Introduction
Hydrogen sulfide (H2S) is an endogenous
gaseous mediator with regulatory roles in neurotransmission,
cardiovascular function and cell metabolism. It also participates
in the regulation of the oxidative balance of the cells, under both
normal physiological conditions, as well as in various diseases
(1–8). Various classes of H2S
donors have been tested in multiple models of inflammation. The
results have revealed that H2S exerts cytoprotective and
anti-inflammatory effects, including the inhibition of multiple
pro-inflammatory signaling pathways and a reduction in the
production of reactive oxygen and nitrogen species (9–24).
The post-translational modification of histones is
one form of epigenetic modifications that alter gene expression
(25). Amino acids present in the
histone tail can be modified by acetylation, methylation,
phosphorylation, ubiquitination and other enzymatic modifications
during RNA synthesis (26).
Histone acetylation is associated with chromatin unfolding, i.e.,
it facilitates gene transcription. On the other hand, histone
deacetylation inhibits gene transcription. Histone methylation can
either inhibit or activate gene transcription, depending on the
localization (27). Histone
methyltransferases (HMTs, enzymes that transfer acetyl groups to
the histone tail at lysine and arginine residues) promote histone
methylation, while histone acetylation is mediated by histone
acetyltransferases (HATs) that exert their effects at lysines of
histones H3 and H4 (26–28). Neither histone methylation nor
acetylation is permanent, as the modifications can be removed by
histone deacetylases (HDACs) and demethylases, respectively, thus
rendering epigenetic regulation a dynamic regulator of gene
transcription (29). In the
present study, we investigated whether H2S acts as a
regulator of chromatin modulation and cytokine production in an
in vitro model of inflammation.
Materials and methods
Cell culture
Tamm-Horsfall protein 1 (THP-1) cells were
maintained in RPMI-1640 supplemented with 2 mm l-glutamine, 100
U/ml penicillin, 100 μg/ml streptomycin and 10% fetal bovine
serum (FBS; Sigma, St. Louis, MO, USA). Ultrapure Escherichia
coli 0111:B4 LPS free of lipoproteins was obtained from
Invitrogen (San Diego, CA, USA). The cells were plated in 22-mm
tissue culture dishes (2×106 cells/dish). Macrophage
differentiation was induced with phorbol myristate acetate (PMA,
100 nM) for 5 h. In one set of experiments (pre-treatment
experiments) the effects of H2S were examined following
a 30-min pre-treatment with sodium hydrosulfide (NaHS, an
H2S donor) (Sigma) at 0.01, 0.1, 0.5 or 1 mM followed by
a washout, followed by incubation with bacterial lipopolysaccharide
(LPS, 1 μg/ml) in 1% FSB RPMI-1640 for 1, 4, 8 or 24 h. In
another set of experiments (co-treatment experiments), NaHS was
administered 30 min prior to the LPS administration, without a
washout. The control cells were maintained in 1% FBS RPMI-1640.
Western blot analysis
The cells were placed in RIPA buffer and sonicated
(3 times for 10 sec each). The supernatants were preserved and the
protein concentration was determined by bicinchoninic acid (BCA)
assay. Protein expression was determined by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) under
reducing conditions. Cell extracts (25 μg/ml) were boiled in
equal volumes of loading buffer (150 mM Tris-HCl, pH 6.8; 4% SDS;
20% glycerol; 15% β-mercaptoethanol; and 0.01% bromophenol blue)
and were electrophoresed on 8–12% polyacrylamide gels. Following
electrophoretic separation, the proteins were transferred onto PVDF
membranes for western blotting. The membranes were blocked with
StartingBlock T20 (PBS) Blocking Buffer (Thermo Scientific,
Waltham, MA, USA) for 1 h. The following primary antibodies were
used: rabbit acetylated histone H3 at the N-terminal tail (06-599;
Millipore, Billerica, MA, USA), trimethyl-histone H3 at lysine
(Lys)9 (17-625; Millipore), trimethyl-histone H3 at Lys27 (17-625;
Millipore), and HRP-conjugated β-actin (Santa Cruz Biotechnology,
Inc., Santa Cruz, CA, USA). The primary antibodies were incubated
overnight at 4°C and the membranes were washed twice in TBST. A
secondary horseradish peroxidase-conjugated antibody (goat
anti-rabbit; Cell Signaling Technology, Danvers, MA, USA) was then
applied at a dilution of 1:5,000 for 1 h. Over a 30-min period, the
blots were washed twice in TBST, after which they were incubated in
enhanced chemiluminescence reagents (SuperSignal Detection kit;
Pierce, Rockford, IL, USA). The band intensity of the original
blots was quantified using GeneTools (Syngene; Synoptics Ltd.,
Cambridge, MA, USA) and was normalized to β-actin expression.
Chromatin immunoprecipitation (ChIP)
Chromatin immunoprecipitation was performed using
the EZ-ChIP kit following the manufacturer’s instructions (17-371;
Millipore). Following stimulation, the THP-1 cells were fixed by
the addition of 37% formaldehyde to a final concentration of 1%.
After 10 min, 10X glycine was added. The cells were washed with
ice-cold phosphate-buffered saline (PBS), collected and centrifuged
for 4 min at 700 × g. The cells were then lysed with SDS lysis
buffer. Chromatin was sheared by sonication (5×10 sec at
approximately 30% of maximum power), centrifuged to pellet debris
and in dilution buffer. Chromatin extracts were pre- cleared for 1
h with a 50% suspension of protein G agarose. Immunoprecipitations
were carried out overnight at 4°C with the following antibodies:
ChIPAb trimethyl-histone H3 at Lys9 (17-625; Millipore), ChIPAb
trimethyl-histone H3 at Lys27 (17-625; Millipore) and acetylated
histone H3 at the N-terminal tail (06-599; Millipore). Immune
complexes were collected with protein G for 1 h and washed 3 times
with high-salt buffer (20 mM Tris at pH 8.0, 0.1% SDS, 1% NP-40, 2
mM EDTA and 0.5 M NaCl) followed by washes in low-salt buffer (50
mM NaCl) and no salt buffer (TE). Immune complexes were extracted
in elution buffer and DNA cross-links were reverted by heating at
65°C for 12 h. Following proteinase K digestion, DNA was extracted
using spin columns following the manufacturer’s instructions. The
following promoter-specific primers were used in the polymerase
chain reactions (PCRs): TNF-α forward, 5′-GATTCTGAGCAAAATA
GCCAGCA-3′ and reverse, 5′-GGCTTCCTTCTTGTTG TGTGT-3′; interleukin-6
(IL-6) forward, 5′-CCTAGTTGT GTCTTGCGATG-3′ and reverse,
5′-GGAGGGGAGATAG AGCTTCT-3′.
Measurement of cytokine production and
HDAC/HAT activity
The medium was collected to determine the levels of
tumor necrosis factor-α (TNF-α) and IL-6 using ELISA kits (R&D
Systems, Minneapolis, MN, USA). HDAC activity was analyzed in the
cell extracts by a colorimetric assay (HDAC activity assay kit
K331-100; BioVision, Mountain View, CA, USA). As negative control,
we added Trichostatin (TSA) to the THP-1 extract at final
concentration of 0.01 mM following the manufacturer’s instructions.
HAT activity was also analyzed in the cell extract using a histone
acetyltransferase activity assay (ab65352). All procedures were
conducted according to the manufacturer’s recommendations.
Statistical analysis
All values are expressed as the means ± standard
error of the mean (SEM) from 5 or 6 repetitions per group for the
biochemistry analysis and 3–4 technical replicates for the western
blot analyses. Statistical analysis was performed using GraphPad
InStat software (GraphPad Software Inc., San Diego, CA, USA).
Comparisons among the experimental groups were carried out by
analysis of variance and Tukey’s post-hoc test. A p-value <0.05
was considered to indicate a statistically significant
difference.
Results
H2S attenuates cytokine
production and modulates HDAC activity
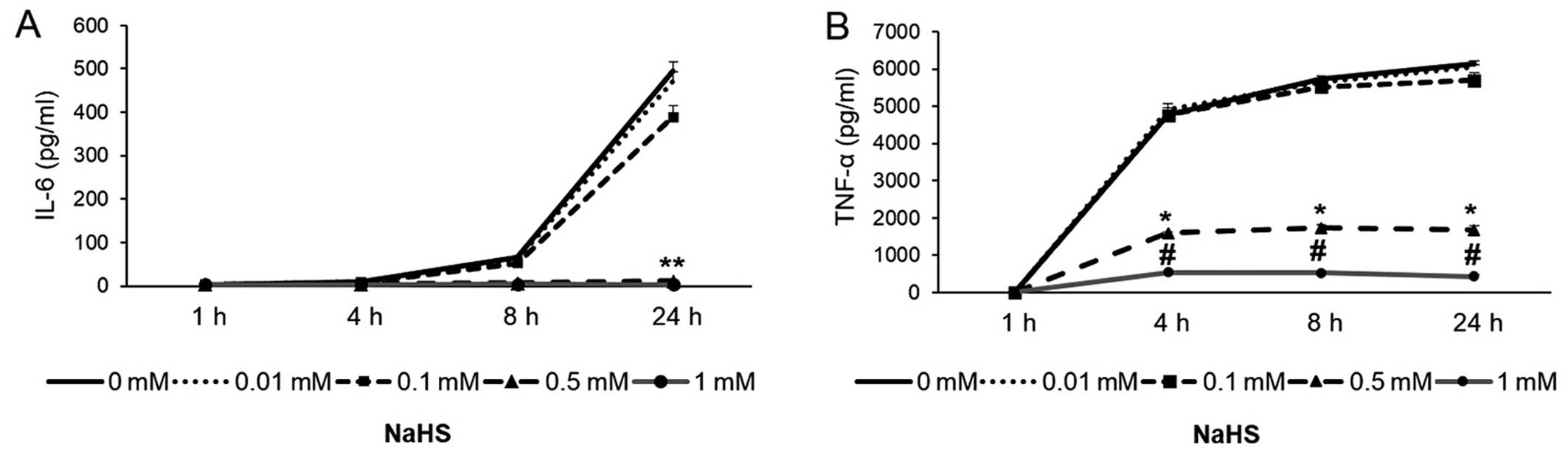
Pre-treatment with NaHS inhibited the LPS-induced
production of IL-6 and TNF-α in a concentration-dependent manner,
as measured by ELISA (Figs. 1 and
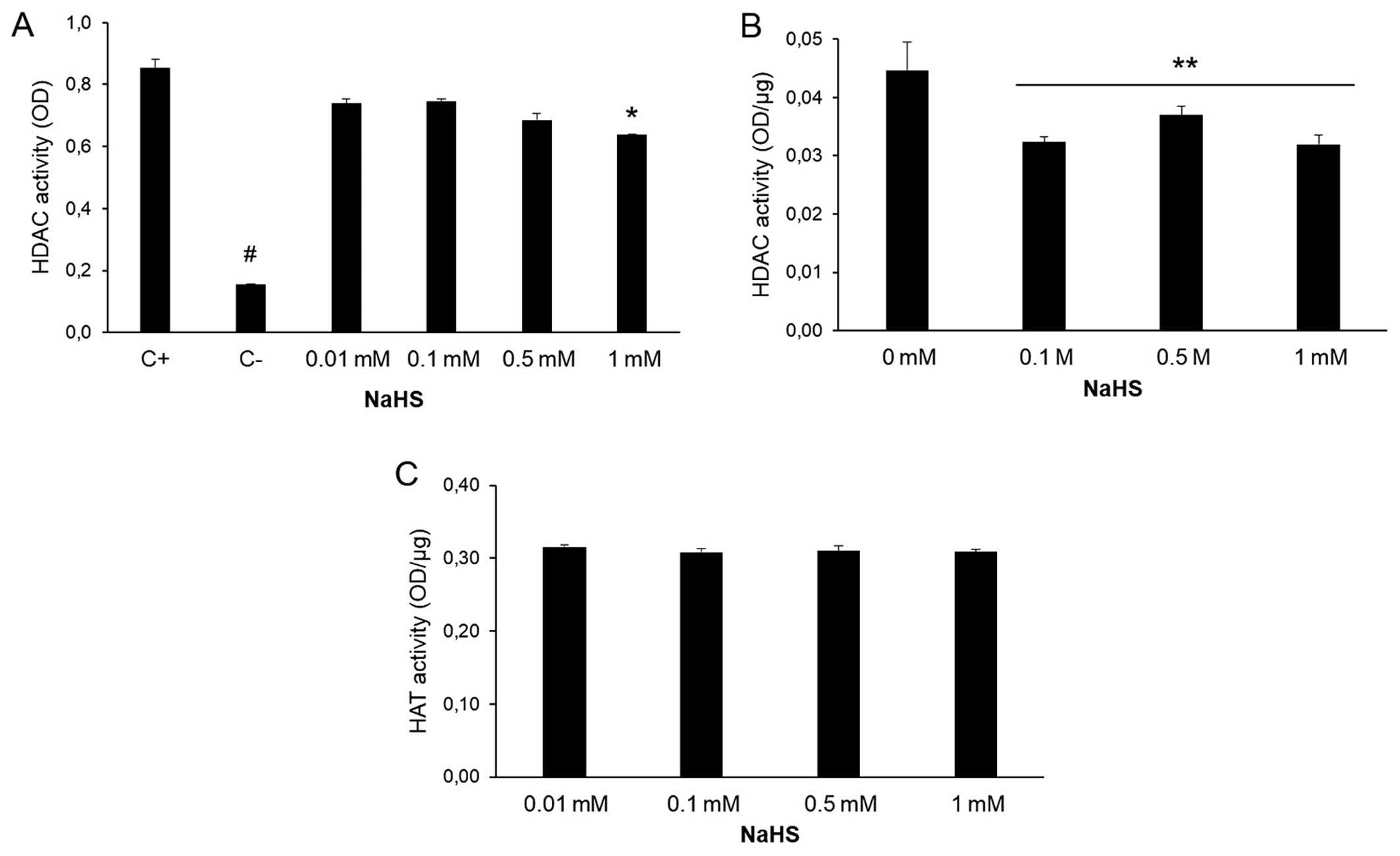
2). The effects of NaHS on HDAC
activity were analyzed in the THP-1 extracts (Fig. 3A) and in the macrophage cultures
(Fig. 3B). HAT activity was
analyzed in the macrophage cultures (Fig. 3C). NaHS reduced the activity of
HDAC in the cell extracts (Fig.
3A). Moreover, the macrophages pre-treated with NaHS for 30 min
exhibited a significant decrease in HDAC activity, as measured at 4
h (Fig. 3B). In contrast to HDAC
activity, HAT activity was not affected by treatment with NaHS
(Fig. 3C). H2S
modulates histone acetylation and methylation, and regulates
histone modifications at the IL-6 and TNF-α promoters. The effects
of NaHS on chromatin were analyzed in the cells pre-treated with
NaHS (0.1, 0.5 and 1 mM for 30 min, followed by stimulation with
LPS (1 μg/ml) for 4 h. Pre-treatment with the H2S
donor increased the acetylation of histone H3 (Fig. 4A) and the methylation at Lys9
(Fig. 4B). The methylation of
histone H3 at Lys27 did not present a statically significant change
(Fig. 4C). The cells were also
analyzed by the chromatin immunoprecipitation method. The chromatin
associated with histone H3 (acetylated, methylated at Lys9 or
Lys27) was precipitated prior to the determination of TNF-α and
IL-6 gene expression. The values shown represent the enrichment of
histone H3 acetylation or methylation compared to the input. The
IL-6 (Fig. 5A) and TNF-α
(Fig. 6A) promoters were
associated with a lower histone H3 acetylation in the
H2S-treated groups. H2S enriched the IL-6
(Fig. 5C) and TNF-α (Fig. 6B) promoters for histone H3
methylated at Lys27 and Lys9, respectively. On the other hand, the
cells that were treated with both NaHS and LPS exhibited an
enrichment in histone H3 methylation at Lys9 in the IL-6 promoter
(Fig. 5B) and at Lys27 in the
TNF-α promoter (Fig. 6C).
Discussion
The results of the present study demonstrate that
H2S modulates the acetylation and methylation of
histones. Based on the known role of these epigenetic alterations
in the regulation of pro-inflammatory mediator production (25–29), we hypothesized that these effects
may contribute to a reduction in the amount of cytokines released
following stimulation with LPS. Moreover, the present findings are
also consistent with the conclusion that H2S, on its
own, induces significant epigenetic alterations.
Given the short half-life of Na2S or NaHS
in aqueous solutions (30), it is
interesting to note that even a temporary stimulus (pre-treatment,
followed by a washout) with the H2S donor NaHS reduces
cytokine production and induces histone modifications. Our results
revealed that the H2S donor increased the total
methylation and acetylation of histone H3. Since HAT activity was
not altered under the same conditions, it can be concluded that the
effect of H2S on histone acetylation is due to the
direct action of H2S on the reduction of HDAC activity,
as demonstrated in cell culture and by a direct biochemical assay.
It can also be concluded that, in turn, H2S reduces the
chromatin openness by decreasing histone acetylation at the IL-6
and TNF-α promoters. On the other hand, H2S (either in
the absence of any pro-inflammatory stimuli or when applied prior
to LPS stimulation) enriches histone H3 methylation at the TNF-α
and IL-6 promoters.
The importance of chromatin remodeling in the
modulation of gene transcription has been investigated in a number
of previous studies (25–29). The locus-specific changes in
histone H3 observed in the present study and the associated
suppression of inflammatory gene transcription may be one of the
mechanisms responsible for the inhibition of cytokine production by
H2S. Histone acetylation is often related to the
chromatin openness, as it weakens the charge attraction between
histones and DNA, leading to the decondensation of chromatin,
thereby facilitating gene transcription. Both cytokine promoters
analyzed in this study exhibited lower acetylation levels at
histone H3 following treatment with NaHS. On the other hand, H3K27
trimethylation brings about transcriptional repression (31), as this epigenetic regulation is
related to the silencing of human polycomb target genes (32). Treatment with the H2S
donor alone or together with LPS increased H3K27 methylation at
both the IL-6 and TNF-α promoters. A similar response was observed
in the methylation of this histone at Lys9, which was enriched at
the promoters analyzed in the groups that received NaHS or HaHS
together with LPS. H3K9 methylation is linked to heterochromatin
and to an endurance of transcriptional repression (33). Furthermore, it has been shown that
H3K9 methylation acts as a regulatory mechanism for inducible
inflammatory genes (33).
It has been demonstrated that, in unstimulated
cells, H3K9 methylation is a mechanism for silencing the
transcription of some genes whose expression rapidly increases
following exposure to stimuli (33). On the other hand, in stimulated
cells, H3K9 methylation may also repress inflammatory gene
transcription (33). We found
that H3K9me was associated with IL-6 but not with TNF-α in
stimulated cells. This difference indicates a mechanism which
allows for the rapid increase in IL-6 expression, as this cytokine
can either function as a pro- or anti-inflammatory cytokin and it
is important to the regulation of other inflammatory mediators
following LPS stimulation.
In conclusion, the present study established a
connection between H2S and epigenetic modulation. Future
research on the mechanisms through which this action is associated
with the various, previously demonstrated effects (12–16) of H2S on gene
transcription, and inflammatory and cell growth signaling is
required. In additition, whether endogenous H2S, which
is similar to exogenous H2S used in the present study,
modulates histones remains to be elucidated. Finally, the results
of the present study remain to be confirmed under in vivo
conditions. While much work remains to be done in this area of
research, on the whole, our findings may prove to be beneficial for
future studies exploring the the effects of H2S on
epigenetic regulation.
Acknowledgments
This study was supported by a US National Institutes
of Health grant (R01GM107846) to C.S.
Abbreviations:
|
BCA
|
bicinchoninic acid
|
|
ChIP
|
chromatin immuno-precipitation
|
|
H2S
|
hydrogen sulfide
|
|
H3K9
|
lysine 9 of histone H3
|
|
HAT
|
histone acetyltransferase
|
|
H3K27
|
lysine 27 of histone H3
|
|
HDAC
|
histone deacetylase
|
|
HMT
|
histone methyltransferase
|
|
IL-6
|
interleukin-6
|
|
LPS
|
lipopolysaccharide
|
|
NaHS
|
sodium hydrosulfide
|
|
PDF
|
polyvinylidene fluoride
|
|
PMA
|
phorbol myristate acetate
|
|
RIPA buffer
|
radioimmunoprecipitation assay
buffer
|
|
SDS
|
sodium dodecyl sulfate
|
|
SEM
|
standard error of the mean
|
|
THP-1
|
Tamm-Horsfall protein 1
|
|
TNF-α
|
tumor necrosis factor-α
|
References
|
1
|
Wang R: The gasotransmitter role of
hydrogen sulfide. Antioxid Redox Signal. 5:493–501. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fiorucci S, Distrutti E, Cirino G and
Wallace JL: The emerging roles of hydrogen sulfide in the
gastrointestinal tract and liver. Gastroenterology. 131:259–271.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Szabo C: Hydrogen sulphide and its
therapeutic potential. Nat Rev Drug Discov. 6:917–935. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Szabo C: Gaseotransmitters: New frontiers
for translational science. Sci Transl Med. 2:59ps542010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Whiteman M, Le Trionnaire S, Chopra M, Fox
B and Whatmore J: Emerging role of hydrogen sulfide in health and
disease: Critical appraisal of biomarkers and pharmacological
tools. Clin Sci (Lond). 121:459–488. 2011. View Article : Google Scholar
|
|
6
|
Wang R: Physiological implications of
hydrogen sulfide: A whiff exploration that blossomed. Physiol Rev.
92:791–896. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Predmore BL, Lefer DJ and Gojon G:
Hydrogen sulfide in biochemistry and medicine. Antioxid Redox
Signal. 17:119–140. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kimura H, Shibuya N and Kimura Y: Hydrogen
sulfide is a signaling molecule and a cytoprotectant. Antioxid
Redox Signal. 17:45–57. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Esechie A, Kiss L, Olah G, et al:
Protective effect of hydrogen sulfide in a murine model of acute
lung injury induced by combined burn and smoke inhalation. Clin Sci
(Lond). 115:91–97. 2008. View Article : Google Scholar
|
|
10
|
Stuhlmeier KM, Broll J and Iliev B:
NF-kappaB independent activation of a series of proinflammatory
genes by hydrogen sulfide. Exp Biol Med (Maywood). 234:1327–1338.
2009. View Article : Google Scholar
|
|
11
|
Osipov RM, Robich MP, Feng J, et al:
Effect of hydrogen sulfide on myocardial protection in the setting
of cardioplegia and cardiopulmonary bypass. Interact Cardiovasc
Thorac Surg. 10:506–512. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Whiteman M, Li L, Rose P, Tan CH,
Parkinson DB and Moore PK: The effect of hydrogen sulfide donors on
lipopoly-saccharide-induced formation of inflammatory mediators in
macrophages. Antioxid Redox Signal. 12:1147–1154. 2010. View Article : Google Scholar :
|
|
13
|
Zhang J, Sio SW, Moochhala S and Bhatia M:
Role of hydrogen sulfide in severe burn injury-induced inflammation
in mice. Mol Med. 16:417–424. 2010.PubMed/NCBI
|
|
14
|
Zuidema MY, Peyton KJ, Fay WP, Durante W
and Korthuis RJ: Antecedent hydrogen sulfide elicits an
anti-inflammatory phenotype in postischemic murine small intestine:
Role of heme oxygenase-1. Am J Physiol Heart Circ Physiol.
301:H888–H894. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Ang SF, Moochhala SM, MacAry PA and Bhatia
M: Hydrogen sulfide and neurogenic inflammation in polymicrobial
sepsis: Involvement of substance P and ERK-NF-κB signaling. PLoS
One. 6:e245352011. View Article : Google Scholar
|
|
16
|
Gao C, Xu DQ, Gao CJ, et al: An exogenous
hydrogen sulphide donor, NaHS, inhibits the nuclear factor κB
inhibitor kinase/nuclear factor κB inhibitor/nuclear factor-κB
signaling pathway and exerts cardioprotective effects in a rat
hemorrhagic shock model. Biol Pharm Bull. 35:1029–1034. 2012.
View Article : Google Scholar
|
|
17
|
Tokuda K, Kida K, Marutani E, et al:
Inhaled hydrogen sulfide prevents endotoxin-induced systemic
inflammation and improves survival by altering sulfide metabolism
in mice. Antioxid Redox Signal. 17:11–21. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wang T, Wang L, Zaidi SR, et al: Hydrogen
sulfide attenuates particulate matter-induced human lung
endothelial barrier disruption via combined reactive oxygen species
scavenging and Akt activation. Am J Respir Cell Mol Biol.
47:491–496. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sen N, Paul BD, Gadalla MM, et al:
Hydrogen sulfide-linked sulfhydration of NF-κB mediates its
antiapoptotic actions. Mol Cell. 45:13–24. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chan MV and Wallace JL: Hydrogen
sulfide-based therapeutics and gastrointestinal diseases:
Translating physiology to treatments. Am J Physiol Gastrointest
Liver Physiol. 305:G467–G473. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aslami H, Beurskens CJ, de Beer FM, et al:
A short course of infusion of a hydrogen sulfide-donor attenuates
endotoxemia induced organ injury via stimulation of
anti-inflammatory pathways, with no additional protection from
prolonged infusion. Cytokine. 61:614–621. 2013. View Article : Google Scholar
|
|
22
|
Rivers JR, Badiei A and Bhatia M: Hydrogen
sulfide as a therapeutic target for inflammation. Expert Opin Ther
Targets. 16:439–449. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Benetti LR, Campos D, Gurgueira SA,
Vercesi AE, Guedes CE, Santos KL, Wallace JL, Teixeira SA,
Florenzano J, Costa SK, Muscará MN and Ferreira HH: Hydrogen
sulfide inhibits oxidative stress in lungs from allergic mice in
vivo. Eur J Pharmacol. 698:463–469. 2013. View Article : Google Scholar
|
|
24
|
Li L, Fox B, Keeble J, et al: The complex
effects of the slow-releasing hydrogen sulfide donor GYY4137 in a
model of acute joint inflammation and in human cartilage cells. J
Cell Mol Med. 17:365–376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hitchler MJ and Domann FE: An epigenetic
perspective on the free radical theory of development. Free Radic
Biol Med. 43:1023–1036. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Fischle W, Wang Y and Allis CD: Histone
and chromatin cross-talk. Curr Opin Cell Biol. 15:172–183. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lachner M and Jenuwein T: The many faces
of histone lysine methylation. Curr Opin Cell Biol. 14:286–298.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Noland BJ, Hardin JM and Shepherd GR:
Histone acetyltransferase activity in synchronized mammalian cells.
Biochim Biophys Acta. 246:263–268. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Fuks F, Hurd PJ, Deplus R and Kouzarides
T: The DNA meth-yltransferases associate with HP1 and the SUV39H1
histone methyltransferase. Nucleic Acids Res. 31:2305–2312. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Papapetropoulos A, Whiteman M and Cirino
G: Pharmacological tools for hydrogen sulphide research: a brief,
introductory guide for beginners. Br J Pharmacol. June 9–2014.Epub
ahead of print. View Article : Google Scholar
|
|
31
|
Schlesinger Y, Straussman R and Keshet I:
Polycomb-mediated methylation on Lys27 of histone H3 pre-marks
genes for de novo methylation in cancer. Nat Genet. 39:232–236.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kirmizis A, Bartley SM, Kuzmichev A, et
al: Silencing of human polycomb target genes is associated with
methylation of histone H3 Lys 27. Genes Dev. 18:1592–1605. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Saccani S and Natoli G: Dynamic changes in
histone H3 Lys 9 methylation occurring at tightly regulated
inducible inflammatory genes. Genes Dev. 16:2219–2224. 2002.
View Article : Google Scholar : PubMed/NCBI
|