Introduction
Sepsis is a systemic inflammatory response syndrome
(SIRS) caused by probable or documented infection, which continues
to pose serious clinical challenges. Sepsis affects individuals of
all ages and is the leading cause of morbidity and mortality for
critically ill patients (1).
Sepsis-induced myocardial dysfunction (SIMD), one of the main
predictors of morbidity and mortality associated with sepsis, is
present in >40% of cases of sepsis (2). SIMD increases the mortality rate of
sepsis by up to 70% (3). A number
of studies on both patients and experimental animals (4–6)
have indicated that mitochondrial dysfunction seems to be related
to the severity and prognosis of sepsis. The heart is rich in
mitochondria, and thus the role of mitochondrial damage in SIMD has
received much attention. Previous studies have indicated that
multiple aspects of mitochondrial dysfunction contribute to SIMD,
such as the overproduction of reactive oxygen species (ROS), the
altered generation of adenosine triphosphate (ATP) and the
disruption of mitochondrial membrane potential (MMP or ΔΨm)
(78)
Uncoupling proteins (UCPs) may constitute a vital
link between ATP and ROS production (9,10);
they are inner mitochondrial membrane proteins that disperse the
mitochondrial proton gradient by translocating H+ across
the inner membrane. Of the 5 UCP homologues, uncoupling protein 2
(UCP2) is ubiquitously expressed; for example, it is expressed in
the liver, brain, pancreas, adipose tissue, immune cells, spleen,
kidneys and heart. In vitro and in vivo, by
modulating MMP, as well as the ATP and ROS levels, the upregulation
of UCP2 plays a neuroprotective role (11–13), while UCP2 knockout mice present
with increased mitochondrial ROS production (9). In addition, UCP2 is involved in the
regulation of other physiological or pathological events, such as
in the formation of atherosclerotic plaque, food intake and
metabolic diseases (14). Given
its central role in regulating mitochondrial ROS production and
cellular energy transduction, we hypothesized that UCP2 may play a
protective role in sepsis. To the best of our knowledge, the
possible role of UCP2 in sepsis has received little attention.
However, some scholars have found that UCP2 deficiency provides
protection in acute liver failure induced by endotoxemic stress
(15) and in the pathogenesis of
experimental leishmaniosis (16).
Nonetheless, whether UCP2 plays a protective role in sepsis needs
to be determined.
In early experiments, we found high levels of UCP2
gene and protein expression in septic myocardial tissue
(unpublished data), which is consistent with the findings of other
studies (17,18). However, the exact role of UCP2 in
myocardial cells under septic conditions remains to be determined.
It remains unclear as to whether UCP2 plays a protective role in
myocardial cells under septic conditions by regulating MMP and the
generation of mitochondrial ROS. We hypothesized that UCP2 may
regulate MMP and mitochondrial function under septic conditions. In
this study, we measured the levels of ROS and ATP production, as
well as the extent of MMP and other relative indicators following
the silencing of UCP2 by RNA interference technology in H9C2
cardiomyocytes under septic conditions.
Materials and methods
Small interfering RNA (siRNA)
transfection
Two siRNAs against UCP2 (siUCP2) and negative
control siRNA (ncRNA) were synthesized by Shanghai GenePharma Co.,
Ltd. (Shanghai, China). The sequences of siRNA1 were as follows:
forward, 5′-GCA CUG UCG AAG CCU ACA A dTdT-3′ and reverse, 5′-UUG
UAG GCU UCG ACA GUG C dTdT-3′. The sequences of siRNA2 were
forward, 5′-CCU CAU GAC AGA CGA CCU C dTdT-3′ and reverse, 5′-GAG
GUC GUC UGU CAU GAG G dTdT-3′. The sequences of ncRNA were forward,
5′-UUC UCC GAA CGU GUC ACG UTT-3′ and reverse, 5′-ACG UGA CAC GUU
CGG AGA ATT-3′. The siRNAs were transfected using Lipofectamine
2000 (Invitrogen Life Technologies, Carlsbad, CA, USA) according to
the manufacturer’s instructions. Briefly, siRNA (final
concentration 80 nmol/l) and Lipofectamine 2000 were firstly
diluted separately in Opti-DMEM medium (Gibco™; Life Technologies,
Grand Island, NY, USA) without antibiotics or serum, and incubated
together for 20 min. The complexes were then added to the H9C2
cells. After 6 h of incubation, the medium was changed as needed.
The silencing efficiency of the 2 siRNAs was tested in experiments
of transient RNA interference; the UCP2 transcript was assayed by
reverse transcription-quantitative PCR (RT-qPCR) 48 h after
infection. siRNA2, which showed the most prominent silencing effect
(69% knockdown efficiency of mRNA) was used for the subsequent
experiments. For further experiments, the H9C2 cells were cultured
following transfection.
Cell culture and treatment
Rat embryonic cardiomyo-blast-derived H9C2 cells
were purchased from the Typical Culture Preservation Commission
Cell Bank, Chinese Academy of Sciences (Shanghai, China). H9C2
cells mimic most of the characteristics of adult cardiomyocytes and
this is an ideal cell line with which to explore the role of UCP2
in the septic myocardium in a cell culture system. The cells were
cultured in DMEM (Gibco-BRL, Beijing, China) supplemented with 10%
fetal calf serum and 5% CO2 at 37°C. The H9C2 cells were
passaged regularly and subcultured to 75% confluence prior to use
in the experiments. In order to simulate sepsis, some cells were
cultured in the presence of 2 μg/ml lipopolysaccharide (LPS,
from Escherichia coli O111:B4; Sigma-Aldrich, St. Louis, MO,
USA) plus 20 μg/ml peptidoglycan G (PepG, from
Micrococcus luteus; Sigma-Aldrich). The experimental design
consisted the following 4 groups: i) the control group, cells were
treated with saline only; ii) the LPS/PepG group, cells treated
with LPS and PepG as described above; iii) the LPS/PepG + siRNA
group, cells transfected with siRNA2 and 24 h later treated with
LPS plus PepG as described above; and iv) the LPS/PepG + ncRNA
group, cells transfected with ncRNA and 24 h later treated with LPS
plus PepG as described above. Further experiments were carried out
24 h following stimulation with LPS plus PepG.
RT-qPCR
To examine the mRNA levels of UCP2, total RNA was
extracted from the H9C2 cells using TRIzol reagent (Invitrogen Life
Technologies) and then reverse transcribed and synthesized into
cDNA using RT-PCR kits (Toyobo Co., Ltd., Osaka, Japan). RT-PCR
amplification reaction was performed in a volume of 10 μl
containing 0.25 μl forward/reverse primers, 5 μl
SYBP-Green PCR Master Mix and 4 μl cDNA. PCR was performed
for 45 cycles of 5 min at 95°C, 15 sec at 95°C, and 30 sec at 60°C.
The threshold cycle (Ct) was obtained from triplicate samples and
averaged. Calculations were based on the ‘ΔΔCt method’ using the
equation R (ratio) = 2−ΔΔCt and standardized by the
housekeeping gene, 18s RNA. The specific primers for UCP2 (Shanghai
GenePharma Co., Ltd.) were: forward, 5′-GGG CAC CTG TGG TGC TAC
CTG-3′ and reverse, 5′-ATG AGC TTT GCC TCC GTC CGC-3′; and those
for 18s RNA were: forward, 5′-CCA TCC AAT CGG TAG TAG C-3′ and
reverse, 5′-GTA ATG GCG GGT CAT AAG-3′.
Western blot analysis
The H9C2 cells were lysed in 2X SDS sample buffer
and the protein concentrations in the super-natants were measured
by BCA Protein assay. An equal amount of protein (30 mg) from each
sample was subjected to 12–15% SDS-PAGE gels and then transferred
onto PVDF filter membranes (Millipore, Billerica, MA, USA). The
membranes were blocked with 5% (w/v) non-fat dried skimmed milk
powder in wash buffer (Tris-buffered saline/1% Tween-20) for 1 h
and subsequently incubated with primary antibodies overnight at a
dilution recommended by the suppliers. The membranes were washed 3
times with wash buffer and then incubated with corresponding
horseradish peroxidase-conjugated secondary antibodies. The protein
signal was developed using ECL substrate (Beyotime Institute of
Biotechnology, Jiangsu, China) according to the manufacturer’s
instructions. The immunoreactive protein bands were visualized
using the In-Vivo Imaging System F (Eastman Kodak Co., Rochester,
NY, USA). The band intensity was quantified using of Gel-Pro
Analyzer 4.0 software.
Measurement of lactate dehydrogenase
(LDH) and creatine kinase (CK) levels
Forty-eight hours after cell treatment, the culture
supernatant was collected for the subsequent measurement of CK and
LDH levels. The release of the cytosolic enzymes, CK and LDH,
indicators of cytotoxicity, reflected a loss of membrane integrity
in the damaged cells and was detected by colorimetric assay. CK and
LDH activity was measured using commercially available kits
(Nanjing Jiancheng Bioengineering Institute, Jiangsu, China),
according to the manufacturer’s instructions. Absorbance was
respectively read at 660 and 440 nm on a multifunctional microplate
reader (SpectraMax M5/M5e; Molecular Devices, Sunnyvale, CA, USA).
The release of CK and LDH was calculated relative to the percentage
of the control group.
Enzyme-linked immunosorbent assay (ELISA)
for the detection of interleukin (IL)-6 and tumor necrosis factor
(TNF)-α
Forty-eight hours after cell treatment, the culture
supernatant was collected for the subsequent measurement of IL-6
and TNF-α expression levels. The culture supernatants were measured
using commercially available ELISA kits (Cusabio Life Science,
Wuhan, China). All procedures were performed strictly as per the
instructions of the manufacturer. The samples were analyzed in
triplicate.
Degree of mitochondrial swelling
To determine the large amplitude swelling of the
H9C2 cells, the isolation of the mitochondria and the cytosol was
performed using the Cell Mitochondria Isolation kit (Beyotime
Institute of Biotechnology). Mitochondrial fractions were separated
by differential centrifugation according to the manufacturer’s
instructions. Briefly, the H9C2 cells were incubated in ice-cold
mitochondrial lyses buffer for 15 min. The cell suspension was then
poured into a glass homogenizer and homogenized for 20 strokes. The
homogenate was subjected to centrifugation at 600 × g for 10 min at
4°C to remove the nuclei and unbroken cells. The supernatant was
then collected and centrifuged again at 11,000 × g for 15 min at
4°C to obtain the mitochondrial fraction. Samples of mitochondria
were dissolved in lysis buffer and subjected to flow cytometry
(FCM). The size [(forward scatter (FSC)] and structure [side
scatter (SSC)] of the mitochondria was determined. The degree of
mitochondrial swelling was quantified as the FSC/SSC ratio, as
previously described (19).
Transmission electron microscopy
(TEM)
The H9C2 cells were col lected and fixed in a
solution containing 3.0% formaldehyde, 1.5% glutaraldehyde in 100
mM cacodylate containing 2.5% sucrose (pH 7.4). The H9C2 cells were
stained with 4% aqueous uranyl acetate, dehydrated, infiltrated and
embedded in epoxyresin. Ultrathin sections (80 nm) were cut and
imaged using a Hitachi transmission electron microscope (H-7500;
Hitachi, Tokyo, Japan).
MMP (or ΔΨm)
ΔΨm was assessed using a laser scanning confocal
microscope (LSCM, FV10i-W; Olympus Corp., Tokyo, Japan) and a flow
cytometer (BD FACSAria; BD Biosciences, Franklin Lakes, NJ, USA)
with
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazole-carbocyanide
iodine (JC-1; Beyotime Institute of Biotechnology) staining. The
H9C2 cells were stained with JC-1 for 20 min at 37°C after 24 h of
incubation with LPS/PepG. Cells on a 35-mm confocal special dish
were scanned using an LSCM, and mitochondrial suspension was
detected by FCM. Fluorescence was read at 488 nm excitation and 530
nm emission for green, and at 540 nm excitation and 590 nm emission
for red. Cells treated with 10 μM carbonyl cyanide
m-chlorophenylhydrazone (CCCP) which can cause the dissipation of
ΔΨm were used as positive controls. The ratio of aggregated JC-1
(red fluorescence) and monomeric JC-1 (green fluorescence)
represented the ΔΨm of H9C2 the cells.
Assay of intracellular ROS
The production of ROS in the H9C2 cells was
fluorometrically monitored using the non-fluorescent probe,
2′,7′-dichlorofluorescein diacetate (DCFH-DA) (Beyotime Institute
of Biotechnology). DCFH-DA passively diffuses into cells and is
deacetylated, changing into the fluorescent compound,
2′,7′-dichlorofluorescein (DCFH). DCFH reacts with ROS to form the
fluorescent product, DCF, which is trapped inside the cells. Cells
in 6-well culture dishes were trypsinized, and collected by
centrifugation. DCFH-DA, diluted to a final concentration of 10
μM with DMEM, was added to the H9C2 cells followed by
incubation at 37°C for 20 min. Following treatment with DCFH-DA,
the H9C2 cells were washed 3 times with PBS. DCF fluorescence was
then read using a multifunctional microplate reader (SpectraMax
M5/M5e; Molecular Devices) at an excitation wavelength of 488 nm
and an emission wavelength of 525 nm. The increase in the value of
the levels of ROS was expressed as a percentage of the control. To
visually observe the changes in ROS production, fluorescence images
were acquired using a fluorescence microscope with 450–490 nm
(excitation) and 520 nm (emission) filters.
Detection of cellular ATP levels
The cellular amount of ATP was measured using a
firefly luciferase-based ATP assay kit (Beyotime Institute of
Biotechnology) according to the manufacturer’s instructions.
Briefly, after 48 h of treatment, the H9C2 cells were treated with
lysis buffer and centrifuged at 12,000 × g for 8 min. In 24-well
plates, 50 μl of each supernatant were mixed with 100
μl ATP detection working dilution. Luminance [in relative
luminance units (RLU)] was measured using a multifunctional
microplate reader (SpectraMax M5/M5e; Molecular Devices). A
standard curve of the ATP concentration was prepared from a known
amount (0.01–10 μM) and the protein concentration in each
group was detected using the Bradford protein assay (Beyotime
Institute of Biotechnology, Jiangsu, China). The ATP levels were
expressed as nmol/mg protein.
Determination of mitochondrial DNA
(mtDNA) copy number by real-time PCR
Total genomic DNA was extracted using a DNA
extraction kit (Qiagen, Hilden, Germany), and the DNA concentration
was measured by optical density. The mtDNA copy number was obtained
according to the manufacturer’s instructions (AceQ™ qPCR
SYBR®-Green Master Mix; Promega Biotech Co., Ltd,
Beijing, China). Briefly, DNA primers were designed to detect Cytb
and 18s RNA at a maximum amplicon length of 150 bp: 18s RNA
forward, 5′-GTAAGTGCGGGTCATAAG-3′ and reverse,
5′-CCATCCAATCGGTAGTAGC-3′; Cytb forward, 5′-GTCGAATGAATTTGAGGGGG-3′
and reverse, 5′-GAGGAGGTGAACGATTGCTAGG-3′. The PCR reaction mixture
contained 2X SYBR-Green PCR Master Mix 10 μl, each primer
0.5 μl, and total genomic DNA 4.0 μl, and
dH2O 5.0 μl. The real-time PCR conditions were 5
min at 95°C and followed by 40 cycles of 15 sec at 95°C and 34 sec
at 60°C. The Ct is the cycle at which the first significant
increase in the fluorescence signal is detected. Relative values
for Cytb and 18s RNA in the samples were used to obtain the ratio
of mtDNA to nuclear DNA (nDNA) in that sample.
Statistical analysis
The experimental values were obtained from 3
independent experiments with a similar pattern and are expressed as
the means ± SD. For the determination of the statistical
differences between the control and treatment groups, we used
ANOVA, followed by post hoc pairwise comparison (LSD) tests for
analysis. Statistical analysis was carried out using the SPSS
software package 20.0. Statistical significance was set at
P<0.05.
Results
Successful transfection of siRNA and
knockdown efficiency of siUCP2
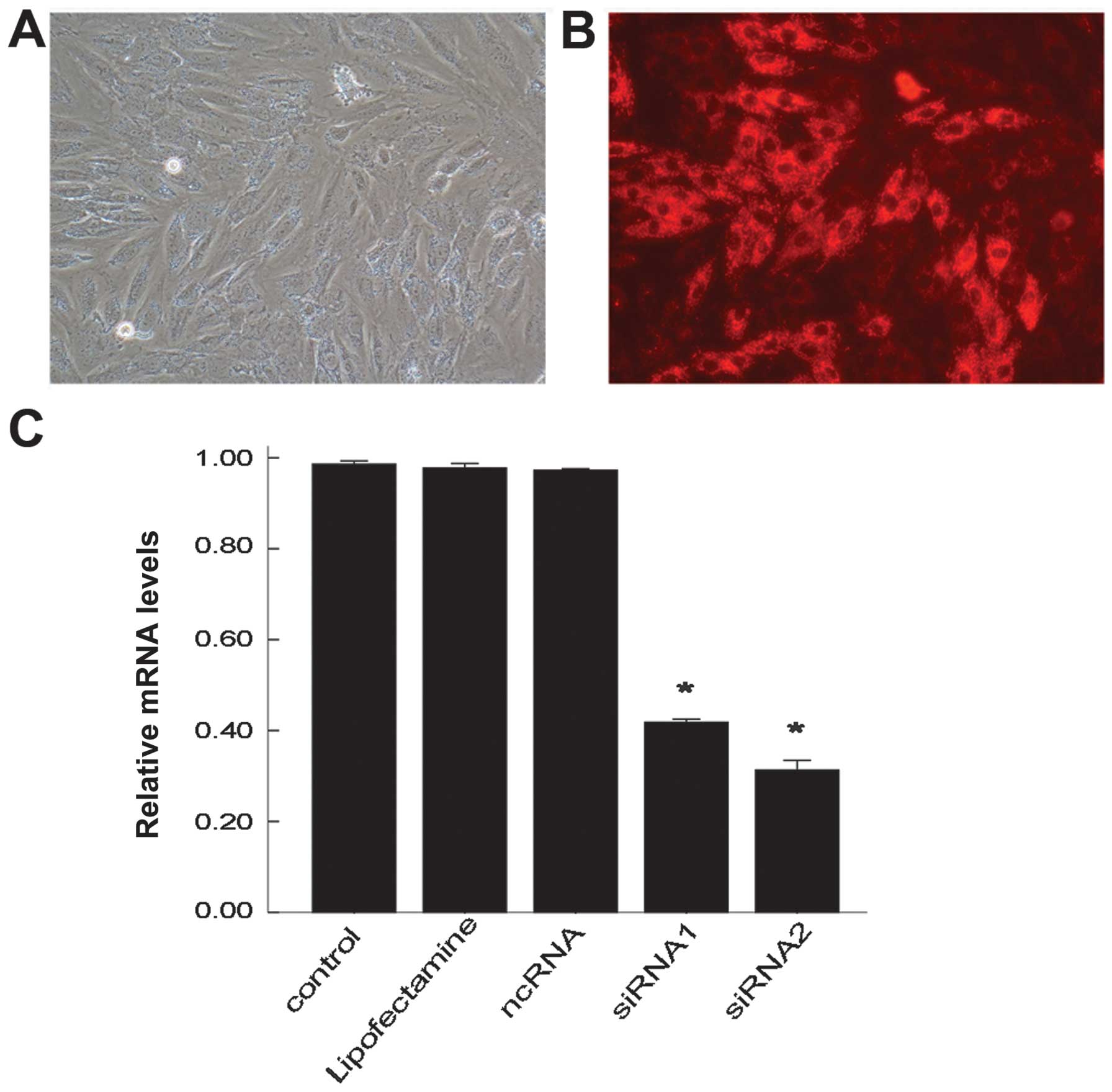
The H9C2 cells were successfully transfected with
siRNA-cy3 using Lipofectamine 2000 and observed under a
fluorescence microscope. The H9C2 cells transfected with siRNA-cy3
all emitted red fluorescence. The transfection efficiency after 24
h was determined through the observation of red fluorescence
(Fig. 1A and B). The transfection
efficiency was 85% which was evaluated in 500 H9C2 cells
randomly.
To examine the function of UCP2 in sepsis, 2 siRNAs
against UCP2 were used to specifically suppress UCP2 mRNA
expression in this study. The H9C2 cells were either left
untransfected (control), transfected with Lipofectamine 2000 (no
siRNA), transfected with ncRNA, or with siRNA1 or 2. After 24 h,
the mRNA levels of UCP2 were determined by RT-qPCR. The mRNA level
of UCP2 was reduced by up to nearly 69% following transfection with
siRNA2 and by 59% following transfection with siRNA1 compared with
the controls (Fig. 1C). There
were no significant changes observed in the UCP2 mRNA level in the
cells transfected with Lipofectamine only or with ncRNA compared
with the control group. siRNA2 was thus selected for use in further
experiments.
Effects of siRNA on the levels of CK,
LDH, IL-6 and TNF-α
The CK and LDH levels are biochemical markers for
the extent of injury to H9C2 cells (20–22). The increased levels of CK and LDH
in the culture supernatant indicate the degree of cell injury.
TNF-α and IL-6 are important pro-inflammatory cytokines in sepsis.
As shown in Fig. 2, following
treatment with LPS/PepG, the levels of CK, LDH, TNF-α and IL-6 in
the cell supernatant notably increased compared with those in the
control group (no treatment; all P<0.05). In other words,
compared with the control group, treatment with LPS/PepG induced a
2-fold increase in CK levels, a 1.5-fold increase in LDH levels, a
1.6-fold increase in IL-6 levels and a 1.5-fold increase in TNF-α
levels. The significant increase in CK, LDH, TNF-α and IL-6 levels
in the culture supernatant indicated that the H9C2 cell model of
sepsis had been successfully constructed. Subsequently, we further
explored the effects of siRNA on H9C2 cells under septic
conditions. Supernatants from the cells treated with LPS/PepG +
siRNA had markedly higher CK, LDH and TNF-α concentrations (all
P<0.05; Fig. 2A, B and D)
compared with the cells treated with LPS/PepG; however, no
statistically significant differences were observed in the IL-6
concentration (P>0.05; Fig.
2C) when compared with the cells treated with LPS/PepG.
Compared with the LPS/PepG group, ncRNA had no effect on the CK,
LDH, TNF-α and IL-6 levels in the culture supernatant (all
P>0.05; Fig. 2).
UCP2 mRNA and protein levels in H9C2
cells under septic conditions
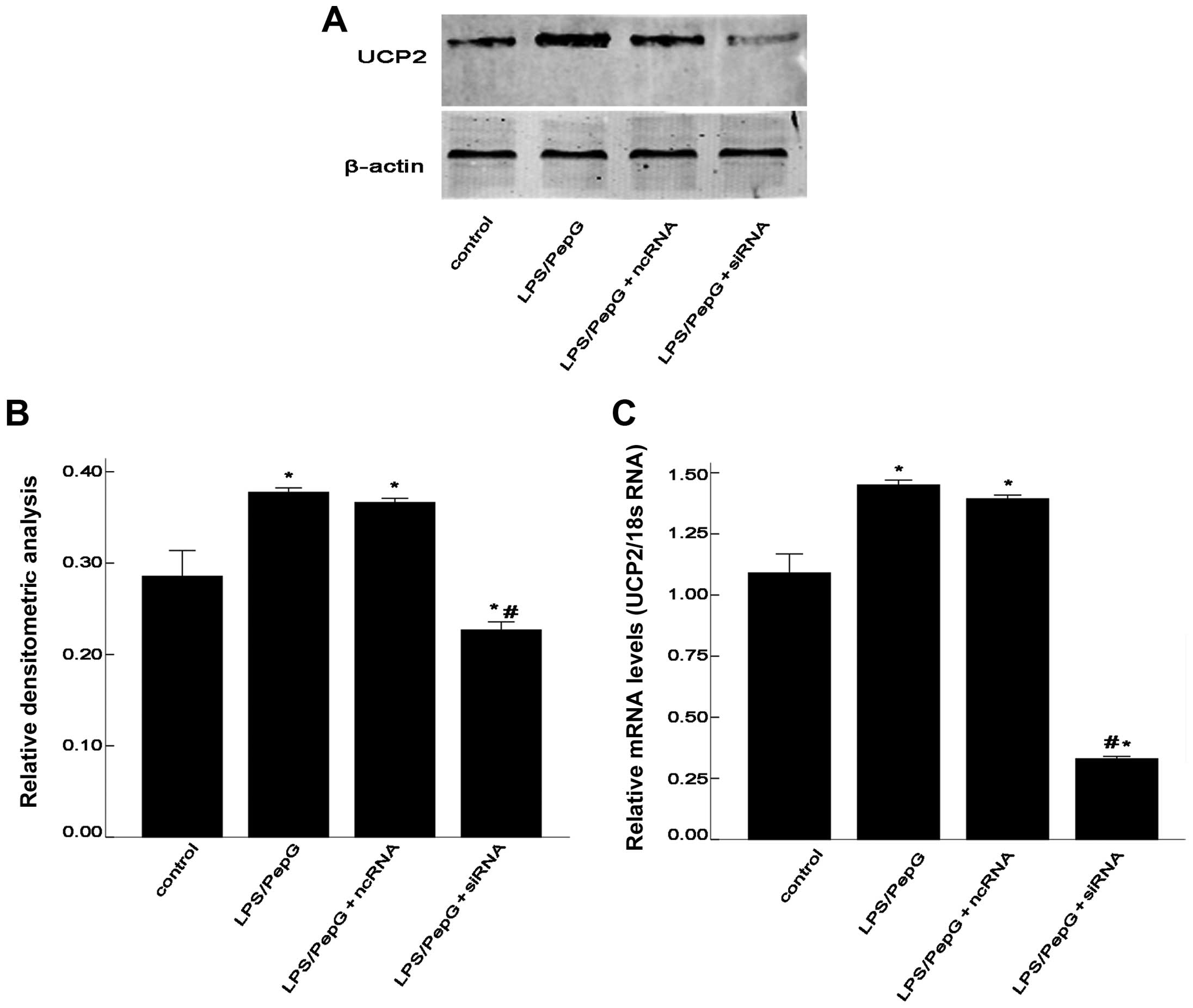
To determine whether sepsis increases the UCP2 mRNA
and protein levels, the H9C2 cells were treated with LPS plus PepG
or saline. After 48 h, total RNA was extracted for RT-qPCR and
total protein was extracted for western blot analysis. As shown in
Fig. 3, the mRNA and protein
expression of UCP2 was enhanced by treatment with LPS/PepG.
Compared with the controls, sepsis caused a 1.4-fold increase in
UCP2 mRNA levels and a 1.3-fold increase in UCP2 protein levels
following treatment with LPS/PepG or saline (P<0.05; Fig. 3). To determine the role of UCP2 in
cardiomyocytes under septic conditions, the septic H9C2 cells were
transfected with siRNA to suppress UCP2 expression. Compared with
the LPS/PepG group, siRNA induced a 0.22-fold decrease in UCP2
protein expression and a 0.59-fold decrease in UCP2 mRNA expression
(P<0.05; Fig. 3). As shown in
Fig. 3, ncRNA had no effect on
UCP2 mRNA or protein expression in the H9C2 cells under septic
conditions (P>0.05).
MMP (or ΔΨm) in H9C2 cells under septic
conditions
JC-1 aggregates in the mitochondria emit red
fluorescence (Fig. 4A and B,
panel a). For a more objective understanding of MMP, confocal
microscopy and FCM were both used to detect ΔΨm. Treatment of the
H9C2 cells with LPS/PepG for 24 h re sulted in the dissipation of
MMP, which was shown as increased green fluorescence (Fig. 4A and B, panel b) by JC-1 staining.
To further elucidate the role of UCP2 in cardiomyocytes under
septic conditions, the H9C2 cells pre-treated with LPS/PepG were
transfected with ncRNA or siRNA. The red fluorescence intensity in
the LPS/PepG group (Fig. 4A and
B, panel b) was weaker than that in the LPS/PepG + siRNA group
(Fig. 4A and B, panel d), but was
similar to that in the LPS/PepG + ncRNA group (Fig. 4A and B, panel c). CCCP renders the
mitochondrial innermembrane permeable to protons and causes the
dissipation of the proton gradient across the inner mitochondrial
membrane (Fig. 4A and B, panel
e). To quantify the changes in MMP, the ratio of red fluorescence
intensity to the green fluorescence intensity as shown by FCM was
determined (Fig. 4C). In the
control group, the ratio was 0.429±0.071, while the
LPS/PepG-treated cells showed a lower ratio (0.222±0.038,
P<0.05, vs. the control group, n=3) indicating the dissipation
of ΔΨm in the H9C2 cells under septic conditions. The H9C2 cells
transfected with LPS/PepG + siRNA demonstrated a rebound in the ΔΨm
(0.563±0.121, P<0.05, vs. LPS/PepG group, n=3), while treatment
with LPS/PepG + ncRNA had no effect on ΔΨm (0.219±0.197, P>0.05,
vs. LPS/PepG group, n=3).
 | Figure 4Effect of siRNA on mitochondrial
membrane potential (MMP or ΔΨm) in H9C2 cells under septic
conditions. (A) Scanning of MMP in H9C2 cells using a confocal
microscope. Green fluorescence represents the monomeric form of the
JC-1 molecule, which appears in the cytosol after mitochondrial
membrane depolarization. Red fluorescence reveals the mitochondrial
aggregate form of JC-1, indicating MMP (magnification, x40). (B)
Evaluation of MMP in H9C2 cells by flow cytometry (FCM). FITC-H,
green; PI-H, red. (A and B) Panel a, control group; panel b,
lipopolysaccharide (LPS)/peptidoglycan G (PepG) group; panel c,
LPS/PepG + negative control siRNA (ncRNA) group; panel d, LPS/PepG
+ siRNA group; panel e, carbonyl cyanide m-chlorophenylhydrazone
(CCCP)-treated cells. (C) The ratio of the red over the green
fluorescence intensity by FCM represents the quantitative MMP in
each group. Values are expressed as the means ± SD.
*P<0.05 vs. control. #P<0.05 vs.
LPS/PepG group. n=3 per group. |
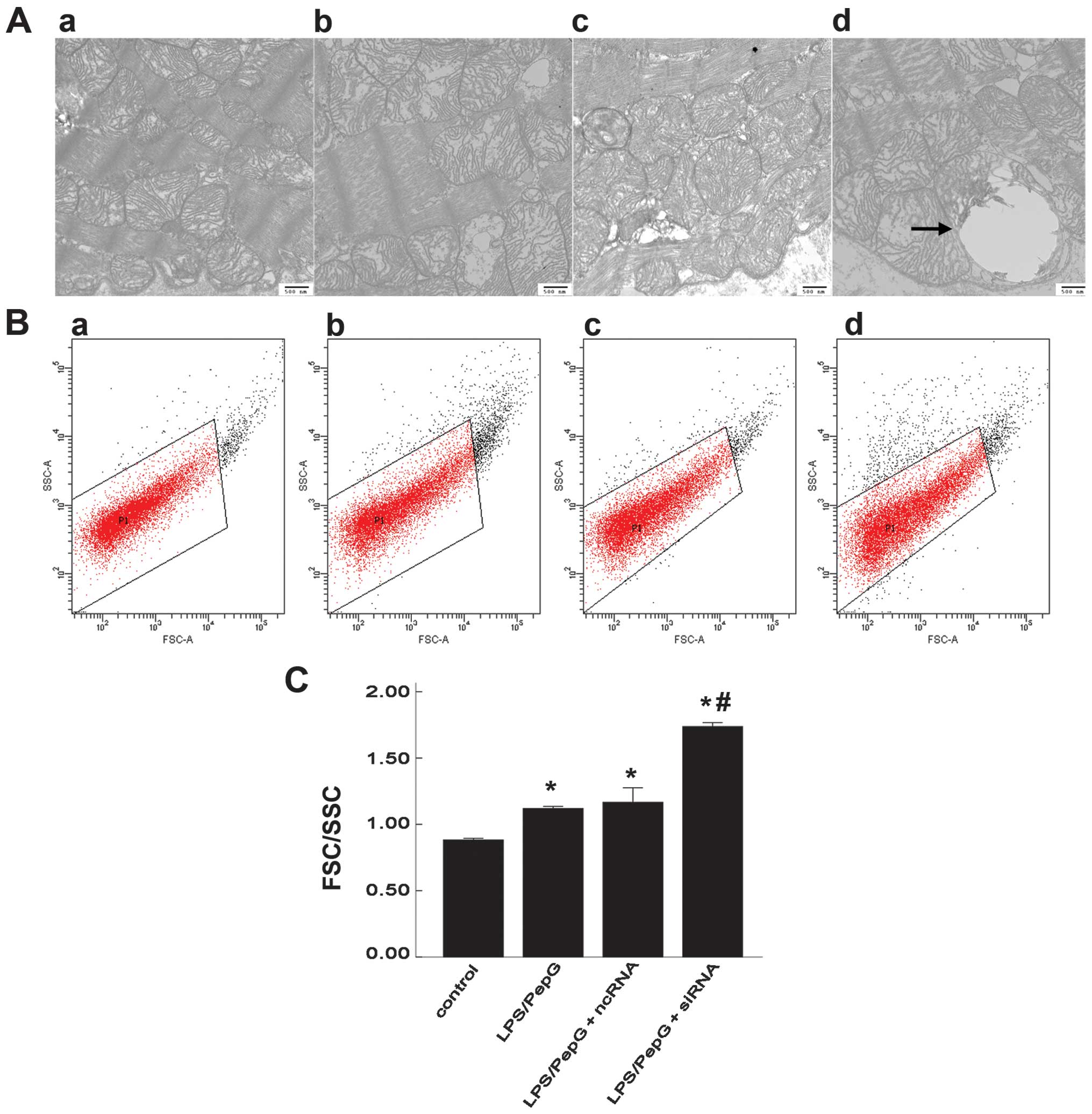
Mitochondrial morphology and swelling in
H9C2 cells under septic conditions
In an aim to understand the mitochondrial structure
more objectively, the mitochondrial ultrastructure was scanned by
TEM and the quantitative degree of mitochondrial swelling was
detected by FCM. Fifteen electron micrographs were prepared for
each specimen and in each micrograph, 15 mitochondria were randomly
selected. The ultrastructure of the mitochondria was normal in the
control group, while mitochondrial swelling and vacuolization, loss
of matrix and the disruption of crests were detected in the
LPS/PepG-treated cells (with or without siRNA and ncRNA
transfection). Compared with the LPS/PepG group, the mitochondrial
ultrastructure was damaged more severely in the LPS/PepG + siRNA
group in which megamitochondria were also frequently observed
(Fig. 5A). Using FCM with
appropriate settings, the FSC-SSC parameters were determined in the
mitochondria. FSC highly correlates with cell size or volume and
SSC is related to granularity and the refractive index (Fig. 5B). The quantitive degree of
mitochondrial swelling was represented by the FSC/SSC ratio. The
FSC/SSC ratio was 1.119±0.016 in the LPS/PepG group and 1.737±0.029
in the LPS/PepG + siRNA group. Both were markedly higher than that
of the control group (0.882±0.012, P<0.05, n=3; Fig. 5C). Compared with the LPS/PepG
group, treatment with LPS/PepG + siRNA induced a 1.6-fold increase
in the FSC/SSC ratio (P<0.05, n=3), while LPS/PepG + ncRNA had
no effect on this ratio (1.160±0.110, P>0.05, vs. LPS/PepG
group, n=3; Fig. 5C).
Effect of siRNA on intracellular ROS and
ATP levels, and mtDNA copy numbers
To elucidate the role of UCP2 in mitochondrial
function, intracellular ROS and cellular ATP levels and mtDNA copy
numbers were detected. DCFH-DA is a fluorescent probe of ROS.
Intracellular ROS production was observed using a fluorescence
microscope and quantified using a multifunctional microplate reader
with DCFH-DA. The DCF fluorescence intensity was significantly
higher in the LPS/PepG, LPS/PepG + ncRNA and LPS/PepG + siRNA
groups than that in the control group (2.50±0.10, 2.49±0.08,
3.28±0.17, respectively vs. 1.00±0.12, F=183.8, P<0.05, n=3;
Fig. 6A and B), indicating an
enhanced production of ROS stimulated by LPS/PepG. The DCF
fluorescence intensity in the LPS/PepG + siRNA group was notably
increased compared with that in the LPS/PepG group (3.28±0.17 vs.
1.00±0.12, P<0.05, n=3; Fig.
6B). To further determine the effects of siRNA on mitochondrial
function, indicators of mitochondrial activity, such as the levels
of cellular ATP and mtDNA copy numbers were determined. After 24 h
of exposure to LPS/PepG, the cellular ATP content was significantly
decreased in the LPS/PepG, LPS/PepG + ncRNA and LPS/PepG + siRNA
groups compared with the control group (2.643±0.076, 2.647±0.065,
2.560±0.092, respectively, vs. 6.220±0.105, F=1322.6, P<0.05,
n=3; Fig. 6C). However, compared
with the LPS/PepG group, ncRNA or siRNA had no effect on the ATP
levels in the H9C2 cells under septic conditions (P>0.05;
Fig. 6C). Cellular mtDNA is also
a sensitive indicator of mitochondrial function. The mtDNA copy
numbers in the LPS/PepG group were significantly lower than those
of the control group (0.233±0.153 vs. 1.000±0.040, P<0.05, n=3;
Fig. 6D). The mtDNA copy numbers
in the LPS/PepG + siRNA group were significantly decreased compared
with those in the LPS/PepG group (0.147±0.152 vs. 0.233±0.153,
P<0.05, n=3; Fig. 6D).
However, the mtDNA copy numbers in the LPS/PepG + ncRNA group did
not differ from those in the LPS/PepG group (0.233±0.201 vs.
0.233±0.153, P>0.05, n=3; Fig.
6D).
Discussion
The principal findings of the current study are as
follows: i) treatment with LPS/PepG severely damaged the H9C2 cells
and initiated an inflammatory response, indicating that the cell
model of sepsis was successfully created; ii) siRNA down-regulated
the expression of UCP2 at the transcriptional and translational
level, indicating that UCP2 deficiency in the cells was
successfully established; and iii) during sepsis, siRNA aggravated
the injury to mitochondrial structure and eventually led to the
dysfunction of the mitochondrion, demonstrating that UCP2 may play
a protective role in the mitochondria in H9C2 cells under septic
conditions. In brief, these findings support the hypothesis that
UCP2 plays an important and protective role in cardiomyocytes under
septic conditions.
This study demonstrates that the cell wall
component, PepG, derived from the pathogenic Gram-positive
bacterium, Micrococcus luteus, synergizes with LPS to damage
H9C2 cells and causes the release of IL-6 and TNF-α in
vitro. Sepsis is not only an unresolved problem in the medical
field, but also a very serious threat to human health. Previous
studies have created a number of models of sepsis in vivo,
but few cellular models of sepsis have been created (23–25). LPS or LPS plus PepG are often used
to induce sepsis in various cells which mimics models of sepsis
in vitro. The significant increase in the levels of CK, LDH,
TNF-α and IL-6 in our study indicated that the H9C2 cell model of
sepsis had been created successfully. UCP2 is a member of the UCP
family and is highly expressed in a number of tissues. Our previous
study demonstrated that UCP2 expression was upregulated in the
septic myocardium (unpublished data). To further determine whether
UCP2 plays a protective role in the myocardium under septic
conditions, we used siRNA to reduce the mRNA level of UCP2 by up to
69% in the current study. Our findings indicated that the silencing
of UCP2 resulted in damage to the H9C2 cells, an increased release
of TNF-α, the disruption of mitochondrial morphology, the rebound
of MMP, enhanced ROS generation, as well as a reduction in cellular
ATP levels and mtDNA copy numbers in the H9C2 cells under septic
conditions.
MMP is dependent on the speed of proton pumping
generated from the transport of electrons and protons. MMP reflects
the performance of the electron transport chain (ETC) and this is
often used as an indicator of the pathological disorder of the
mitochondrion. As previously demonstrated, cells treated with LPS
show a disruption of the MMP, which is a critical event in lethal
cell damage (26,27). In this study, treatment with
LPS/PepG induced the disruption of the MMP in the H9C2 cells and
the silencing of UCP2 resulted in a marked rebound of MMP under
septic conditions, which was consistent with the results of our
previous study using a model of sepsis (unpublished data) and other
models in vitro (27,28). The meaning of uncoupling is the
collapse of the MMP by proton leakage through the mitochondrial
membrane. UCPs, including UCP2 can disperse mitochondrial proton
gradient to stabilize the inner MMP, so they are closely related to
the MMP. For example, under MPP+ toxicity, UCP4
overexpression preserves ATP levels and MMP (29); the overexpression of UCP5 in
neuronal cells can preserve the MMP, ATP levels and cell survival
(30). Previous studies have
demonstrated that MMP and ROS interact with each other (11,12,28).
Mitochondrial sources of ROS are considered as a
basic cause of oxidant damage to the heart during sepsis, and they
may underlie the pivotal mechanisms related to cardioprotection in
the myocardium (31). Previous
studies (32–34) have demonstrated that a slight
degree of depolarization within the inner membrane of the
mitochondria may play a protective role by attenuating ROS
production. In the present study, the silencing of UCP2 by siRNA
was associated with a relative increase in ROS production and a
significant rebound in MMP during sepsis, which is consistent with
uncoupling mechanisms (35). In
UCP2 knockout mice, increased MMP production has been shown to
cause vascular remodeling partially through increased ROS
production (28). In vivo,
previous neuronal studies have found that the overexpression of
human UCP2 is associated with increased uncoupling and decreased
ROS production, indicating that UCP2 plays a protective role
(11,12,36). Our present finding that the
silencing of UCP2 leads an increase in MMP and ROS production
during sepsis suggests that UCP2 plays a protective effect in
septic cardiomyocytes.
Compared with nDNA, mtDNA is more susceptible to
oxidative damage, as the DNA repair capacity in the mitochondria is
incomplete and mitochondria are an important source of ROS
(37). ROS can oxidatively impair
mitochondrial mtDNA (38,39) and trigger mtDNA deletions which
can damage the synthesis of the mtDNA-encoded proteins of
respiratory chain complexes I–V (40). In the current study, treatment
with LPS/PepG resulted in damage to mtDNA and the overproduction of
ROS which was not reversed by siUCP2, indicating that UCP2 may be a
protective factor in cardiomyocytes under septic contitions. In
vivo, LPS had been shown to cause mtDNA damage (41) which can activate the immune system
and may contribute to SIRS and compromise organ function in a
number of diseases (42–44). Research on the association between
UCP2 and mtDNA in sepsis is limited, and the possible explanation
of our finding is that the silencing of UCP2 through the alteration
in ROS production, uncoupling activity and MMP, eventually leads to
the deletion of mtDNA under septic conditions.
Apart from ROS and mtDNA, cellular ATP is another
indicator of mitochondrial function. The mitochondria are the major
source of ATP, which is the only universal energy-yielding currency
in cells. ATP synthesis relies on the coupling of electron
transport through the ETC to the proton motive force (45)
The coupling procedure is regulated by proton
leakage through the mitochondrial inner membrane which is partly
mediated by UCP2. However, the precise mechanisms through which
UCP2 modulates this process continue to be a matter of debate. It
has been demonstrated that UCP2 activity may lead to decreased ATP
availability (46–49), although others believe that the
overexpression of UCP2 results in elevated levels of tissue ATP
(50–52). The association between UCP2 and
ATP ramains debatable. In this study, sepsis damaged the H9C2 cells
and led to low levels of ATP, while the silencing of UCP2 had no
significant effect on the ATP levels (P>0.05). The alternative
explanation may be that LPS/PepG is far more important than UCP2 in
affecting the ATP level or UCP2 primarily has no effect on ATP in
H9C2 cells under septic conditions.
To obtain more credible results, TEM was used for
qualitative analysis and FCM was used for quantitative analysis.
Our findings suggest that, under septic conditions, mitochondrial
dysfunction increases oxidative stress, induces the collapse of the
MMP and the deletion of mtDNA, which can, in turn, lead to
mitochondrial swelling and damage to mitochondrial morphology. The
silencing of UCP2 aggravated the degree of mitochondrial swelling
and damage to mitochondrial morphology, indicating that UCP2 may be
a protective factor in cells under septic conditions. There may be
other explanations for the changes in mitochodrial swelling and
morphology observed in this study. For example, the change in the
Ca2+ concentration (53), intracellular ion levels (54), the status of mitochondrial pore
(55,56), which regrettably were not detected
in the current study; these may also affect the mitochondrial
morphology and function. It is a pity that we only used siRNA for
the study of UCP2 function in cardiomyocytes under septic
conditions. To further clarify the association between UCP2 and
mitochondrial function and to confirm the hypothesis that UCP2 may
play a protective role during sepsis, the effects of the
overexpression of UCP2 in H9C2 cells should be examined.
In brief, our study demonstrates that treatment with
LPS/PepGn induces mitochondrial dysfunction in H9C2 cells and that
the silencing of UCP2 by siRNA aggravates the damage to
mitochondrial morphology and function, indicating that UCP2 may
play a protective role in cardiomyocytes under septic
conditions.
Acknowledgments
The current study was supported by the National
Natural Science Foundation of China (grant no. 81272070).
Abbreviations:
|
SIRS
|
systemic inflammatory response
syndrome
|
|
SIMD
|
sepsis-induced myocardial
dysfunction
|
|
ROS
|
reactive oxygen species
|
|
UCPs
|
uncoupling proteins
|
|
UCP2
|
uncoupling protein 2
|
|
siRNA
|
small interfering RNA
|
|
LPS
|
lipopolysaccharide
|
|
PepG
|
peptidoglycan G
|
|
ncRNA
|
negative control siRNA
|
|
LDH
|
lactate dehydrogenase
|
|
CK
|
creatine kinase
|
|
FCM
|
flow cytometry
|
|
TEM
|
transmission electron microscopy
|
|
MMP or ΔΨm
|
mitochondrial membrane potential
|
|
DCFH-DA
|
2′,7′-dichlorofluorescein
diacetate
|
|
DCFH
|
2′,7′-dichlorofluorescein
|
|
mtDNA
|
mitochondrial DNA
|
|
nDNA
|
nuclear DNA
|
|
ETC
|
electron transport chain
|
|
JC-1
|
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazole-carbocyanide
iodine
|
References
|
1
|
Weycker D, Akhras KS, Edelsberg J, Angus
DC and Oster G: Long-term mortality and medical care charges in
patients with severe sepsis. Crit Care Med. 31:2316–2323. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Fernandes CJ Jr, Akamine N and Knobel E:
Cardiac troponin: A new serum marker of myocardial injury in
sepsis. Intensive Care Med. 25:1165–1168. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Blanco J, Muriel-Bombín A, Sagredo V,
Taboada F, Gandía F, Tamayo L, Collado J, García-Labattut A,
Carriedo D, Valledor M, et al Grupo de Estudios y Análisis en
Cuidados Intensivos (G.R.E.C.I.A.): Incidence, organ dysfunction
and mortality in severe sepsis: A Spanish multicentre study. Crit
Care. 12:R1582008. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Soriano FG, Nogueira AC, Caldini EG, Lins
MH, Teixeira AC, Cappi SB, Lotufo PA, Bernik MM, Zsengellér Z, Chen
M and Szabó C: Potential role of poly(adenosine
5′-diphosphate-ribose) polymerase activation in the pathogenesis of
myocardial contractile dysfunction associated with human septic
shock. Crit Care Med. 34:1073–1079. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Romero-Bermejo FJ, Ruiz-Bailen M,
Gil-Cebrian J and Huertos-Ranchal MJ: Sepsis-induced
cardiomyopathy. Curr Cardiol Rev. 7:163–183. 2011. View Article : Google Scholar
|
|
6
|
Parrillo JE, Parker MM, Natanson C,
Suffredini AF, Danner RL, Cunnion RE and Ognibene FP: Septic shock
in humans. Advances in the understanding of pathogenesis,
cardiovascular dysfunction, and therapy. Ann Intern Med.
113:227–242. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Levy RJ: Mitochondrial dysfunction,
bioenergetic impairment, and metabolic down-regulation in sepsis.
Shock. 28:24–28. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Watts JA, Kline JA, Thornton LR, Grattan
RM and Brar SS: Metabolic dysfunction and depletion of mitochondria
in hearts of septic rats. J Mol Cell Cardiol. 36:141–150. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Arsenijevic D, Onuma H, Pecqueur C,
Raimbault S, Manning BS, Miroux B, Couplan E, Alves-Guerra MC,
Goubern M, Surwit R, et al: Disruption of the uncoupling protein-2
gene in mice reveals a role in immunity and reactive oxygen species
production. Nat Genet. 26:435–439. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Krauss S, Zhang CY and Lowell BB: A
significant portion of mitochondrial proton leak in intact
thymocytes depends on expression of UCP2. Proc Natl Acad Sci USA.
99:118–122. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Diano S, Matthews RT, Patrylo P, Yang L,
Beal MF, Barnstable CJ and Horvath TL: Uncoupling protein 2
prevents neuronal death including that occurring during seizures: A
mechanism for preconditioning. Endocrinology. 144:5014–5021. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Mattiasson G, Shamloo M, Gido G, Mathi K,
Tomasevic G, Yi S, Warden CH, Castilho RF, Melcher T,
Gonzalez-Zulueta M, et al: Uncoupling protein-2 prevents neuronal
death and diminishes brain dysfunction after stroke and brain
trauma. Nat Med. 9:1062–1068. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
13
|
Conti B, Sugama S, Lucero J,
Winsky-Sommerer R, Wirz SA, Maher P, Andrews Z, Barr AM, Morale MC,
Paneda C, et al: Uncoupling protein 2 protects dopaminergic neurons
from acute 1,2,3,6-methyl-phenyl-tetrahydropyridine toxicity. J
Neurochem. 93:493–501. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Donadelli M, Dando I, Fiorini C and
Palmieri M: UCP2, a mitochondrial protein regulated at multiple
levels. Cell Mol Life Sci. 71:1171–1190. 2014. View Article : Google Scholar
|
|
15
|
Le Minh K, Kuhla A, Abshagen K, Minor T,
Stegemann J, Ibrahim S, Eipel C and Vollmar B: Uncoupling protein-2
deficiency provides protection in a murine model of endotoxemic
acute liver failure. Crit Care Med. 37:215–222. 2009. View Article : Google Scholar
|
|
16
|
Carrión J, Abengozar MA, Fernández-Reyes
M, Sánchez-Martín C, Rial E, Domínguez-Bernal G and
González-Barroso MM: UCP2 deficiency helps to restrict the
pathogenesis of experimental cutaneous and visceral leishmaniosis
in mice. PLoS Negl Trop Dis. 7:e20772013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Faggioni R, Shigenaga J, Moser A, Feingold
KR and Grunfeld C: Induction of UCP2 gene expression by LPS: A
potential mechanism for increased thermogenesis during infection.
Biochem Biophys Res Commun. 244:75–78. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Roshon MJ, Kline JA, Thornton LR and Watts
JA: Cardiac UCP2 expression and myocardial oxidative metabolism
during acute septic shock in the rat. Shock. 19:570–576. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lecoeur H, Langonné A, Baux L, Rebouillat
D, Rustin P, Prévost MC, Brenner C, Edelman L and Jacotot E:
Real-time flow cytometry analysis of permeability transition in
isolated mitochondria. Exp Cell Res. 294:106–117. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Han XZ, Gao S, Cheng YN, Sun YZ, Liu W, et
al: Protective effect of naringenin-7-O-glucoside against oxidative
stress induced by doxorubicin in H9c2 cardiomyocytes. Biosci
Trends. 6:19–25. 2012.PubMed/NCBI
|
|
21
|
Zhu L, Wei T, Chang X, He H, Gao J, et al:
Effects of salidroside on myocardial injury in vivo in vitro via
regulation of Nox/NF-kappaB/AP1 pathway. Inflammation. Feb
15–2015.Epub ahead of print. View Article : Google Scholar
|
|
22
|
Wang SG, Xu Y, Xie H, Wang W and Chen XH:
Astragaloside IV prevents lipopolysaccharide-induced injury in H9C2
cardiomyocytes. Chin J Nat Med. 13:127–132. PubMed/NCBI
|
|
23
|
Wray GM, Foster SJ, Hinds CJ and
Thiemermann C: A cell wall component from pathogenic and
non-pathogenic gram-positive bacteria (peptidoglycan) synergises
with endotoxin to cause the release of tumour necrosis
factor-alpha, nitric oxide production, shock, and multiple organ
injury/dysfunction in the rat. Shock. 15:135–142. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lowes DA, Thottakam BM, Webster NR, Murphy
MP and Galley HF: The mitochondria-targeted antioxidant MitoQ
protects against organ damage in a
lipopolysaccharide-peptido-glycan model of sepsis. Free Radic Biol
Med. 45:1559–1565. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Lowes DA, Almawash AM, Webster NR, Reid VL
and Galley HF: Melatonin and structurally similar compounds have
differing effects on inflammation and mitochondrial function in
endothelial cells under conditions mimicking sepsis. Br J Anaesth.
107:193–201. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Cregan SP, Dawson VL and Slack RS: Role of
AIF in caspase-dependent and caspase-independent cell death.
Oncogene. 23:2785–2796. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kim YC, Song SB, Lee MH, Kang KI, Lee H,
Paik SG, Kim KE and Kim YS: Simvastatin induces caspase-independent
apoptosis in LPS-activated RAW264.7 macrophage cells. Biochem
Biophys Res Commun. 339:1007–1014. 2006. View Article : Google Scholar
|
|
28
|
Pak O, Sommer N, Hoeres T, Bakr A,
Waisbrod S, Sydykov A, Haag D, Esfandiary A, Kojonazarov B, Veit F,
et al: Mitochondrial hyperpolarization in pulmonary vascular
remodeling. Mitochondrial uncoupling protein deficiency as disease
model. Am J Respir Cell Mol Biol. 49:358–367. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chu AC, Ho PW, Kwok KH, Ho JW, Chan KH,
Liu HF, Kung MH, Ramsden DB and Ho SL: Mitochondrial UCP4
attenuates MPP+ - and dopamine-induced oxidative stress,
mitochondrial depolarization, and ATP deficiency in neurons and is
interlinked with UCP2 expression. Free Radic Biol Med. 46:810–820.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kwok KH, Ho PW, Chu AC, Ho JW, Liu HF, Yiu
DC, Chan KH, Kung MH, Ramsden DB and Ho SL: Mitochondrial UCP5 is
neuroprotective by preserving mitochondrial membrane potential, ATP
levels, and reducing oxidative stress in MPP+ and
dopamine toxicity. Free Radic Biol Med. 49:1023–1035. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Halestrap AP, Clarke SJ and Khaliulin I:
The role of mitochondria in protection of the heart by
preconditioning. Biochim Biophys Acta. 1767:1007–1031. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Korshunov SS, Skulachev VP and Starkov AA:
High protonic potential actuates a mechanism of production of
reactive oxygen species in mitochondria. FEBS Lett. 416:15–18.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Kowaltowski AJ, Castilho RF and Vercesi
AE: Mitochondrial permeability transition and oxidative stress.
FEBS Lett. 495:12–15. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Brookes PS: Mitochondrial H(+) leak and
ROS generation: An odd couple. Free Radic Biol Med. 38:12–23. 2005.
View Article : Google Scholar
|
|
35
|
Lambert AJ and Brand MD: Superoxide
production by NADH:ubiquinone oxidoreductase (complex I) depends on
the pH gradient across the mitochondrial inner membrane. Biochem J.
382:511–517. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Andrews ZB, Horvath B, Barnstable CJ,
Elsworth J, Yang L, Beal MF, Roth RH, Matthews RT and Horvath TL:
Uncoupling protein-2 is critical for nigral dopamine cell survival
in a mouse model of Parkinson’s disease. J Neurosci. 25:184–191.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yakes FM and Van Houten B: Mitochondrial
DNA damage is more extensive and persists longer than nuclear DNA
damage in human cells following oxidative stress. Proc Natl Acad
Sci USA. 94:514–519. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Wieland P and Lauterburg BH: Oxidation of
mitochondrial proteins and DNA following administration of ethanol.
Biochem Biophys Res Commun. 213:815–819. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Larosche I, Lettéron P, Berson A, Fromenty
B, Huang TT, Moreau R, Pessayre D and Mansouri A: Hepatic
mitochondrial DNA depletion after an alcohol binge in mice:
Probable role of peroxynitrite and modulation by manganese
superoxide dismutase. J Pharmacol Exp Ther. 332:886–897. 2010.
View Article : Google Scholar
|
|
40
|
Coleman WB and Cunningham CC: Effects of
chronic ethanol consumption on the synthesis of polypeptides
encoded by the hepatic mitochondrial genome. Biochim Biophys Acta.
1019:142–150. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Choumar A, Tarhuni A, Lettéron P,
Reyl-Desmars F, Dauhoo N, Damasse J, Vadrot N, Nahon P, Moreau R,
Pessayre D and Mansouri A: Lipopolysaccharide-induced mitochondrial
DNA depletion. Antioxid Redox Signal. 15:2837–2854. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal
T, Junger W, Brohi K, Itagaki K and Hauser CJ: Circulating
mitochondrial DAMPs cause inflammatory responses to injury. Nature.
464:104–107. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Kohler C, Radpour R, Barekati Z,
Asadollahi R, Bitzer J, Wight E, Bürki N, Diesch C, Holzgreve W and
Zhong XY: Levels of plasma circulating cell free nuclear and
mitochondrial DNA as potential biomarkers for breast tumors. Mol
Cancer. 8:1052009. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cossarizza A, Pinti M, Nasi M, Gibellini
L, Manzini S, Roat E, De Biasi S, Bertoncelli L, Montagna JP, Bisi
L, et al: Increased plasma levels of extracellular mitochondrial
DNA during HIV infection: A new role for mitochondrial
damage-associated molecular patterns during inflammation.
Mitochondrion. 11:750–755. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Hosler JP, Ferguson-Miller S and Mills DA:
Energy transduction: Proton transfer through the respiratory
complexes. Annu Rev Biochem. 75:165–187. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Parton LE, Ye CP, Coppari R, Enriori PJ,
Choi B, Zhang CY, Xu C, Vianna CR, Balthasar N, Lee CE, et al:
Glucose sensing by POMC neurons regulates glucose homeostasis and
is impaired in obesity. Nature. 449:228–232. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Chavin KD, Yang S, Lin HZ, Chatham J,
Chacko VP, Hoek JB, Walajtys-Rode E, Rashid A, Chen CH, Huang CC,
et al: Obesity induces expression of uncoupling protein-2 in
hepatocytes and promotes liver ATP depletion. J Biol Chem.
274:5692–5700. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Serviddio G, Bellanti F, Tamborra R, et
al: HNE induces UCP2 expression and proton leak to limit oxidative
stress but reduces ATP synthesis in non-alcoholic steatohepatitis.
Free Radic Res. 41:S55. 2007.
|
|
49
|
Brar SS and Martin WJ II: Bleomycin
upregulates UCP2 mRNA expression, decreases ATP levels and induces
apoptosis in alveolar epithelial cells. American Thoracic Society
International Conference Abstracts 179: C68; Alveolar Epithelium.
abs. A5002. 2009
|
|
50
|
Andrews ZB, Diano S and Horvath TL:
Mitochondrial uncoupling proteins in the CNS: In support of
function and survival. Nat Rev Neurosci. 6:829–840. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Andrews ZB, Liu ZW, Walllingford N, Erion
DM, Borok E, Friedman JM, Tschöp MH, Shanabrough M, Cline G,
Shulman GI, et al: UCP2 mediates ghrelin’s action on NPY/AgRP
neurons by lowering free radicals. Nature. 454:846–851. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Cheng G, Polito CC, Haines JK, Shafizadeh
SF, Fiorini RN, Zhou X, Schmidt MG and Chavin KD: Decrease of
intracellular ATP content downregulated UCP2 expression in mouse
hepatocytes. Biochem Biophys Res Commun. 308:573–580. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Di Lisa F, Menabò R, Canton M, Barile M
and Bernardi P: Opening of the mitochondrial permeability
transition pore causes depletion of mitochondrial and cytosolic
NAD+ and is a causative event in the death of myocytes
in postischemic reperfusion of the heart. J Biol Chem.
276:2571–2575. 2001. View Article : Google Scholar
|
|
54
|
Hunter FE Jr, Gebicki JM, Hoffsten PE,
Weinstein J and Scott A: Swelling and lysis of rat liver
mitochondria induced by ferrous ions. J Biol Chem. 238:828–835.
1963.PubMed/NCBI
|
|
55
|
Baines CP, Song CX, Zheng YT, Wang GW,
Zhang J, Wang OL, Guo Y, Bolli R, Cardwell EM and Ping P: Protein
kinase Cepsilon interacts with and inhibits the permeability
transition pore in cardiac mitochondria. Circ Res. 92:873–880.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Clarke SJ, McStay GP and Halestrap AP:
Sanglifehrin A acts as a potent inhibitor of the mitochondrial
permeability transition and reperfusion injury of the heart by
binding to cyclophilin-D at a different site from cyclosporin A. J
Biol Chem. 277:34793–34799. 2002. View Article : Google Scholar : PubMed/NCBI
|