Introduction
Restenosis remains a challenge to percutaneous
coronary intervention (PCI) and coronary artery bypass graft (CABG)
for patients with coronary artery diseases (CHD) (1,2).
The proliferation and migration of vascular smooth muscle cells
(VSMCs) has been considered the major cause for underlying
restenosis after PCI or CABG (3,4).
To the best of our knowledge, the activation of mitogen-activated
protein kinase (MAPK) and phosphoinositide-3 kinase (PI3K)
signaling pathways mediated by Ras protein play a pivotal role in
the proliferation of VSMCs in the presence of vascular injury
(5,6). Consequently, we hypothesized that
blockage of Ras protein activation may be a target for therapeutic
strategy for vascular restenosis after PCI and CABG.
Using the systematic evolution of ligands by
exponential enrichment (SELEX) technique, several aptamers with
high affinity and specificity for target proteins were previously
selected from a library of randomized sequences in vitro,
including single-stranded DNA (ssDNA) or RNA oligo-nucleotides
(7–9). For instance, Bell et al
(10) developed an
oligonucleotide designated as NX1838 (2′-fluoropyrimidine,
RNA-based oligonucleotide/40-kDa-PEG) that inhibited vascular
endothelial growth factor 165 (VEGF165)-mediated cell response
in vitro by directly targeting the VEGF165 factor. In
addition, an oligonucleotide inhibiting multiple interferon-γ
signaling transduction was isolated, which could block the binding
of interferon-γ to its receptor (11). However, few studies have been
carried out to develop a specific aptamer binding directly to the
Ras protein.
In this study, a Ras protein-targeted ssDNA aptamer
designated as Ras-a1 was identified, and its potential function in
the proliferation and migration of human saphenous VSMCs was
investigated. Our study indicated that Ras-a1 inhibited the
proliferation and migration of VSMCs, and suppressed the
phosphorylation of proteins involved in the MEK, ERK1/2 and Akt
signaling pathways.
Materials and methods
Materials
Dulbecco’s modified Eagle’s medium with high glucose
(DMEM-HG), Opti-MEM I medium, Dulbecco’s phosphate-buffered saline
(PBS), glutamine, trypsin-EDTA, and penicillin/streptomycin were
purchased from Invitrogen (Carlsbad, CA, USA). Fetal bovine serum
(FBS) was purchased from Thermo Scientific (Logan, UT, USA). Cell
Counting kit-8 (CCK-8) was purchased from Dojindo Laboratories
(Kumamoto, Japan). The Transwell assay with 8-μm pore
polycarbonate membrane was purchased from Corning Inc. (Corning,
NY, USA). Rabbit polyclonal anti-phospho-MEK1/2, rabbit polyclonal
anti-phospho-ERK1/2, rabbit polyclonal anti-phospho-Akt, mouse
polyclonal anti-GAPDH, mouse polyclonal anti-Ras, mouse polyclonal
anti-MEK1/2, mouse polyclonal anti-ERK1/2, and mouse polyclonal
anti-Akt antibodies were purchased from Cell Signaling Technology
(Beverly, MA, USA).
Cell culture
VSMCs were cultured in DMEM-HG medium supplemented
with 10% FBS, 100 U/ml penicillin and 100 μg/ml
streptomycin. Cultures were maintained at 37°C in a humidified 95%
air and 5% CO2 atmosphere. The primary cells were
obtained from three patients with stenosis. All experiments were
performed in cell passages of 4–8. Written informed consent was
obtained from each patient. The study protocols were approved by
the Ethics Committee of Renmin Hospital of Wuhan University.
Random DNA library and primers
The DNA library was produced using the sequence,
5′-GGGAGCTCAGAATAAACGCTCAA-N35-TTCGACATGAGGCCCGGATC-3′,
where central N35 represented the random incorporation
of A, G, C and T at each position. The initial dsDNA random library
was generated by PCR amplification using forward,
5′-GGGAGCTCAGAATAAACGCTCAA-3′ and the biotin-labelled reverse
5′-GATCCGGGCCTCATGTCGAA-3′ primers.
In vitro selection of Ras ssDNA
aptamers
For the selection of Ras ssDNA aptamers in
vitro, Ras protein (100 pmol) and sterile dsH2O (100
μl) were incubated with 100 μl coating buffer (0.05
mol/l NaHCO3, PH 9.6) in 96-well plates for 18 h at 4°C.
After washing with PBS, the wells were blocked with 200 μl
3% bovine serum albumin (BSA) for 45 min at 37°C. Subsequently, 100
μl random ssDNA pools were incubated with 100 μl
binding buffer (20 mM Hepes pH 7.35, 120 mM NaCl, 5 mM KCl, 1 mM
MgCl2, and 1 mM CaCl2) for 45 min at 37°C.
The supernatants were then incubated for another 45 min at 37°C.
Following the removal of the unbound sequences, well-retained ssDNA
was eluted by adding the elution buffer (20 mM Tris-HCl, 4 mol/l
guandine thiocyanate, and 1 mM DTT, pH 8.3) and incubated at 80°C
for 8 min. Ethanol was then added to precipitate the ssDNA
products.
The isolated ssDNA was amplified by PCR reactions
performed in a total volume of 20 μl of PCR mixtures,
containing 10 μl of 2X PCR buffer, 0.4 mM dNTPs, 10 pmol
forward primers, 10 pmol reverse biotinylated primers, 1 pmol
template, and 0.5 units Taq DNA polymerase. The PCR conditions
were: denaturation at 94°C for 3 min, followed by 24 cycles of
denaturation at 94°C for 40 sec, annealing at 60°C for 30 sec, and
extension at 72°C for 30 sec. PCR products were electrophoresed on
a 3% agarose gel and then purified by a UNIQ-10 DNA Extraction kit
(Sangon, Shanghai, China) according to the manufacturer’s
instructions.
The ssDNA pool was obtained by separating the dsDNA
PCR product with streptavidin-coated magnetic beads (M-280
Dynabeads; Invitrogen, Karlsruhe, Germany). Briefly, 50 μg
dsDNA PCR products were incubated with 10 mg beads in a tube on a
rotator for 30 min. Subsequently, the tube was placed on a magnet
for 3 min to obtain the biotinylated dsDNA. The coated beads were
washed three times in binding and washing buffer (10 mM Tris-HCl, 1
mM EDTA, 2 M NaCl). Alkaline denaturation was performed with 0.15 M
NaOH for 2 min. Subsequently, the supernatant was collected, and
the selected ssDNA aptamer for the following SELEX round was
successfully obtained. The repeated selection and amplification
were performed until no significant increase was evident in the
binding rates and affinities.
Binding affinity assays between the
aptamer pools and Ras protein
The ELISA assay was used to examine the direct
binding of aptamer to Ras protein. Ras protein was used to coat
Nunc 96-well plates and incubated overnight at 4°C. After washing
with PBS, the wells were blocked with 3% BSA for 45 min at 37°C.
The obtained biotinylated aptamers (up to 100 nM) were allowed to
bind with Ras protein for 45 min at 37°C. The plates was washed
again, and streptavidin-HRP conjugate was applied as a secondary
detection reagent. The plate was washed, and tetramethyl-benzidine
substrate was added. For color development, a wavelength of 450 nm
was used for the light absorbance measured by using an ELISA reader
(Lumitron Ltd., Petach-Tikva, Israel). The dissociation constant
(Kd) was calculated according to the formula Y = Bmax x
X/(Kd+X), where X was the concentration, Y the mean optical
density, and Bmax the maximum optical density.
Cloning, sequencing and structure
analysis of selected aptamers
The selected aptamers from the 11th round of SELEX
were purified and linked with pMD18-T vector, and then transformed
into Escherichia coli DH5α strains. Plasmid DNA was isolated
from individual clones, purified and analyzed by sequencing. A
total of 45 individual aptamers were obtained. The primary and
secondary structures of individual aptamers were analyzed based on
their sequences using the DNAMAN 5.29 software (Lynnon Corp.,
Quebec, QC, Canada).
Transfection of Ras-a1
Prior to transfection, the cells were washed twice
with Opti-MEM I medium and resuspended in Opti-MEM I medium until a
final concentration of 2.0×106 cells/ml. After the
addition of Ras-a1 (20 μg/ml), the cells were cooled on ice
for 10 min. Subsequently, 400 μl cell suspension was
transferred to a 0.4-cm-gap cuvette (BTX, Holliston, MA, USA).
Subsequently, electroporation was performed using one pulse of 200
V electric field and 20 msec pulse length using ECM 830 Electro
Square Porator (BTX). The cells treated using only electroporation
were set as the control. Following electroporation, the cells were
incubated at 37°C in a humidified 95% air and 5% CO2
atmosphere for 30 min. For the assessment of cell viability, 50
μl cell suspension obtained from each cuvette was
transferred to a 96-well plate with 1×104 cells per well
and cultured for 12 h. To assess the efficiency of transfection,
200 μl cell suspension from each cuvette was plated in a
24-well plate containing 1 ml DMEM-HG medium supplemented with 10%
FBS, followed by incubation for 6 h.
Cell proliferation assay
The proliferation of VSMCs was determined using a
CCK-8 kit according to the manufacturer’s instructions. Briefly,
the cells were collected 30 min after electroporation. The cells
were washed using PBS, and resuspended in DMEM-HG medium
supplemented with 10% FBS. Subsequently, 100 μl cell
suspension was added into each well of the 96-well plate until a
concentration of 1×105 cells/ml. The cells were then
collected at 12, 24, 36 and 48 h, respectively. At each time-point,
10 μl CCK-8 reagent was added to each well and incubated for
another 2 h prior to measuring the optical density (OD) in each
well at 450 nm with a VICTOR3 Multilabel Counter model 1420
(Elmer-Perkin, Boston, MA, USA). The inhibitory rates were
calculated using the formula: Inhibitory rate (%) = 1-OD (Ras-a1
cells)/OD (control cells) ×100.
Cell migration assay
The migration of VSMCs was measured using Transwell
chambers with 8.0-μm pore polycarbonate membranes as
previously described (12).
Briefly, the cells were harvested 30 min after electroporation.
After washing with PBS, the cells were resuspended in serum-free
DMEM supplemented with 0.2% BSA. Cell suspension (100 μl)
was then transferred into the upper chamber at a concentration of
1×105 cells/ml, with the lower chambers filled with 600
μl DMEM-HG medium supplemented with 10% FBS. The chambers
were incubated in a cell culture incubator for 6, 12, 18 and 24 h
at 37°C, respectively. At each time-point, the cells on the upper
surface of the polycarbonate membrane were gently removed, while
the cells on the reverse side of the membrane were fixed with 4%
paraformaldehyde for 15 min and stained with DAPI for 10 min. Cell
migration was quantified by counting the number of cells on the
reverse side of the polycarbonate membrane under an IX51 converted
fluorescence microscope (Olympus Corporation, Tokyo, Japan). Five
fields were randomly selected in each membrane for the cell count.
The experiments were performed at least in triplicate.
Western blot analysis
MEK1/2, ERK1/2 and Akt protein expression in VSMCs
was determined using western blot analysis as previously described
(13). Briefly, the cultured
VSMCs were lysed at 4°C in lysis buffer containing 100 mmol/l
Tris-HCl pH 7.4, 150 mmol/l NaCl, 1% NP-40, 1% sodium deoxycholate,
5 mmol/l EDTA, 10 μg/ml leupeptin, 10 μg/ml
aprotinin, 10 μg/ml pepstain, 1 mmol/l PMSF, 10 mmol/l
β-glycerophosphate, 1 mmol/l Na3VO4 and 10
mmol/l NaF. Subsequently, 50 μg proteins were separated via
10% SDS-PAGE and transferred onto nitrocellulose membranes (Pall
Corporation, Port Washington, NY, USA). The membranes were then
blocked in Tris-buffered saline (TBS) containing 5% non-fat milk
for 2 h. The mixture was then incubated in the primary antibody
including rabbit poly-clonal anti-phospho-MEK1/2 (1:1,000),
anti-phospho-ERK1/2 (1:2,000), anti-phospho-Akt (1:1,000), mouse
polyclonal anti-GAPDH (1:500), anti-Ras (1:1,000), anti-MEK1/2
(1:1,000), anti-ERK1/2 (1:2,000), or anti-Akt (1:1,000) antibody in
TBST containing 1% BSA overnight at 4°C. The membranes were then
washed and incubated in IRDye 800 CW goat anti-rabbit/mouse IgG
secondary antibodies (1:10,000) for detection. The same membrane
was probed for GAPDH for the loading control. Intensity of the
protein band was determined using an Odyssey Infrared Imaging
System (LI-COR Biotechnology, Lincoln, NE, USA).
Statistical analysis
Data are expressed as the means ± standard deviation
(SD). ANOVA or the unpaired Student’s t-test was used to determine
the inter-group difference. P<0.05 was considered statistically
significant.
Results
Isolation of high-affinity Ras protein
aptamers
In this study, Ras protein was used as a selection
target, while random ssDNA library was used as screening ligands.
Sterile dsH2O was used for the counter selection. After
11 rounds of selection, different pools of aptamers were obtained
using SELEX. The aptamers of each pool and the PCR products were
identified using electrophoresis on a 3% agarose gel. The length of
the dsDNA ranged 40–50 bp (Fig.
1A).
As revealed by the ELISA assay, the binding
affinities increased gradually from 51.5 to 18.3 nM (Kd). The
highest binding affinity was noted in the 11th pool with a Kd of
18.3 nM. No further improvement was observed in the binding
affinities obtained after 11 rounds of selection. Subsequently, the
products selected through the 11th pool were linked to the pMD18-T
vector, based on which the individual clones of single aptamers
were selected and sequenced. The consensus regions within the
individual aptamers are shown in Table I. As shown in Tables II and III, a total of 45 individual aptamers
were obtained. For the binding affinities between individual
aptamers and Ras, Ras-a1 showed the highest affinity with a Kd of
15.3 nM. The sequence of binding affinities between the aptamer
pools was as follows: Ras-a1 >11th pool >10th pool >9th
pool >8th pool >6th pool >4th pool >Ras-a27 >1st
pool (Table II). DNAMAN 5.29
software (Lynnon Corp.) was used to predict the secondary structure
of Ras-a1, which showed that the binding sites to the Ras protein
were localized at the terminal loop of a stem-loop structure
(Fig. 1B).
 | Table IDoses of aptamer pools and Ras
proteins in each selection. |
Table I
Doses of aptamer pools and Ras
proteins in each selection.
| Round | ssDNA (pmol) | Ras protein
(pmol) | ssDNA/Ras |
|---|
| 1 | 1,000 | 100 | 10 |
| 2 | 500 | 50 | 10 |
| 3 | 200 | 20 | 10 |
| 4 | 100 | 10 | 10 |
| 5 | 100 | 7.5 | 13 |
| 6 | 50 | 3.75 | 13 |
| 7 | 50 | 2.5 | 20 |
| 8 | 30 | 1.5 | 20 |
| 9 | 30 | 1.2 | 25 |
| 10 | 20 | 0.8 | 25 |
| 11 | 20 | 0.6 | 33 |
| Table IIThe OD and Kd values of different
aptamers. |
Table II
The OD and Kd values of different
aptamers.
| Aptamer | 1st P | 4th P | 6th P | 8th P | 9th P | 10th P | 11th P | Ras-a1 | Ras-a27 |
|---|
| OD | 0.220 | 0.498 | 0.702 | 0.917 | 1.012 | 0.988 | 1.080 | 1.213 | 0.459 |
| Kd (nM) | 51.5 | 47.4 | 39.9 | 24.8 | 19.8 | 19.6 | 18.3 | 15.3 | 48.3 |
| Table IIIThe results of cloning and
sequencing. |
Table III
The results of cloning and
sequencing.
| Aptamer | Random sequence
(5′→3′) |
|---|
| Family 1 |
| 1 |
TCGTCGGATCGACGCGGTTTAGTGAGTGTGCGTGG |
| 2 |
TCGTCGGATCGACGCGGTTTAGTGAGTGTGCGTGG |
| 3 |
GCTGAAGCAGGGTTGAGTGATAAATGGTGCGTCGG |
| 4 |
GGGCAAGCGTCGTGGACAGGAAGAAGTATGTGGTA |
| 5 |
GTAAGTGTGTGGTGATTTGGTATATGTAGGCGTCG |
| Family 2 |
| 6 |
GGGGGGGTCGGGAGGTCAGGTTAGGGGGGGTGTTG |
| 7 |
GAGGGGCGGAGTATCGGGGGGGGGGGAGGGGGCGG |
| 8 |
GAAAGAAGGGGTTGTTGTGAGGGGGGTGGTGGTGG |
| 9 |
GCAAAGATTGGGGGGATGATTGAAATAAGCGTGTG |
| 10 |
GCGGGGGTGGTTGGGCTGGGGGATGTGGGGTGGTG |
| 11 |
TGGGGACGGGGGTATGGGAGGGGGGTGGTGGTGGA |
| 12 |
GCTAATGCCGTATGCGCAAGGATGACACACGGGGG |
| 13 |
AAGAGTGAGATGATGGATAATGCAAACTGGGGGTG |
| 14 |
AGTGCTGCTTGGGGGTTCGTAGTTCGAGTGCTAGG |
| 15 |
CGATGGTGTGGTTGGTGGGGGAGTGCGACGCCTGA |
| 16 |
TAAGCGCGAGGGGGTATAGGTGTATGTAGCGTGAG |
| Family 3 |
| 17 |
GAGGGATAGGAGTGGTGTTGTTCGTTGTTGCATGG |
| 18 |
GTGGAGTATTGGTAGTGATAGAGGGCTTAGGCGGG |
| 19 |
GGAGGGGATGACGAGAGCAATCGGCGTGCAAGGGA |
| 20 |
GGGAGTTGGCGAATAAGCTTTAGTTGGGAGGGATG |
| 21 |
TGGTTGAGGGGAGGCAGAGGGAGGTATAAGGTATG |
| 22 |
ACCGGGCGGGTTGCAAGATGGTGAGGGTGCGGGTG |
| 23 |
GGAGGGGAAGAAGACGCGAACATGAAAGATGCGTG |
| Family 4 |
| 24 |
GAGACAGAGCTACGGAGGTGAGATATCAGCATGTG |
| 25 |
TTGAGCGGGTAATTGGAGTATGTGTGTGTAGGTGG |
| 26 |
TACTGCAAGAGTGAAAATGTGGTTAGTGAGGTCGG |
| 27 |
GATGATGTGGGGAGTGGAGTGTAATGGAGAGTGAG |
| 28 |
GGCGGATGTGAGTTGTTATGAGTTGCAGAGTGGAG |
| 29 |
GGAGTGTGTTGGGGTGCTATTTGGTTGTATGTGTG |
| Family 5 |
| 30 |
GGTGAGGAGAAGCGGTAGGTGAAATGGAAGTAAGG |
| 31 |
AGCAAAGGAAGACAGGGGAAGTGGTGAAGTTGACG |
| 32 |
AGAGGATAGTGATGGACGGTGAAGTGCGTGGGTGG |
| 33 |
AGGGGAAGTGGTGATCGGATAGGACAAAGGAAGTG |
| Family 6 |
| 34 |
GAGGTATGCACGATGGGAGTGAAACTAACGGGGAG |
| 35 |
TAAGGGAAAGTGTAGTAAGGAAAGTTGAGTGGGAG |
| 36 |
CGTGGGAGCTGGAACATATTGCTGAGGAAGTTCGT |
| Family 7 |
| 37 |
GCCTCGTGGCGATAAAGGTTGGGTAGGTTGGGTCC |
| 38 |
GAGGTTGGGCTGGAATAAGATTGGTTGTGGGTAGG |
| 39 |
TGCGAGAAGCGCGGAAAAGGGTAGGCACGGAAGCG |
| Family 8 |
| 40 |
GCGTGGTAAGGTCGGCAGCAGGGCGCAGTTGGAGG |
| 41 |
TGCCGGAACCGGCAGGGAAGGTGTGTGGGTGTGCA |
| 42 |
GCGGAGGAAGGTAGAGACGATGGCTATAGCGATAG |
| Family 9 |
| 43 |
GTCACATCGAACAACGGGCGTAATTCGTGTCTGGG |
| 44 |
GAAAGACGGAGGCGTGTAGGAAAGTGGAGTGAGCG |
| Family 10 |
| 45 |
TCTTTCTATTTTCTGTTGGTTTTTGTTGTGTGTTT |
Electrotransfer of Ras-a1 into VSMC
To observe the electrotransfer of Ras-a1 into VSMCs,
Cy3-labeled Ras-a1 was visualized using a fluorescence microscope.
The transfection efficiency was defined as the ratio of
electroporated cells labeled with Cy3 red fluorescence in the dark
field to the cells in the bright field (Fig. 2). The transfection efficiency was
(82.11±6.03%) with a survival rate of (83.04±1.74%).
Ras-a1 inhibits VSMC proliferation
The proliferation of VSMCs was significantly
inhibited in cells transfected with Ras-a1 compared with that of
control. The inhibition rates obtained at 12, 24, 36 and 48 h were
25, 40, 29 and 23%, respectively (Fig. 3B). The maximal inhibitory rate was
achieved at 24 h, and prolonged treatment resulted in no greater
growth inhibition (Fig. 3).
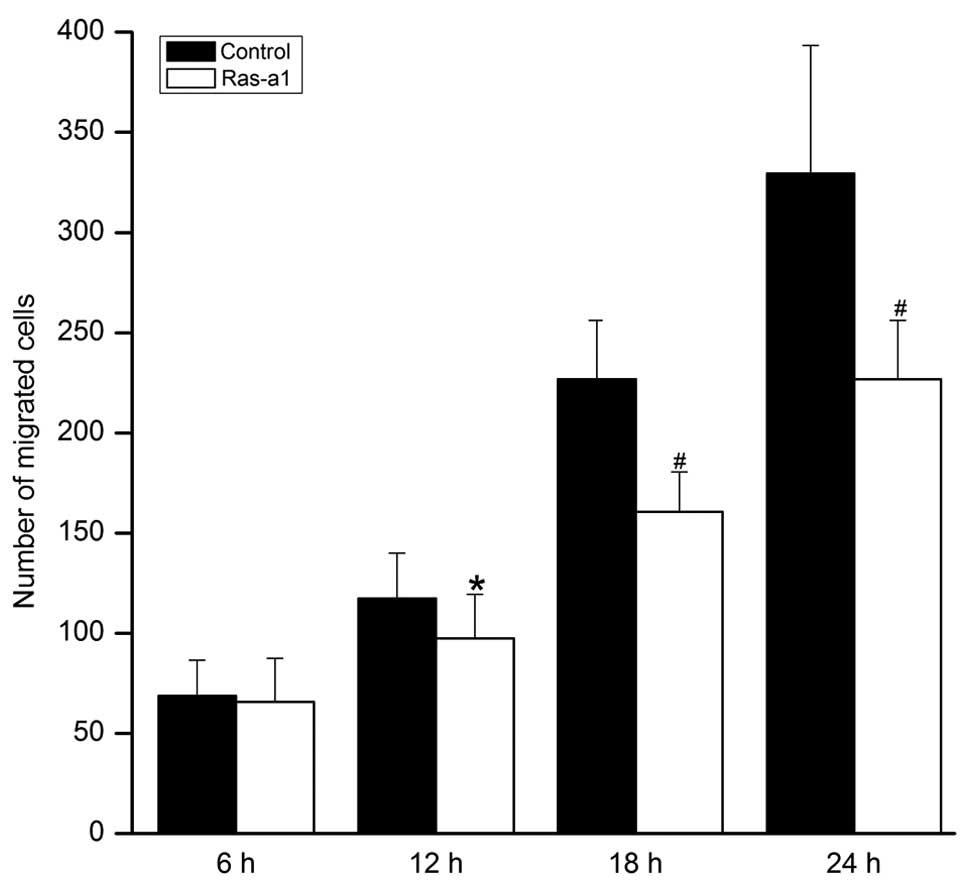
Ras-a1 inhibits VSMC migration
Cell migration of VSMCs was significantly inhibited
in Ras-a1 cells in a time-dependent manner. Compared with the
control group, no statistical difference was noted in the number of
migrated cells in the Ras-a1-transfected group at 6 h (65.86±21.75
vs. 68.73±17.8, P>0.05). However, compared with the control
group, statistical differences were noted in the cellular migration
of the Ras-a1 transfected group at 12 h (97.53±21.89 vs.
117.47±22.66, P<0.05), 18 h (160.60±19.91 vs. 226.80±29.29,
P<0.01), and 24 h (228.73±28.31 vs. 329.53 ±63.73, P<0.01),
respectively (Fig. 4).
Effects of Ras-a1 on MEK1/2, ERK1/2 and
Akt phosphorylation in VSMC
To investigate the effect of Ras-a1 on the MAPK and
PI3K signaling pathways, we measured the phosphorylation of MEK1/2,
ERK1/2 and Akt proteins using western blot analysis. As shown in
Fig. 5, a significant decrease
was detected in the phosphorylation of MEK1/2, ERK1/2 and Akt
proteins in Ras-a1-transfected cells compared with the control
(P<0.05). By contrast, the expression of Ras protein was not
altered by Ras-a1 transfection. These data suggested that Ras-a1
inhibited the proliferation and migration of VSMCs by modulating
the signal transduction of MAPK and PI3K pathways.
Discussion
Restenosis, defined as the reoccurrence of the
luminal narrowing of a vessel, remains a challenge to the long-term
success of therapeutic approaches to CHD such as PCI and CABG. It
has been reported that VSMC proliferation and migration was
proportional to the extent of vascular injury (14). Therefore, we aimed to develop a
potential therapeutical strategy for neointimal hyperplasia and
subsequent restenosis by inhibiting the proliferation and migration
of VSMCs.
Extensive studies have been performed to investigate
the potential application of aptamer in disease diagnosis and
treatment. Aptamers with high affinity and specificity have been
developed to target peptides, proteins, drugs, organic and
inorganic molecules and even whole cells (7–9).
Accumulating evidence has indicated that certain aptamers showed
great therapeutic efficacy both in vitro and in vivo.
Pegaptanib, the first therapeutic aptamer approved by the FDA for
the treatment of age-related macular degeneration, acts by
inhibiting VEGF165 (15,16). This aptamer is primarily
responsible for pathological ocular neovascularization and
increasing vascular permeability. OPN-R3, an RNA aptamer against
human osteopontin, has been reported to significantly decrease
local progression and distant metastases (17).
Few studies have been performed regarding the
interruption or inhibition of the proliferation and migration of
VSMCs by interrupting the Ras signals using aptamers. Sedding et
al (18) demonstrated that
3-deazaadenosine (c3Ado) may prevent VSMC proliferation and
neointima formation by interrupting Ras signaling. In addition,
using recombinant adenoviruses containing a constitutively active
mutant (AdRasV12) to infect rat common carotid arteries following
balloon injury, Jin et al (19) showed that the neointimal formation
was significantly elevated compared with the negative control at
week 2. In the present study, we initially isolated ssDNA aptamers
that bound specifically to Ras proteins using SELEX. During the
in vitro selection, blank control groups were set in order
to eliminate false positives. Different pools of aptamers were
obtained and the highest binding affinity was identified in the
11th pool. A total of 45 individual aptamers were thus obtained,
among which Ras-a1 had the highest affinity with a Kd of 15.3 nM.
The biological activity of Ras protein was silenced by ssDNA
aptamer Ras-a1, and the proliferation and migration of VMSCs was
significantly reduced.
To investigate the mechanism of Ras-a1-mediated
inhibition of VSMC proliferation and migration, we examined the
effects of Ras-a1 on the expression and activation of Ras, MEK1/2,
ERK1/2 and Akt, respectively. It has been well established that
ERK1/2 and PI3K/Akt signaling pathways play key roles in the
regulation of VSMC proliferation and migration (20,21). According to previous findings, the
activation of Ras induced by growth factors through direct binding
with the receptors contributed to the activation of the downstream
components of MEK and PI3K, which activate ERK1/2 and Akt,
respectively (22). As a result,
various downstream effectors controlling the proliferation and
migration of VSMCs were triggered. Our results revealed that the
phosphorylation of Ras, MEK1/2, ERK1/2 and Akt showed a significant
decrease in Ras-a1-transfected VSMCs within 24 h. Consequently, we
hypothesized that Ras-a1 inhibits the phosphorylation and
activation of Ras, resulting in the subsequent blocked activation
of the downstream signaling proteins in the Ras-MEK-ERK1/2 and
Ras-PI3K-Akt pathways. Therefore, the migration and proliferation
of VSMCs was blocked due to the transduction failure of
extracellular signals into the nucleus.
In conclusion, a novel Ras ssDNA aptamer designated
as Ras-a1 with high specificity and affinity to Ras protein was
isolated using SELEX. It inhibited the proliferation and migration
of VSMCs by downregulating the phosphorylation of Ras protein and
interrupting the signal transduction in Ras-MEK1/2-ERK1/2 and
PI3K/Akt pathways. The results demonstrate Ras-a1 may be used as an
effective strategy to prevent restenosis after PCI and CABG.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (no. 30872537) and the
Fundamental Research Funds for the Central Universities (no.
2042014kf0117).
References
|
1
|
Shahabuddin S, Sami SA, Ansari JA, et al:
Coronary artery bypass grafting after percutaneous coronary
intervention. J Coll Physicians Surg Pak. 22:340–341.
2012.PubMed/NCBI
|
|
2
|
Abbate A, Biondi-Zoccai GG, Agostoni P,
Lipinski MJ and Vetrovec GW: Recurrent angina after coronary
revascularization: a clinical challenge. Eur Heart J. 28:1057–1065.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang Z, Zhang X, Chen S, et al: Lithium
chloride inhibits vascular smooth muscle cell proliferation and
migration and alleviates injury-induced neointimal hyperplasia via
induction of PGC-1α. PLoS One. 8:55471–55482. 2013. View Article : Google Scholar
|
|
4
|
Louis SF and Zahradka P: Vascular smooth
muscle cell motility: From migration to invasion. Exp Clin Cardiol.
15:e75–e85. 2010.
|
|
5
|
Guo X, Chen KH, Guo Y, Liao H, Tang J and
Xiao RP: Mitofusin 2 triggers vascular smooth muscle cell apoptosis
via mitochondrial death pathway. Circ Res. 101:1113–1122. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ramos KS: H-RAS controls phenotypic
profiles of vascular smooth muscle cells and the pathogenesis of
vascular proliferative disorders. Circ Res. 104:1139–1141. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kong HY and Byun J: Nucleic acid aptamers:
new methods for selection, stabilization, and application in
biomedical science. Biomol Ther (Seoul). 21:423–434. 2013.
View Article : Google Scholar
|
|
8
|
Chen CH, Chernis GA, Hoang VQ and Landgraf
R: Inhibition of heregulin signaling by an aptamer that
preferentially binds to the oligomeric form of human epidermal
growth factor receptor-3. Proc Natl Acad Sci USA. 100:9226–9231.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cerchia L, Duconge F, Pestourie C, et al:
Neutralizing aptamers from whole-cell SELEX inhibit the RET
receptor tyrosine kinase. PLoS Biol. 3:e1232005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bell C, Lynam E, Landfair DJ, Janjic N and
Wiles ME: Oligonucleotide NX1838 inhibits VEGF165-mediated cellular
responses in vitro. In Vitro Cell Dev Biol Anim. 35:533–542. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee PP, Ramanathan M, Hunt CA and Garovoy
MR: An oligonucleotide blocks interferon-gamma signal transduction.
Transplantation. 62:1297–1301. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Torres RA, Drake DA, Solodushko V, et al:
Slingshot isoform-specific regulation of cofilin-mediated vascular
smooth muscle cell migration and neointima formation. Arterioscler
Thromb Vasc Biol. 31:2424–2431. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Izumiya Y, Kim S, Izumi Y, et al:
Apoptosis signal-regulating kinase 1 plays a pivotal role in
angiotensin II-induced cardiac hypertrophy and remodeling. Circ
Res. 93:874–883. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Indolfi C, Esposito G, Di Lorenzo E, et
al: Smooth muscle cell proliferation is proportional to the degree
of balloon injury in a rat model of angioplasty. Circulation.
92:1230–1235. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Deissler HL and Lang GE: Effect of VEGF165
and the VEGF aptamer pegaptanib (Macugen) on the protein
composition of tight junctions in microvascular endothelial cells
of the retina. Klin Monbl Augenheilkd. 225:863–867. 2008.In German.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Apte RS: Pegaptanib sodium for the
treatment of age-related macular degeneration. Expert Opin
Pharmacother. 9:499–508. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mi Z, Guo H, Russell MB, Liu Y, Sullenger
BA and Kuo PC: RNA aptamer blockade of osteopontin inhibits growth
and metastasis of MDA-MB231 breast cancer cells. Mol Ther.
17:153–161. 2009. View Article : Google Scholar
Sedding DG, Tröbs M, Reich F, et al:
3-Deazaadenosine prevents smooth muscle cell proliferation and
neointima formation by interfering with Ras signaling. Circ Res.
104:1192–1200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sedding DG, Tröbs M, Reich F, Walker G,
Fink L, Haberbosch W, Rau W, Tillmanns H, Preissner KT, Bohle RM,
et al: 3-Deaza-adenosine prevents smooth muscle cell proliferation
and neointima formation by interfering with Ras signaling. Circ
Res. 104:1192–1200. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Jin G, Chieh-Hsi Wu J, Li YS, Hu YL, Shyy
JY and Chien S: Effects of active and negative mutants of Ras on
rat arterial neointima formation. J Surg Res. 94:124–132. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Isenović ER, Kedees MH, Tepavcevic S, et
al: Role of PI3K/AKT, cPLA2 and ERK1/2 signaling pathways in
insulin regulation of vascular smooth muscle cells proliferation.
Cardiovasc Hematol Disord Drug Targets. 9:172–180. 2009. View Article : Google Scholar
|
|
21
|
Shen YJ, Zhu XX, Yang X, et al: Cardamonin
inhibits angio-tensin II-induced vascular smooth muscle cell
proliferation and migration by downregulating p38 MAPK, Akt, and
ERK phos-phorylation. J Nat Med. 3:623–629. 2014. View Article : Google Scholar
|
|
22
|
Shah BH, Neithardt A, Chu DB, Shah FB and
Catt KJ: Role of EGF receptor transactivation in phosphoinositide
3-kinase-dependent activation of MAP kinase by GPCRs. J Cell
Physiol. 206:47–57. 2006. View Article : Google Scholar
|