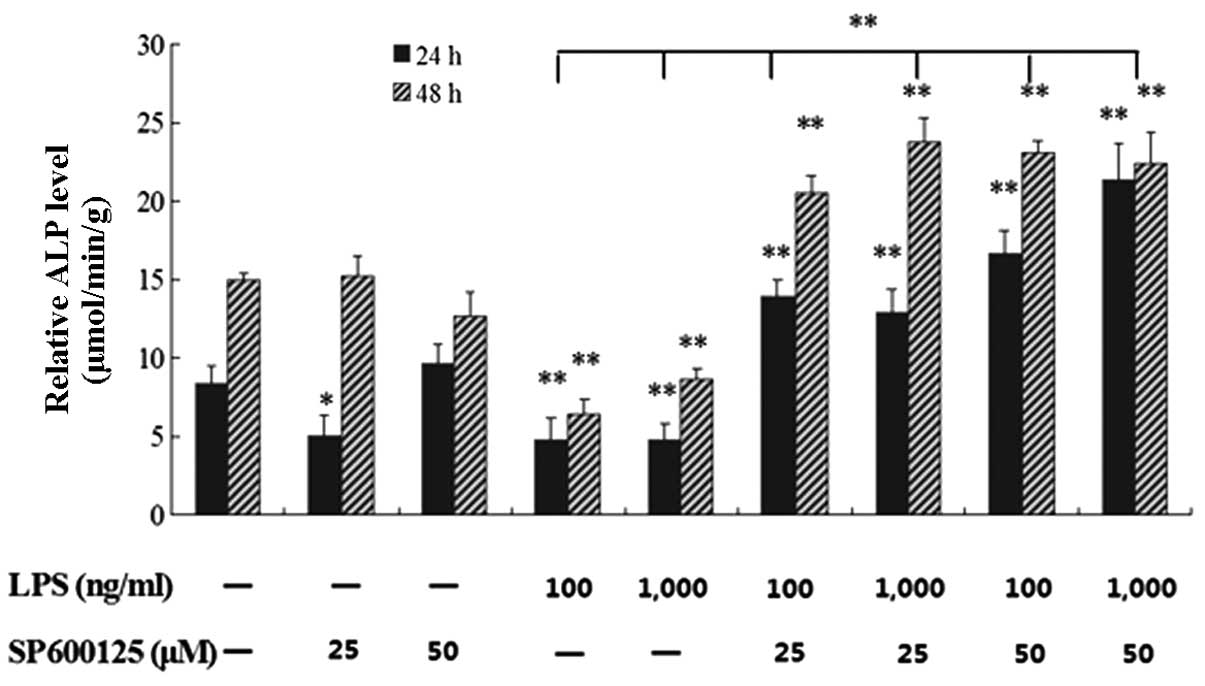
Effect of SP600125 on ALP activity in
MC3T3-E1 cells suppressed by LPS
Following pretreatment with 25 and 50 μM
SP600125 or without SP600125 for 2 h, the cells were treated with
100 and 1,000 ng/ml LPS or without LPS in the presence of SP600125
for 1 and 2 days. The downregulated ALP activity in MC3T3-E1 cells
induced by LPS was significantly restored by SP600125 compared to
the non-treated culture at 1 and 2 days (P<0.01) (Fig. 2).
Effect of SP600125 on caspase-3 activity
in MC3T3-E1 cells induced by LPS
Cells were pretreated with 25 and 50 μM
SP600125 or without SP600125 for 2 h, and then with 100 and 1,000
ng/ml LPS or without LPS in the presence of SP600125 for 1 day. The
upregulated caspase-3 activity in MC3T3-E1 cells induced by LPS was
significantly downregulated by SP600125 compared to the non-treated
culture at 1 day (P<0.01) (Fig.
3).
Effect of SP600125 on mRNA expression
levels of osteoblast-specific genes in MC3T3-E1 cells suppressed by
LPS
Effect of SP600125 on mRNA expression levels of
osteoblast-specific genes such as Runx2, ALP,
BSP, OSX, OCN and Col1α1 in MC3T3-E1
cells stimulated with LPS was determined by the qPCR experiment.
The results showed that SP600125 has restored the downregulated
expression of early-stage osteoblast marker genes such as
Runx2, ALP and BSP in MC3T3-E1 cells induced
by LPS compared to the non-treated culture at 1 day. However, the
expression of late-stage genes such as OSX, OCN and
Col1α1 was not affected (Fig.
4A and C). This result suggested that SP600125 may play a
protective role in early-stage osteoblast differentiation in
MC3T3-E1 cells induced by LPS.
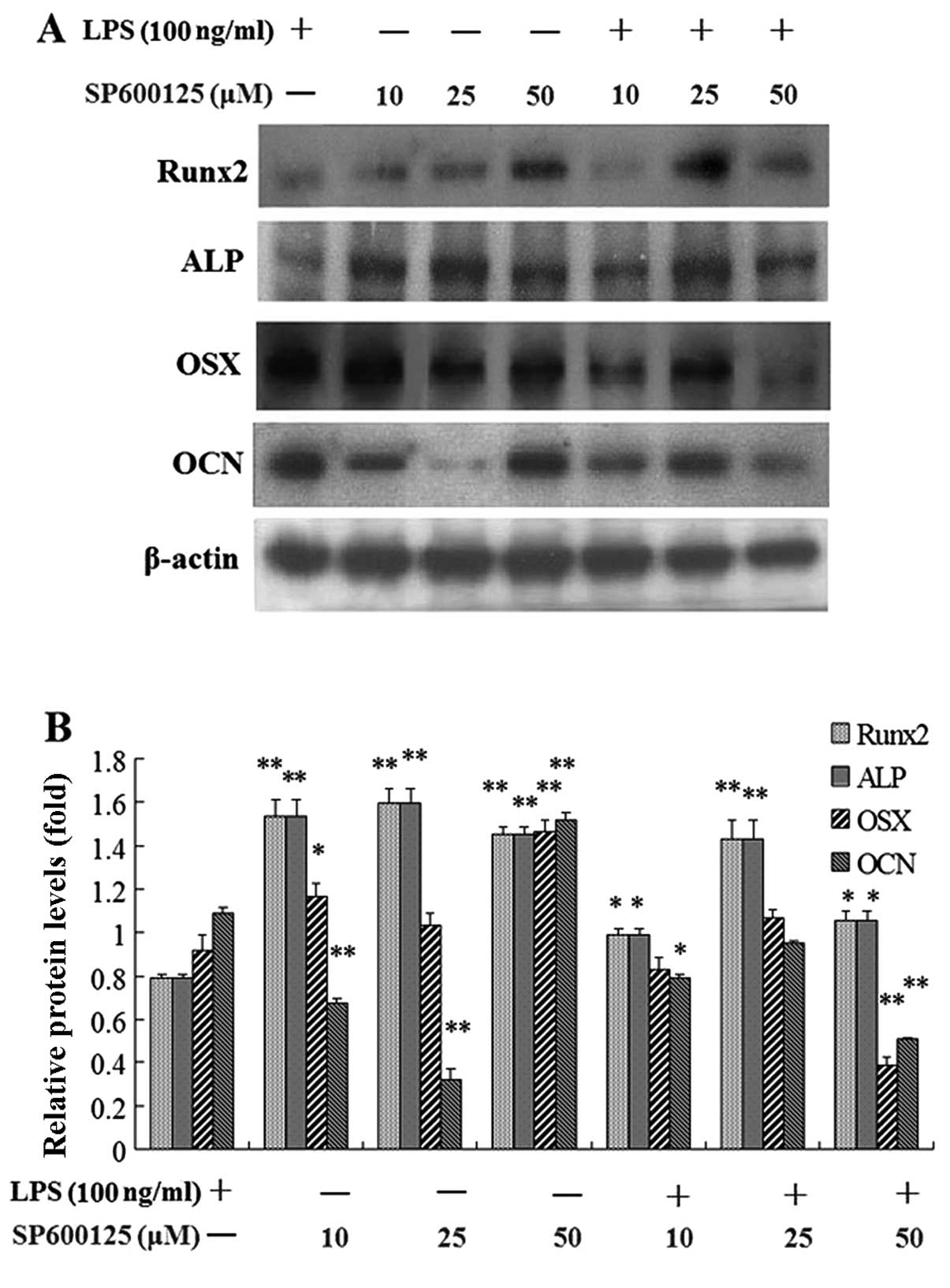
Effect of SP600125 on protein levels of
osteoblast-specific markers in MC3T3-E1 cells suppressed by
LPS
Following pretreatment with 10, 25 and 50 μM
SP600125 or without SP600125 for 2 h, the cells were treated with
100 and 1,000 ng/ml LPS or without LPS in the presence of SP600125
for 1 day. The protein expression of early-stage osteoblast markers
such as Runx2 and ALP in MC3T3-E1 cells inhibited by LPS was
significantly enhanced by SP600125 compared to the non-treated
culture at 1 day, whereas the protein expression of late-stage
marker genes such as OSX and OCN was not affected by
SP600125 (Fig. 5).
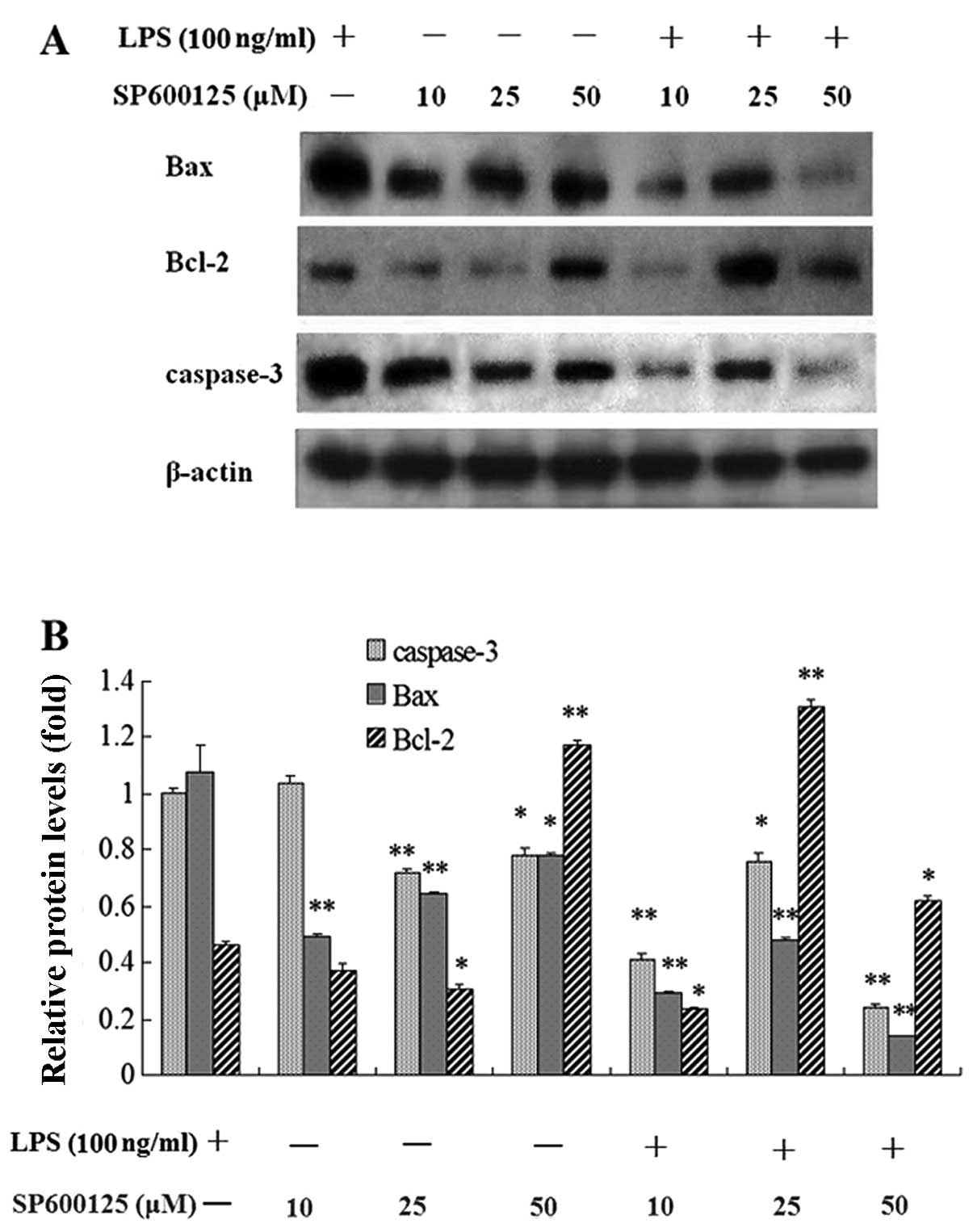
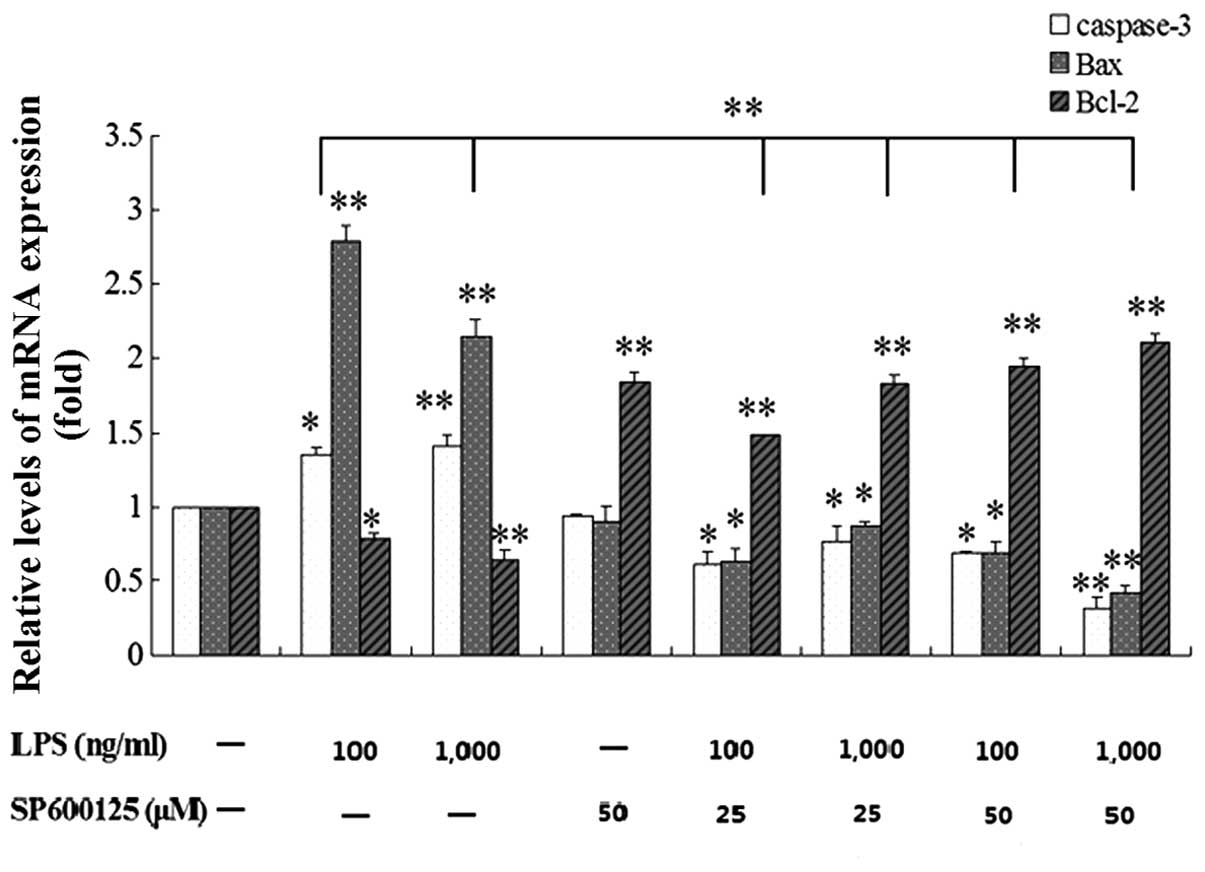
Effect of SP600125 on mRNA and protein
expression levels of Bax, Bcl-2 and caspase-3 in MC3T3-E1 cells
induced by LPS
Following pretreatment with 10, 25 and 50 μM
SP600125 or without SP600125 for 2 h, the cells were treated with
100 and 1,000 ng/ml LPS or without LPS in the presence of SP600125
for 1 and 2 day. The upregulated mRNA and protein expression levels
of caspase-3 and Bax in MC3T3-E1 cells induced by LPS were
significantly decreased by SP600125 compared to the non-treated
culture, whereas the downregulated expression of Bcl-2 was
significantly enhanced by SP600125 (Figs. 6–8).
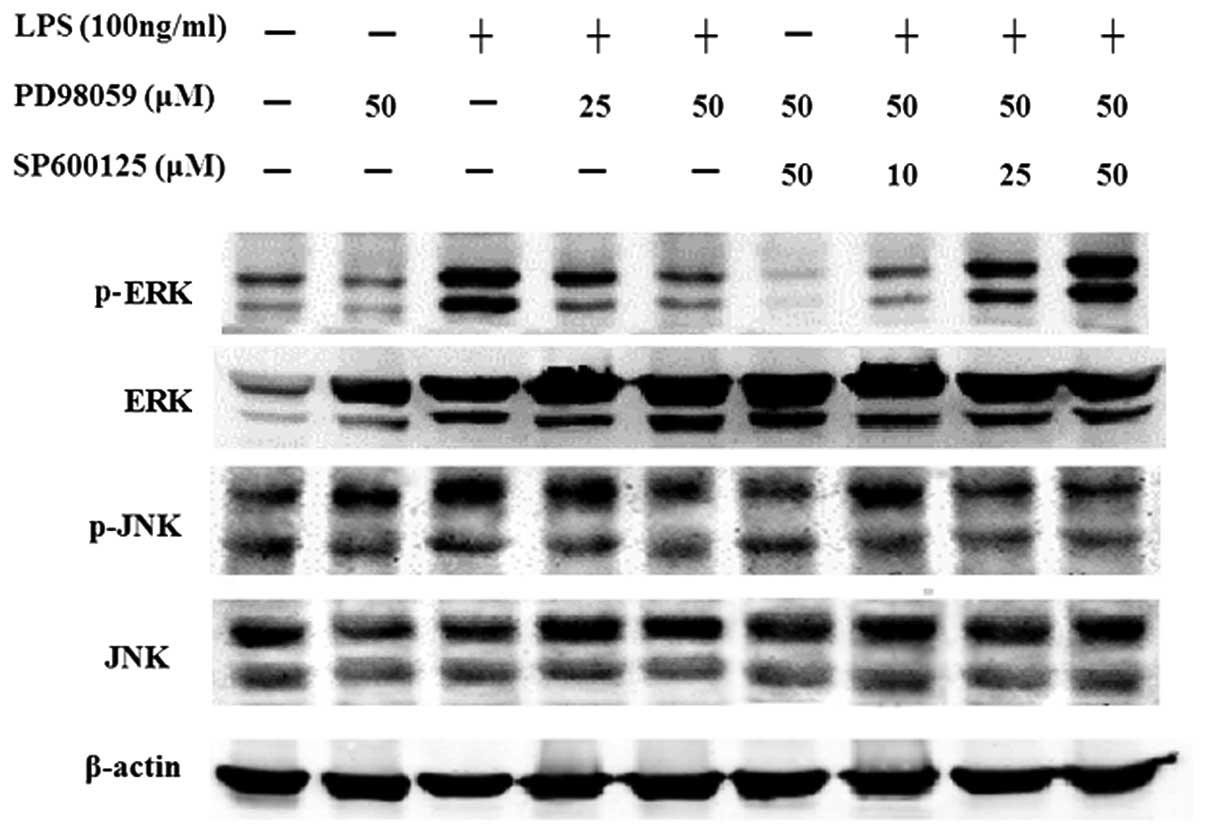
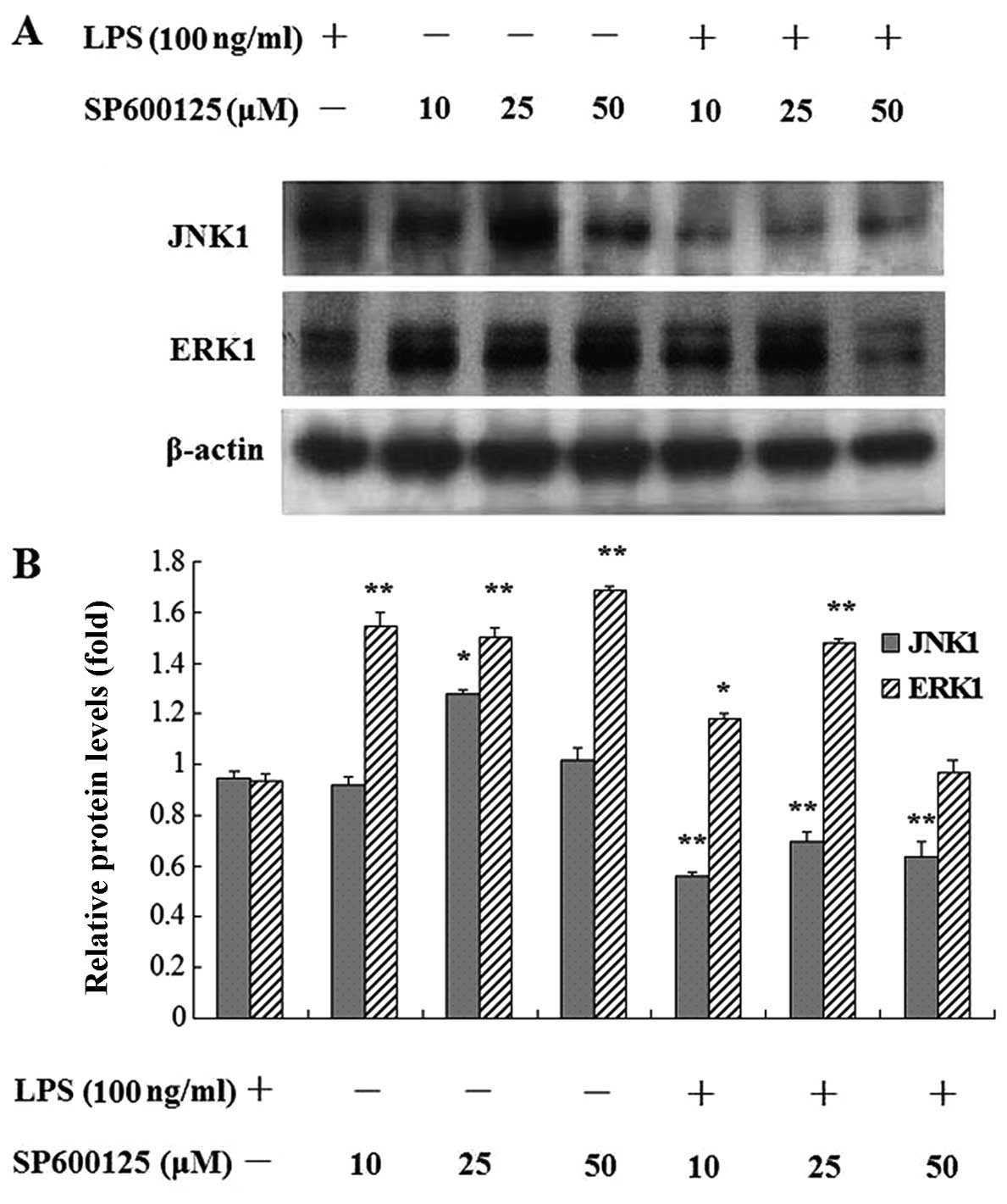
Effect of SP600125 on the protein
expression of MAPKs in MC3T3-E1 cells induced by LPS
After pretreatment with PD98059 alone, or treatment
with PD98059 for 2 h followed by treatment SP600125 for 2 h, the
cells were treated with or without LPS for 30 min or 1 day in the
presence of MAPK inhibitors. The results showed that PD98059
attenuated phosphorylation of ERK1/2 induced by LPS. Additional
treatment with SP600125 showed that phosphorylation of ERK1/2 was
significantly enhanced. SP600125 significantly enhanced the protein
expression of ERK1, but decreased JNK1 expression in MC3T3-E1 cells
induced by LPS after SP600125 treatment for 24 h (Figs. 9 and 10). SP600125 >10 μM was shown
to completely decrease the protein expression of JNK1 compared to
the non-treated cultures in MC3T3-E1 cells stimulated with LPS.
Discussion
Excessive bone resorption in chronic inflammatory
diseases such as septic arthritis, osteomyelitis, and infected
orthopedic implant failure is at least partially caused by
bacteria-induced activation of inflammatory responses (2). LPS, a pro-inflammatory glycolipid
component of the gram-negative bacteria cell wall, is mediated by
gram-negative bacterial bone destruction (3–8).
Moreover, LPS inhibits osteoblast differentiation and function
in vitro (9–12). Therefore, identification of
potential drugs that can restore osteoblast function remains a
fundamental objective in the prevention of bone destruction in
infective bone diseases. In a previous study we found LPS triggered
apoptosis and inhibited osteoblast differentiation through
activation of the JNK pathways (19). In this study, we evaluated the
effect of JNK inhibition by SP600125 on apoptosis and
differentiation in MC3T3-E1 osteoblasts stimulated with LPS. The
results show that SP600125 reduced LPS-induced osteo-blast
apoptosis and restored the early-stage differentiation of
osteoblasts inhibited by LPS through MAPK signaling.
Induction of osteoblast apoptosis caused
bacteria-induced bone destruction. We evaluated the effect of
SP600125 on the apoptosis of MC3T3-E1 cells induced by LPS. The
results showed that SP600125 significantly restored the cell
metabolism and downregulated the mRNA and protein expression levels
of Bax and caspase-3 as well as caspase-3 activity of MC3T3-E1
cells upregulated by LPS, but increased Bcl-2 expression of
MC3T3-E1 cells downregulated by LPS. These results indicate that
SP600125 reduced osteoblast apoptosis of MC3T3-E1 cells induced by
LPS. Thus, we suggest that JNK inhibition by SP600125 reduces
apoptosis of osteoblasts through the mitochondrial pathway.
Bone formation is a highly regulated process
including the differentiation of mesenchymal stem cells to
osteoblasts, preosteoblasts and osteoblasts. Runx2, ALP and BSP
have been suggested to be involved in the early-stage molecular
events of osteoblast differentiation. By contrast, OSX, downstream
of Runx2, as well as OCN and Col1α1, have been shown to be involved
in the late-stage of osteoblast differentiation and bone formation
(18,21–26). The reverse effect of SP600125 on
LPS-induced inhibition of osteoblast differentiation has been
confirmed by evaluating SP600125 on mRNA and protein expression
levels of osteoblast-associated genes (18). In this study, SP600125
significantly restored the expression of mRNA and protein of
early-stage osteoblast differentiation genes such as Runx2,
ALP and BSP in MC3T3-E1 cells suppressed by LPS.
However, the expression of late-stage markers such as OSX,
OCN and Col1α1 was not affected by SP600125. Our
results indicate that the reverse effect of SP600125 on LPS-induced
inhibition of osteoblast differentiation may be due to restoration
of osteoblast-associated genes.
MAP kinases are activated by various stresses
including LPS and influence apoptosis either positively or
negatively (27). In many cell
types, JNKs contribute to the induction of apoptosis, whereas ERK
inhibits apoptotic processes (28–30). In a previous study, treatment with
LPS enhanced the protein levels of phosphorylation of ERK1/2 and
JNK, while pretreatment with JNK inhibitor SP600125 attenuated the
phosphorylation of JNK and ERK1/2 enhanced by LPS. In this study,
pretreatment with SP600125 for 1 day decreased the protein level of
JNK1 in MC3T3-E1 cells stimulated with LPS, but enhanced protein
levels of ERK1. Furthermore, pretreatment with SP600125 and PD98059
decreased the protein level of phosphorylation of JNK but enhanced
the protein levels of phosphorylation of ERK1/2 in MC3T3-E1 cells
stimulated with LPS. Cross-activation of ERK to JNK has been
reported with JNK being the final downstream mediator for cell
proliferation and differentiation (31). Our results confirmed that
inactivation of JNK activity impaired the ERK-JNK interaction,
thereby prolonging the activation of ERK, which was mostly involved
in early-stage osteoblast differentiation in LPS-treated MC3T3-E1
cells, but did not affect the late-stage genes which were involved
in bone mineralization. The present study also confirms that JNK
inhibition may restore the early inhibition of osteoblast
differentiation induced by LPS through MAPK signaling (18).
In conclusion, our data suggest that SP600125 may
reduce LPS-induced osteoblast apoptosis and restore the early-stage
differentiation of osteoblasts inhibited by LPS through MAPK
signaling. These findings suggest therapeutic agents that inhibit
JNK1 may be used to restore osteoblast function in bacteria-induced
bone diseases.
Acknowledgments
This study was supported by a research grant from
the National Natural Science Foundation of China (grant no.
81371981), the School Fund of Luohe Medical College (no.
2012-DF001) and the Fund of Bureau of Science and Technology of
Luohe City (no. 2013–001).
References
|
1
|
Roodman GD: Advances in bone biology: the
osteoclast. Endocr Rev. 17:308–332. 1996.PubMed/NCBI
|
|
2
|
Nair SP, Meghji S, Wilson M, Reddi K,
White P and Henderson B: Bacterially induced bone destruction:
mechanisms and misconceptions. Infect Immun. 64:2371–2380.
1996.PubMed/NCBI
|
|
3
|
Orcel P, Feuga M, Bielakoff J and De
Vernejoul MC: Local bone injections of LPS and M-CSF increase bone
resorption by different pathways in vivo in rats. Am J Physiol.
264:E391–E397. 1993.PubMed/NCBI
|
|
4
|
Chiang CY, Kyritsis G, Graves DT and Amar
S: Interleukin-1 and tumor necrosis factor activities partially
account for calvarial bone resorption induced by local injection of
lipopolysaccharide. Infect Immun. 67:4231–4236. 1999.PubMed/NCBI
|
|
5
|
Itoh K, Udagawa N, Kobayashi K, et al:
Lipopolysaccharide promotes the survival of osteoclasts via
Toll-like receptor 4, but cytokine production of osteoclasts in
response to lipopolysaccharide is different from that of
macrophages. J Immunol. 170:3688–3695. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Islam S, Hassan F, Tumurkhuu G, et al:
Bacterial lipopolysaccharide induces osteoclast formation in RAW
264.7 macrophage cells. Biochem Biophys Res Commun. 360:346–351.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Mörmann M, Thederan M, Nackchbandi I,
Giese T, Wagner C and Hänsch GM: Lipopolysaccharides (LPS) induce
the differentiation of human monocytes to osteoclasts in a tumour
necrosis factor (TNF) alpha-dependent manner: a link between
infection and pathological bone resorption. Mol Immunol.
45:3330–3337. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zou W and Bar-Shavit Z: Dual modulation of
osteoclast differentiation by lipopolysaccharide. J Bone Miner Res.
17:1211–1218. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kadono H, Kido J, Kataoka M, Yamauchi N
and Nagata T: Inhibition of osteoblastic cell differentiation by
lipopolysaccharide extract from Porphyromonas gingivalis. Infect
Immun. 67:2841–2846. 1999.PubMed/NCBI
|
|
10
|
Xing Q, Ye Q, Fan M, Zhou Y, Xu Q and
Sandham A: Porphyromonas gingivalis lipopolysaccharide inhibits the
osteoblastic differentiation of prosteoblasts by activating Notch1
signaling. J Cell Physiol. 225:106–114. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bandow K, Maeda A, Kakimoto K, Kusuyama J,
Shamoto M, Ohnishi T and Matsuguchi T: Molecular mechanisms of the
inhibitory effect of lipopolysaccharide (LPS) on osteoblast
differentiation. Biochem Biophys Res Commun. 402:755–761. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ochi H, Hara Y, Tagawa M, Shinomiya K and
Asou Y: The roles of TNFR1 in lipopolysaccharide-induced bone loss:
dual effects of TNFR1 on bone metabolism via osteoclastogenesis and
osteoblast survival. J Orthop Res. 28:657–663. 2010.
|
|
13
|
Kyriakis JM and Avruch J: Mammalian
mitogen-activated protein kinase signal transduction pathways
activated by stress and inflammation. Physiol Rev. 81:807–869.
2001.PubMed/NCBI
|
|
14
|
Davis RJ: Signal transduction by the JNK
group of MAP kinases. Cell. 103:239–252. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Chang L and Karin M: Mammalian MAP kinase
signalling cascades. Nature. 410:37–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sabapathy K, Hochedlinger K, Nam SY, Bauer
A, Karin M and Wagner EF: Distinct roles for JNK1 and JNK2 in
regulating JNK activity and c-Jun-dependent cell proliferation. Mol
Cell. 15:713–725. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu J, Minemoto Y and Lin A: c-Jun
N-terminal protein kinase 1 (JNK1), but not JNK2, is essential for
tumor necrosis factor alpha-induced c-Jun kinase activation and
apoptosis. Mol Cell Biol. 24:10844–10856. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Matsuguchi T, Chiba N, Bandow K, Kakimoto
K, Masuda A and Ohnishi T: JNK activity is essential for Atf4
expression and late-stage osteoblast differentiation. J Bone Miner
Res. 24:398–410. 2009. View Article : Google Scholar
|
|
19
|
Guo C, Yuan L, Wang JG, et al:
Lipopolysaccharide (LPS) induces the apoptosis and inhibits
osteoblast differentiation through JNK pathway in MC3T3-E1 cells.
Inflammation. 37:621–631. 2014. View Article : Google Scholar
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
21
|
Kern B, Shen J, Starbuck M and Karsenty G:
Cbfa1 contributes to the osteoblast-specific expression of type I
collagen genes. J Biol Chem. 276:7101–7107. 2001. View Article : Google Scholar
|
|
22
|
Komori T, Yagi H, Nomura S, Yamaguchi A,
Sasaki K, Deguchi K, et al: Targeted disruption of Cbfa1 results in
a complete lack of bone formation owing to maturational arrest of
osteoblasts. Cell. 89:755–764. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kim HK, Cho SG, Kim JH, Doan TK, Hu QS,
Ulhaq R, et al: Mevinolin enhances osteogenic genes (ALP, type I
collagen and osteocalcin), CD44, CD47 and CD51 expression during
osteogenic differentiation. Life Sci. 84:290–295. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Thunyakitpisal P, Alvarez M, Tokunaga K,
et al: Cloning and functional analysis of a family of nuclear
matrix transcription factors (NP/NMP4) that regulate type I
collagen expression in osteoblasts. J Bone Miner Res. 16:10–23.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nakashima K, Zhou X, Kunkel G, Zhang Z,
Deng JM, Behringer RR and de Crombrugghe B: The novel zinc
finger-containing transcription factor osterix is required for
osteoblast differentiation and bone formation. Cell. 108:17–29.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kurata H, Guillot PV, Chan J and Fisk NM:
Osterix induces osteogenic gene expression but not differentiation
in primary human fetal mesenchymal stem cells. Tissue Eng.
13:1513–1523. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Pearson G, Robinson F, Beers Gibson T, et
al: Mitogen-activated protein (MAP) kinase pathways: regulation and
physiological functions. Endocr Rev. 22:153–183. 2001.PubMed/NCBI
|
|
28
|
Xia Z, Dickens M, Raingeaud J, Davis RJ
and Greenberg ME: Opposing effects of ERK and JNK-p38 MAP kinases
on apoptosis. Science. 270:1326–1331. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Park GB, Kim YS, Lee HK, Song H, Cho DH,
Lee WJ and Hur DY: Endoplasmic reticulum stress-mediated apoptosis
of EBV-transformed B cells by cross-linking of CD70 is dependent
upon generation of reactive oxygen species and activation of p38
MAPK and JNK pathway. J Immunol. 185:7274–7284. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Fister S, Günthert AR, Aicher B, Paulini
KW, Emons G and Gründker C: GnRH-II antagonists induce apoptosis in
human endometrial, ovarian, and breast cancer cells via activation
of stress-induced MAPKs p38 and JNK and proapoptotic protein Bax.
Cancer Res. 69:6473–6481. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Pedram A, Razandi M and Levin ER:
Extracellular signal regulated protein kinase/Jun kinase cross-talk
underlies vascular endothelial cell growth factor-induced
endothelial cell proliferation. J Biol Chem. 273:26722–26728. 1998.
View Article : Google Scholar : PubMed/NCBI
|