Introduction
Neutrophils are the first cell type recruited to
infected tissue; they sterilize the wound, clearing out invading
bacteria through phagocytosis and subsequent killing by the release
of reactive oxygen species (ROS) (1,2).
Activated neutrophils also secrete numerous cytokines, chemokines,
proteolytic enzymes stored in preformed granules, and
pro-inflammatory products of arachidonic acid (prostaglandin E2 and
leukotrienes), which collectively serve to recruit additional
immune cells, remove cell debris and fine-tune the adaptive immune
response (1,2). Although these functions of
neutrophils are crucial components of normal wound healing,
exaggerated or long-term neutrophil activity can contribute to
tissue damage as a result of the uncontrolled release of ROS into
the extracellular milieu (1,2).
Under normal conditions, resting neutrophils have a short half-life
and undergo apoptosis in the circulation after 6–9 h (3). However, when neutrophils reach a
site of inflammation, apoptosis is delayed by inflammatory
cytokines present in the tissues, which not only provide additional
time for completion of their microbicidal function, but also
exaggerate inflammation and tissue injury (4). Thus, neutrophils function as a
‘double-edged sword’. In order to prevent these adverse effects due
to the release of proteolytic enzymes, such as elastase and
myeloperoxidase and ROS, neutrophils should either be removed
quickly from the inflamed tissue or their recruitment should be
tightly controlled. Neutrophil recruitment is critical to pulmonary
inflammatory responses associated with acute lung injury (ALI)
(5,6). In our previous studies utilizing
animal models of sepsis, ALI, and gut ishchemia-reperfusion (I/R)
we observed an enhanced neutrophil infiltration at multiple organs,
particularly into the lungs, liver and intestines, which led to the
disruption of endothelial barrier function and promoted
extravascular host tissue damage during uncontrolled inflammation
(7-9). Conversely, the use of therapeutic
regimens that restrict tissue neutrophil infiltration may help the
host to overcome serious diseases in which abnormal neutrophil
infiltration is a major concern.
Neutrophil trafficking into pulmonary tissue and air
spaces in response to a gradient of chemoattractant is essential
for their localization at sites of infection and inflammation to
execute their functions (1).
Neutrophil migration into the lungs is mediated by a complex
cascade of rolling, adhesion and transendothelial migration,
involving a number of factors that have already been well defined
(10). Neutrophil infiltration
into the lungs is mediated by a local production of chemokines
released by macrophages, as well as other cell types in response to
inflammation (11,12). The levels of CXC chemokines, such
as interleukin-8 (IL-8) are significantly elevated in the
bronchoalveolar lavage fluid (BALF) of patients with acute
respiratory distress syndrome (ARDS), and increased IL-8 levels
have been shown to be associated with increased neutrophil
infiltration (13,14). In rodents, the IL-8 homologue,
CINC-1/2, and MIP-2 regulate neutrophil recruitment into the lungs
during experimental ALI through the chemokine receptor, CXCR2
(15,16). CXCR2 is a member of the G
protein-coupled receptor (GPCR) superfamily and is expressed in
neutrophils, monocytes and T cells (17). CXCR2 mediates neutrophil
chemotaxis in response to tissue injury and many types of
infections (17,18). G protein-coupled receptor kinase 2
(GRK2) has emerged as a key regulator of GPCR and other plasma
membrane receptors triggered by chemotactic messengers (19). It has been demonstrated that the
expression, localization and function of CXCR2 in polymorphonuclear
leukocytes (PMNs) are tightly regulated by intracellular GRK2
(20–22). Upon activation, GRK2
phosphorylates CXCR2 and causes receptor desensitization and
internalization, leading to the downregulation of neutrophil
chemotaxis (20–22). Increasing evidence points to the
occurrence of complex mechanisms modulating the subcellular
localization, activity and expression levels of GRK2, which reveals
new functional interactions of this kinase with various cellular
proteins and transduction cascades (23). It has also been reported that GRK2
co-localizes with the mitogen-activated protein (MAP) kinases, and
its activity and bidirectional regulation are mediated through the
MAP kinase pathways (23–25).
Milk fat globule-epidermal growth factor-factor 8
(MFG-E8) is a secretory glycoprotein with bivalent binding activity
to αvβ3-integrin and acidic phospholipids,
such as phosphatidylserine (PS) capable of carrying out versatile
functions in cell physiology, such as the recognition of target
cells and membrane vesicles by phagocytes (26,27), the development of male
reproductive organs and cells (28), cell reorganization in mammary
gland development and involution (29,30), and the regulation of inflammatory
responses, such as macrophage activation (31,32) and neutrophil infiltration
(8). It was named after its
origin and structural properties, i.e., its origin in milk fat
globule and its sequence homology to epidermal growth factor
(EGF)-like domains of Drosophila Notch protein and
C-terminal domains of human coagulation factors VIII and V
(F5/8-type C domain). In our previous studies, we observed a
significant decrease in MFG-E8 expression in the immune reactive
organs following sepsis, ALI and gut I/R injury, and exogenous
treatment with recombinant murine MFG-E8 (rmMFG-E8) markedly
improved survival by attenuating systemic inflammation and the
infiltration of neutrophils at vital organs (8,9,33).
Therefore, in the present study, we aimed to elucidate the pivotal
mechanisms through which MFG-E8 regulates neutrophil migration in
response to the chemoattactant, IL-8. Based on our hypothesis, we
demonstrate that the treatment of the human neutrophil-like cell
line, HL-60, with recombinant human MFG-E8 (rhMFG-E8) results in a
decreased migration ability towards IL-8. We further clarified the
pivotal role of MFG-E8 in the αvβ3-integrin
mediated downregulation of neutrophil migration by modulating the
surface expression of CXCR2 through GRK2-dependent pathways. We
also deduced a novel and previously unexplored mechanism involving
the MAP kinase pathways in the effects of MFG-E8 on the inhibition
of neutrophil migration. Importantly, the present findings identify
an additional role of MFG-E8 in inhibiting neutrophil infiltration
through MAP kinase-dependent pathways. Thus, this may prove to be
an effective therapeutic strategy in the treament of diseases in
which enhanced neutrophil infiltration is a major concern.
Materials and methods
HL-60 cell culture and
differentiation
HL-60 human promyelocytic leukemia cells, obtained
from the American Type Culture Collection (ATCC; Manassas, VA, USA)
were cultured in a T-25 cell culture flask at a density of
2×105 cells/ml in 15 ml RPMI-1640 medium supplemented
with 10% fetal bovine serum (FBS), and penicillin and streptomycin.
The cells were kept in 37°C incubator under humidified conditions
containing 5% CO2. The cells were grown to a density of
1×106 cells/ml, at which time they were passaged by
seeding into a new flask at 2×105 cells/ml. In order to
induce the differentiation of the HL-60 [differentiated HL-60
(dHL-60)] cells, 1×105 cells/ml at the mid-log growth
phase were grown in a T-25 flask in 15 ml of RPMI-1640 medium
containing 190 µl of 100% dimethyl sulfoxide (DMSO)
supplemented with 10% FBS, and penicillin/streptomycin antibiotics
for a period of 5-7 days, as previously described (34), which induced their differentiation
into PMNs.
Stimulation of dHL-60 cells with
rhMFG-E8
The expression, purification and functional
characterization of rhMFG-E8 were performed according to a
previously described protocol (35). In brief, a 1,095 bp fragment
encoding the mature region of human MFG-E8 was obtained by
polymerase chain reaction amplification and cloned into the
SalI and NotI site of the pET-28a(+) vector (Novagen,
Inc., Madison, WI, USA) downstream of the phage T7 RNA polymerase
promoter. The plasmid was transformed into E. coli BL21
(DE3) cells grown at 37°C in 2YT medium (Invitrogen Life
Technologies, Grand Island, NY, USA) with kanamycin overnight.
rhMFG-E8 protein production was induced by the addition of
isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration
of 1.0 mM and cell growth continued for 5 h at 25°C. The cells were
harvested by centrifugation at 6,000 rpm and the induced rhMFG-E8
protein was purified according to the manufacturer’s instructions
(Novagen, Inc.). The rhMFG-E8 fractions were pooled and the
endotoxin of the protein solution was removed by phase separation
using Triton X-114. The content of lipopolysaccharide (LPS) in the
sample was determined using the Limulus Amebocyte Lysate assay kit
(BioWhittaker, Walkersville, MD, USA). The purity of rhMFG-E8 was
evaluated by SDS-PAGE on a 10–20% Tris-HCl gel and visualized using
the GelCode Blue Stain Reagent (Pierce Biotechnology, Inc.,
Rockford, IL, USA). The final product was concentrated by Amicon
ultra-15 centrifugal filter devices to the designed concentration
and stored at -20°C. For the stimulation of the dHL-60 cells with
rhMFG-E8, a total of 1.5×106 cells/ml was placed into
1.5 ml microcentrifuge tubes in serum-free Opti-MEM (Invitrogen
Life Technologies) and then stimulated with rhMFG-E8 at a dose of
500 ng/ml for the indicated period of time. Subsequently,
experiments were carried out for the assessment of cell migration,
and intracellular signal transduction by western blot analysis and
flow cytometry.
In vitro cell migration assay
The migration assays were conducted in a modified
24-well (3.0 µm) Boyden chamber (BD Biosciences, San Jose,
CA, USA). Following differentiation, the dHL-60 cells
(3×105) were pre-treated with either rhMFG-E8 (125–1,000
ng/ml) or PBS for 2 h, and then plated in the Boyden chamber
inserts and medium with 50 ng/ml of recombinant human IL-8 (rhIL-8;
R&D Systems, Minneapolis, MN, USA) was placed in the outer
compartment which served as a chemoattractant. After 1.5 h of
incubation, the upper surface of the filter was swabbed with
cotton-tipped applicators to remove non-migratory cells. The
migrated cells were fixed with 4% paraformaldehyde (PFA) and
stained with propidium iodide (PI) (1 µg/ml). A total of 6
random microscopic fields per well were counted.
Flow cytometric analysis
To examine the surface CXCR2 and intracellular GRK2
expression levels, the dHL-60 cells (1.5×106 cells)
treated with rhMFG-E8 (500 ng/ml) for 2 h were first
surface-stained with PE-CXCR2 (BioLegend, San Diego, CA, USA), and
subsequently, to determine intracellular GRK2 expression, the cells
were fixed and permeabilized with IntraPrep (Beckman Coulter,
Fullerton, CA, USA), followed by staining with FITC-GRK2 antibodies
(Abcam, Cambridge, MA, USA). After washing, the stained cells were
analyzed using a FACSVerse flow cytometer (BD Biosciences). Data
were analyzed by FlowJo software (FlowJo, LLC, Ashland, OR, USA)
with 15,000 events per sample. Isotype controls and Fc Receptor
Blocker (both from BioLegend) were used for all the samples.
Western blot analysis
The dHL-60 cells (1.5×106/ml) were placed
into 1.5 ml microfuge tubes with Opti-MEM and then stimulated with
either rhMFG-E8 (500 ng/ml) or PBS for different periods of time.
Following incubation, the cells were centrifuged at 200 x g for 5
min and the supernatants were removed. The cell pellet was then
lysed by the addition of 80–100 µl of loading buffer
containing 0.5 M Tris-HCl, pH 6.8, 16% glycerol, 10% SDS and 1%
Bromophenol Blue. The lysate was then heated to 95°C for 5 min and
an equal volume (20 µl) of each lysate per lane was loaded
onto a 4–12% Bis-Tris gel (Invitrogen Life Technologies) and
transferred onto a 0.2-µm nitrocellulose membrane
(Invitrogen Life Technologies). The membrane was incubated
overnight at 4°C with the primary antibodies as obtained from
respective vendors: rabbit anti-GRK2 monoclonal antibody (Cat. no.
ab32558; Abcam), rabbit anti-phospho-p38 (Cat. no. 9211),
anti-phospho-extracellular signal-regulated kinase (ERK)1/2 (Cat.
no. 9101), and the total antibodies for p38 (Cat. no. 9212) and
ERK1/2 (Cat. no. 4695), (all from Cell Signaling Technology,
Danvers, MA, USA), at a 1:1,000 dilution, reacted with
peroxidase-conjugated goat anti-rabbit secondary antibody (Cat. no.
4030-05; SouthernBiotech, Birmingham, AL, USA) at a 1:10,000
dilution at room temperature for 2 h, and washed 5 times in TBST.
The immunoblot was washed, stripped off and reprobed with mouse
anti-β-actin antibody (Cat. no. A2228; Sigma-Aldrich, St. Louis,
MO, USA) as a loading control. The resulting signals were detected
by ECL (GE Healthcare, Buckinghamshire, UK), and the band
intensities were assessed by densitometry using ImageJ software, as
previously described (36).
In vitro neutralization of
αv-integrin
For the in vitro neutralization of the
αv-integrin receptor, a total of 1.5×106
dHL-60 cells were placed into 1.5 ml microfuge tubes containing 1
ml of Opti-MEM. The cells were then pre-treated with 1 µg/ml
of each of the IgG isotype control or anti-αv-integrin
neutralizing antibody (both from BioLegend) for 1 h at 37°C.
Subsequently, the cells were stimulated with rhMFG-E8 (500 ng/ml)
or PBS for different periods of time and then analyzed by flow
cytometry or western blot analysis.
Inhibition of p38 and ERK using chemical
inhibitors
The dHL-60 cells were placed into 1.5 ml microfuge
tubes at a density of 1.5×106 cells/ml in Opti-MEM. The
cells were then pre-treated with the p38 inhibitor, SB203580 and
the ERK inhibitor, PD98059 (both from Tocris Bioscience,
Ellisville, MO, USA), at a concentration of 10 µM for each
for 1 h at 37°C. Following incubation, the cells were stimulated
with rhMFG-E8 or PBS for 2 h followed by the assessment of CXCR2
and GRK2 expression by flow cytometry and western blot
analysis.
Statistical analysis
All data are expressed as the means ± SE and
compared by one-way ANOVA and the Student-Newman-Keul (SNK) test.
The Student’s t-test was used when only 2 groups were compared.
Differences in values were considered significant if P<0.05.
Results
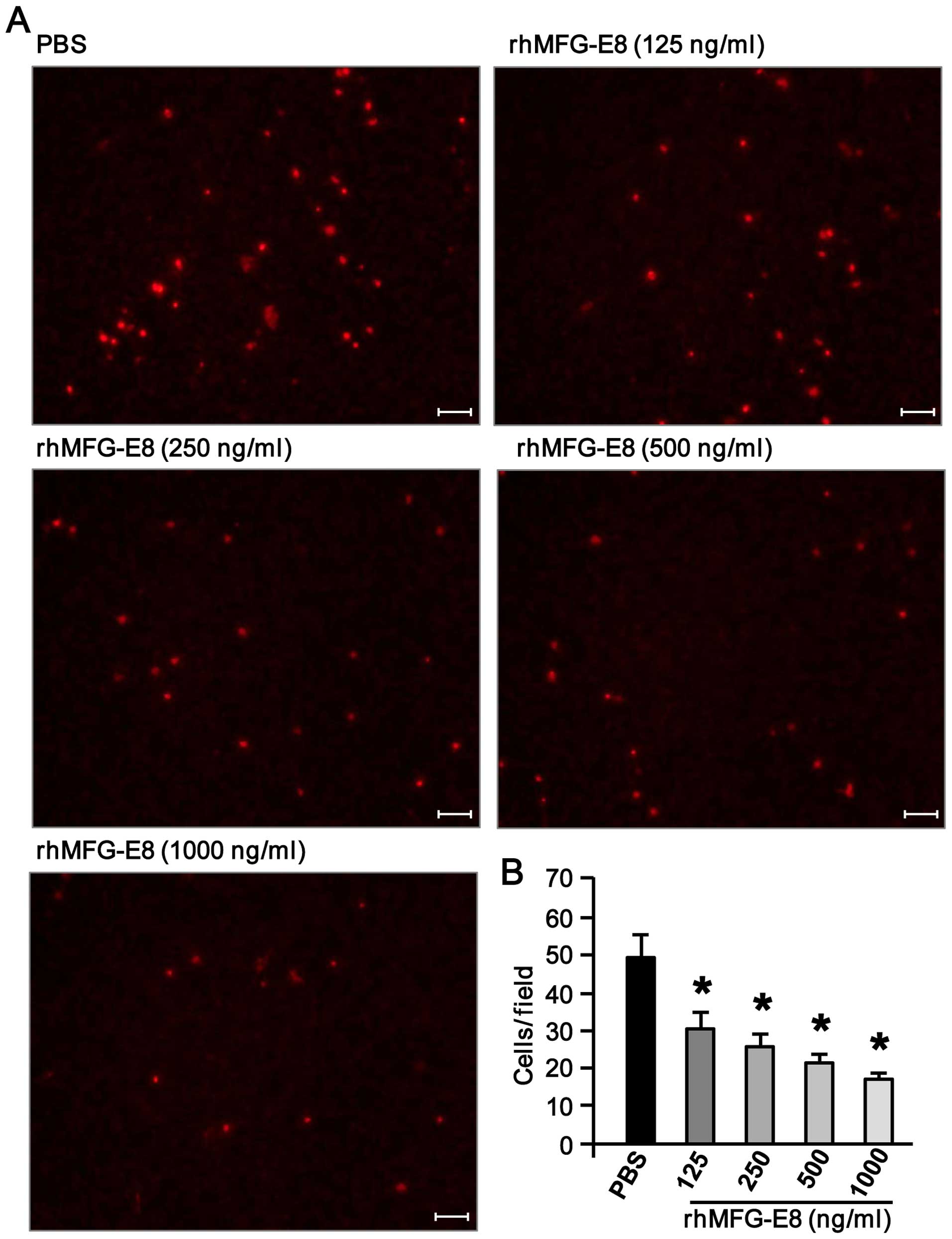
rhMFG-E8 inhibits dHL-60 cell migration
in a dose-dependent manner
To examine the effects of rhMFG-E8 on neutrophil
migration, the dHL-60 cells were pre-treated with various
concentrations of rhMFG-E8 for 2 h. The cells were then allowed to
proceed for migration towards rhIL-8 as a chemoattractant using a
Boyden chamber. As shown in Fig. 1A
and B, pre-treatment of the dHL-60 cells with rhMFG-E8 led to a
significantly decrease in their migration towards rhIL-8 in a
dose-dependent manner. Since the most notable decrease in their
migration occurred following stimulation with rhMFG-E8 at the doses
of 500 and 1,000 ng/ml, among these 2 doses we decided to utilize
the lesser dose of 500 ng/ml of rhMFG-E8 protein for the subsequent
in vitro experiments.
rhMFG-E8 downregulates CXCR2 surface
expression by upregulating intracellular GRK2 expression in dHL-60
cells
CXCR2, a surface receptor for the chemokine, IL-8,
plays a crucial role in IL-8-dependent neutrophil migration
(15,16,37). The dHL-60 cells stimulated with
rhMFG-E8 showed a significant downregulation in CXCR2 expression at
their cell surface (Fig. 2A and
B), which was further linked to the decrease in neutrophil
migration following stimulation with rhMFG-E8. Since intracellular
GRK2 serves as a negative regulator of surface CXCR2 expression in
neutrophils, we wished to assess intracellular GRK2 expression in
the dHL-60 cells following stimulation with rhMFG-E8. Of note, we
observed a significant upregulation in intracellular GRK2
expression in the dHL-60 cells stimulated with rhMFG-E8, as
revealed by flow cytometric analysis (Fig. 2C and D). Consistently, western
blot analysis of GRK2 protein expression also revealed the
reproducible findings of its upregulation upon rhMFG-E8 stimulation
in a time-dependent manner (Fig.
2E), suggesting that the inhibition of CXCR2 expression in
dHL-60 cells may be mediated through the upregulation of GRK2
expression.
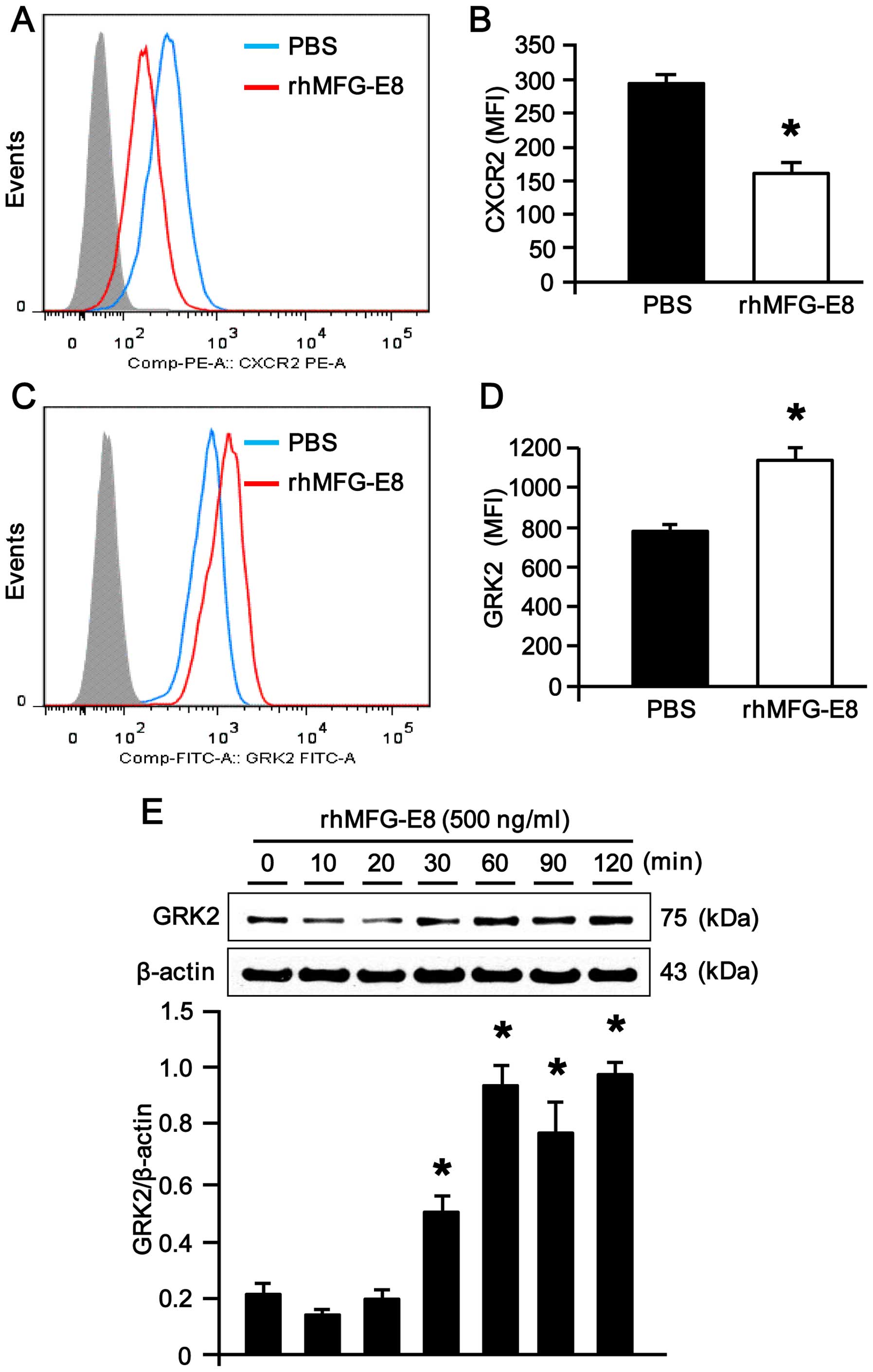 | Figure 2Expression of CXCR2 and G
protein-coupled receptor kinase 2 (GRK2) in recombinant human milk
fat globule-epidermal growth factor-factor 8 (rhMFG-E8)-treated
neutrophils. (A) Differentiated HL-60 (dHL-60) cells
(1.5×106 cells) treated with rhMFG-E8 (500 ng/ml) for 2
h were surface-stained with PE-CXCR2 and then subjected to flow
cytometric analysis. Data were analyzed by Flowjo software with
15,000 events per sample. Isotype controls and Fc receptor blocker
were used for all the samples. The representative histograms for
PBS and rhMFG-E8-treated dHL-60 cells obtained from 3 independent
experiments are shown. (B) Bar diagram representing the mean
fluorescence intensities (MFI) of the PBS- and rhMFG-E8-treated
samples are shown. Data are expressed as the means ± SE (n=3
samples/group), obtained from 3 independent experiments.
*P<0.05 vs. PBS treatment. (C and D) To examine the
expression of intracellular GRK2 levels, dHL-60 cells
(5×106 cells) treated with rhMFG-E8 (500 ng/ml) for 2 h
were first surface-stained with PE-CXCR2, and then to examine
intracellular GRK2 expression, cells were fixed and permeabilized
with Intraprep, followed by staining with FITC-GRK2 antibodies.
After washing, the stained cells were subjected to flow cytometry
using a FACSVerse flow cytometer. Appropriate isotype controls and
Fc receptor blocker were used for all the samples. Representative
histogram and the bar diagrams indicating the MFI of PBS and
rhMFG-E8-treated samples are shown. Data are expressed as the means
± SE (n=3 samples/group), obtained from 3 independent experiments.
*P<0.05 vs. PBS treatment. (E) Differentiated HL-60
cells (1.5×106/ml) were placed into 1.5 ml microfuge
tubes with Opti-MEM and then stimulated with either rhMFG-E8 (500
ng/ml) or PBS for different periods of time. Following incubation,
the cell lysates were harvested and then subjected to western blot
analysis using rabbit anti-GRK2 monoclonal antibody. Results were
normalized to β-actin as an internal control and are expressed as
the fold induction in comparison to the 0 min time point. Data are
expressed as the means ± SE (n=3 samples/group), obtained from 3
independent experiments. *P<0.05 vs. 0 min. |
rhMFG-E8 upregulates MAP kinase
phosphorylation through αvβ3-integrin
To determine whether rhMFG-E8 alters MAP kinase
phosphorylation, the dHL-60 cells were treated with rhMFG-E8 for
different periods of time and we then measured the p38 and ERK
phosphorylation levels by western blot analysis. As shown in
Fig. 3A and B, the dHL-60 cells
stimulated with rhMFG-E8 showed a significant upregulation in p38
and ERK phosphorylation in a time-dependent manner with the highest
induction in their phosphorylation observed at the 20- and 30-min
time points; after these time points, their phosphorylation
decreased to basal levels. Since
αvβ3-integrin recognizes MFG-E8, we wished to
determine whether this integrin is involved in the MFG-E8-mediated
signal transduction of p38 and ERK phosphorylation in the dHL-60
cells. For this purpose, we first treated the dHL-60 cells with the
neutralizing antibody for αv-integrin or IgG isotype
antibody followed by stimulation with rhMFG-E8, which evidently
revealed that the promoting effects of rhMFG-E8 on p38 and ERK
phosphorylation were notably diminished (Fig. 3C), indicating the role of
αvβ3-integrin in transducing MFG-E8-mediated
downstream signaling and MAP kinase activation.
rhMFG-E8 modulates CXCR2 and GRK2
expression through αvβ3-integrin
To determine the involvement of
αvβ3-integrin in the rhMFG-E8-mediated
alteration in CXCR2 and GRK2 expression, the dHL-60 cells were
pre-treated with anti-αv-integrin antibody to block the
MFG-E8 receptor for the transmission of downstream signaling. As
shown in Fig. 4A, pre-treatment
of the cells with anti-αv-integrin antibody neutralized
the rhMFG-E8-induced downregulation in the surface expression of
CXCR2, while a significant downregulation in CXCR2 surface
expression was observed in the cells treated with the IgG isotype
control. Similarly, the expression of GRK2 was induced in the
rhMFG-E8-stimulated dHL-60 cells pre-treated with the IgG isotype
control; conversely the rhMFG-E8-induced upregulation in GRK2
expression was diminished in the cells pre-treated with
anti-αv-integrin (Fig.
4B). Taken together, these data clearly indicate that the
MFG-E8-mediated down-regulation of CXCR2 and the upregulation of
GRK2 expression are transmitted through the
αvβ3-integrin pathway.
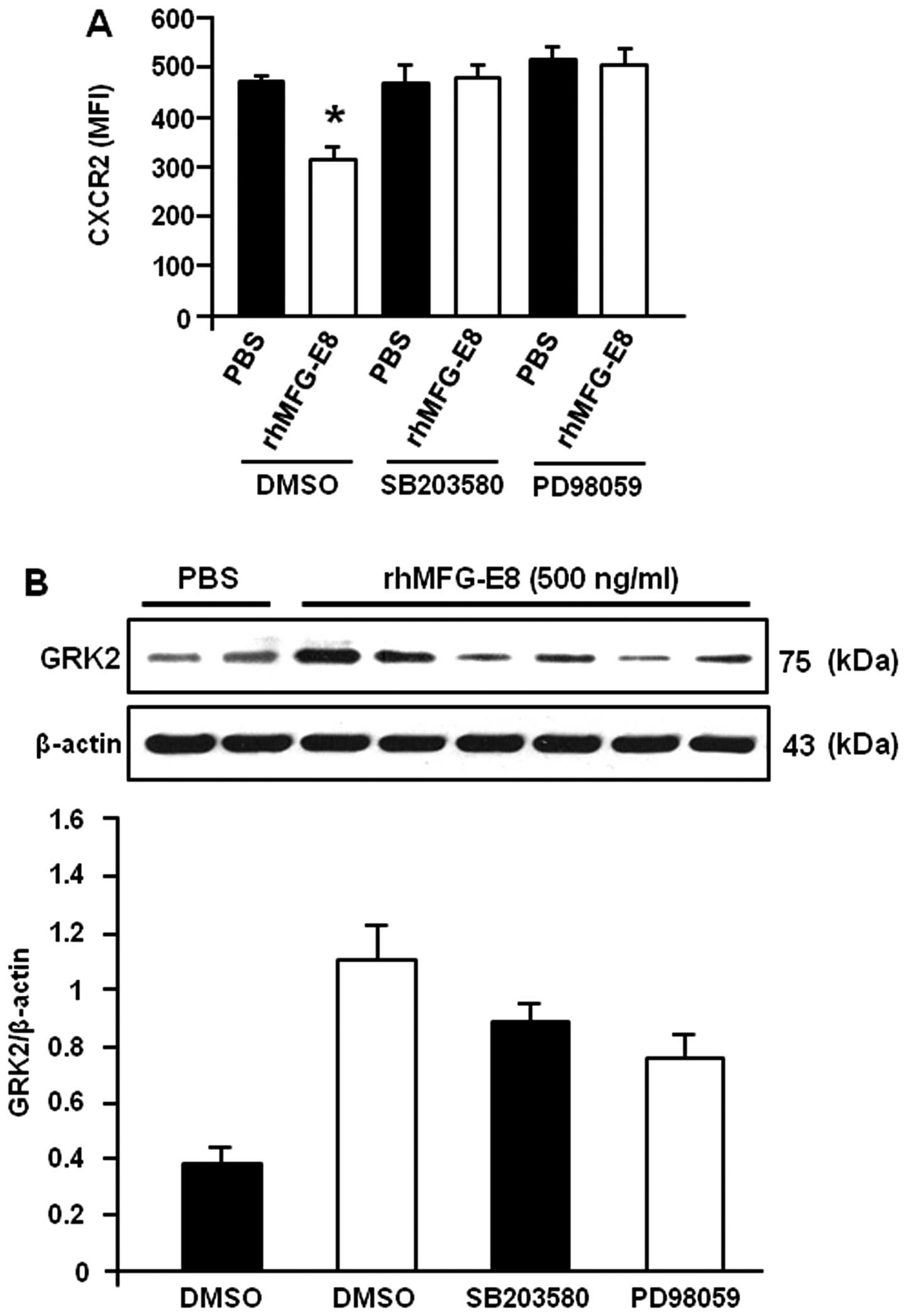
MAP kinase inhibitors neutralize the
rhMFG-E8-induced inhibition of CXCR2 and enhancement of GRK2
expression
To determine the role of MAP kinases in the
rhMFG-E8-mediated inhibition of CXCR2 expression, the dHL-60 cells
were pre-treated with MAP kinase inhibitors and the effects of
rhMFG-E8 on CXCR2 expression were then evaluated. As shown in
Fig. 5A, pre-treatment of the
cells with the specific inhibitors of p38 and ERK diminished the
negative regulatory effects of rhMFG-E8 on CXCR2 which led to the
downregulation of its surface expression, indicating the
involvement of MAP kinases in rhMFG-E8-mediated signaling.
Similarly, the rhMFG-E8-induced upregulation of GRK2 was also
diminished when the p38 and ERK molecules were blocked by using
their specific inhibitors as compared to the DMSO control (Fig. 5B). These data clearly indicate
that the rhMFG-E8-mediated downstream signaling which downregulates
CXCR2 and upregulates GRK2 is mediated through MAP kinase
activation.
Inhibition of αv-integrin and
MAP kinases diminishes the rhMFG-E8-mediated downregulation in
neutrophil migration
In order to determine the involvement of
αvβ3-integrin and MAP kinases in the
rhMFG-E8-mediated inhibition of neutrophil migration, the dHL-60
cells were pre-treated with anti-αv-integrin antibody
and the MAP kinase inhibitors, and the effects of rhMFG-E8 on
neutrophil migration were antibody evaluated. As shown in Fig. 6A and B, pre-treatment of the cells
with anti-αv-integrin antibody and MAP kinase inhibitors
markedly reversed the inhibitory effects of rhMFG-E8 on neutrophil
migration. Collectively, these findings clearly indicate that
MFG-E8 inhibits neutrophil migration by downregulating surface
CXCR2 expression through the upregulation of the expression of the
intracellular negative regulator, GRK2, and this event is mediated
by αvβ3-integrin-mediated MAP kinase
activation (Fig. 6C).
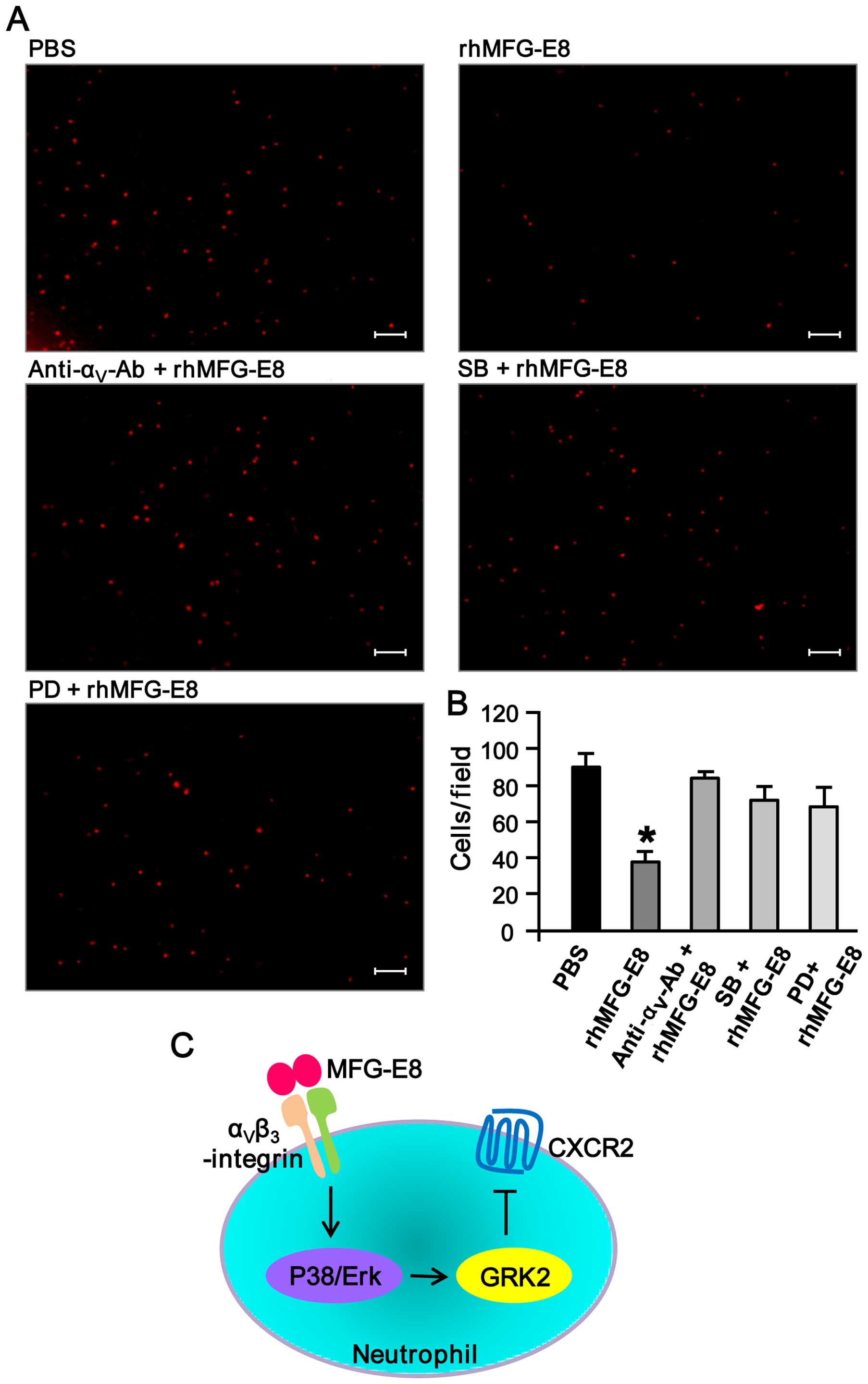 | Figure 6Treatment with
anti-αv-integrin antibody and mitogen-activated protein
(MAP) kinase inhibitors counteracts recombinant human milk fat
globule-epidermal growth factor-factor 8 (rhMFG-E8)-mediated
downregulation of neutrophil migration. (A) Differentiated HL-60
(dHL-60) cells (3×105) were pre-stimulated with 1
µg/ml of each of the IgG isotype control,
anti-αv-integrin neutralizing antibody, the p38
inhibitor, SB203580 (SB), of the ERK inhibitor, PD98059 (PD), at a
concentration of 10 µM for 1 h at 37°C in their respective
1.5 ml microfuge tubes. The cells were then stimulated with
rhMFG-E8 (500 ng/ml) or PBS for 2 h, and then plated in 500
µl volume in the Boyden chamber inserts. The outer
compartment of the inserts contained 500 µl of RPMI medium
with 50 ng/ml of recombinant human interleukin-8 (IL-8) as a
chemoattractant. After 1.5 h of incubation, the upper surface of
the filter was swabbed with cotton-tipped applicators to remove
non-migratory cells. Migrated cells were fixed with 4%
paraformaldehyde (PFA) and stained with propidium iodide (PI) (1
µg/ml). A total of 6 random microscopic fields per well were
counted. Scale bar, 100 µm. (B) The average number of
migrated dHL-60 cells are plotted in a bar diagram where the
results are expressed as the means ± SE obtained from 6
fields/group of 3 independent experiments. *P<0.05
vs. PBS. (C) Mechanistic finding. The secretory glycoprotein,
MFG-E8 binds to its receptor, αvβ3-integrin,
and transduces downstream signaling of MAP kinase (p38 and ERK)
activation. The activated MAP kinases then upregulate G
protein-coupled receptor kinase 2 (GRK2) which in turn negatively
regulates the surface exposure of CXCR2, thereby attenuating
neutrophil migration. |
Discussion
Neutrophils are the first line of defense against
tissue infection, trauma, stress insults and injury. These cells
play a key role in the defense against bacterial, fungal and viral
infections and a growing body of evidence suggests that neutrophils
may also represent a critical link between the innate and adaptive
immune system (1). Therefore, the
migration of neutrophils to infected tissue and secondary lymphoid
organs is critical for effective immune responses to the majority
of pathogens; however, uncontrolled migration can lead to tissue
damage and chronic inflammation (2). In our previous studies, we reported
an enhanced infiltration of neutrophils into the vital organs
following sepsis, ALI, renal, and gut I/R injury, causing severe
inflammation and tissue damage. However, the deleterious events
caused by excessive neutrophil accumulation were ameliorated by
exogenous treatment with recombinant MFG-E8 which attenuated
neutrophil migration and infiltration into tissues (8,9,38).
Although we have initially elucidated the
involvement of the the downregulation of the IL-8 receptor, CXCR2,
at the neutrophil cell surface due to the upregulation of its
intracellular negative regulator, GRK2, in rmMFG-E8-treated murine
bone marrow-derived neutrophils (8), in this study, in order to reveal the
mechanisms through which MFG-E8 attenuates neurophil migration, we
utilized the human neutrophil cell line, HL-60, and stimulated the
cells with rhMFG-E8 and focused on evaluating the upstream
signaling components which may result in the modulation of
GRK2/CXCR2 signaling. We observed a marked induction in the levels
of p-p38 and ERK MAP kinases following stimulation of the HL-60
cells with rhMFG-E8. To confirm this novel link between MFG-E8 and
MAP kinases, we utilized a two-step blocking strategy: i)
neutralization of the αvβ3-integrin
heterodimer by the anti-αv-integrin antibody abrogated
the rhMFG-E8-induced increase in the levels of MAP kinases; and ii)
inhibition of MAP kinases by their inhibitors diminished the
inhibitory effects of rhMFG-E8 on neutrophil migration through the
modulation of GRK2/CXCR2, thus suggesting the involvment of these
two novel pathways in the MFG-E8-mediated downregulation of human
neutrophil migration.
MFG-E8 has two functional domains: the N-terminal
EGF domain that bind to αvβ3-integrin of most
hematopoietic cells, and the C-terminal discoidin domains that
recognizes the PS in apoptotic cells (26). In this study, we considered
αvβ3-integrin as the gateway for
MFG-E8-mediated signal transduction. Structurally,
αvβ3-integrin is a heterodimeric
transmembrane receptor formed by the non-covalent association of
the α and β subunits (39). In
this study, we demonstrated that the blocking
αv-integrin in neutrophils effectively diminished the
effects of rmMFG-E8 on IL-8-mediated HL-60 cell migration through
the activation of MAP kinases and the modulation of GRK2/CXCR2
expression, indicating the involvement of αv-integrin in
mediating MFG-E8 activity. Similar to our results demonstrating
that the blocking of αv-integrin abrogates MFG-E8
signaling, Cheyuo et al also adopted the same approach to
block only αv-integrin for the functional assessment of
MFG-E8-mediated anti-inflammatory and anti-apoptotic roles in
cerebral ischemic injury (40).
Furthermore, our findings identify the integrin signaling pathway
as another critical factor in controlling GRK2-mediated CXCR2
downreg ulation.
It is already known that MFG-E8 was first discovered
as a scavenging factor to promote the phagocytosis of apoptotic
cells by macrophages through the formation of a bridge between them
(26). However, MFG-E8 has
several immunological and physiological functions. Apart from
participating in the phagocytic clearance of apoptotic cells,
MFG-E8 directly attenuates pro-inflammatory milieu by inhibiting
nuclear factor (NF)-κB in in vivo and in vitro
systems (31,41). In addition, MFG-E8 has recently
been reported to have growth-promoting functions, where it promotes
intestinal epithelial cell regeneration through the PKCε-mediated
pathway (42). Furthermore,
MFG-E8 is also known to promote AKT and Twist-dependent malignant
melanoma progression (43) and
ERK-mediated sperm-egg interaction (44), indicating its roles in
manipulating intracellular signaling required for cell
proliferation and their interaction with each other. Although the
involvement of MAP kinases in controlling neutrophil migration has
been well elucidated, to the best of our knowledge, there is no
published study to date utilizing MFG-E8 for the activation of MAP
kinases, which are linked to neutrophil migration. In this study,
indicative of its role in immune cell migration, we observed a
decrease in neutrophil migration as a result of the increased
phosphorylation of p38 and ERK MAP kinases, which led to the
downregulation of CXCR2 surface expression through the
GRK2-dependent pathway. However, a recent study suggested that the
different components of MAP kinases have distinct regulatory roles
in neutrophil migration (25);
the authors revealed the ‘stop’ and ‘go’ signal in the context of
regulating neutrophil migration utilizing ERK and p38
phosphorylation, respectively. Based on their findings, the
activation of p38 promoted, whereas the phosphorylation of ERK
inhibited neutrophil migration. In accordance with their findings,
in this study, using rhMFG-E8, we also found similar results in the
context of the upregulation of ERK phosphorylation which may
inhibit neutrophil migration. However, the difference between their
study and ours is that in our study, rhMFG-E8 mediated the
upregulation of p38 phosphorylation, inhibiting neutrophil
migration, while in their study, p38 phosphorylation promoted
neutrophil migration. However, in our study we tried to exclude
this divergence by utilizing the p38 inhibitor, SB203580, which
diminished the rhMFG-E8-induced down-regulation of neutrophil
migration, hence suggesting a negative regulatory role of p38 in
IL-8-mediated neutrophil migration upon stimulation with rhMFG-E8.
The study carried out by Liu et al is to some extent
different from our study in that they utilized fMLP as a
chemoattractant and revealed the regulatory mechanism of neutrophil
migration by treating dHL-60 cells directly with fMLP, and although
the phosphorylation of p38 and ERK was induced, the feedback
inhibition was initiated only by phospho-ERK (25). The involvement of MAP kinases in
neutrophil migration can also be explained by another study in
which pre-treatment of neutrophils with TLR ligands attenuated
neutrophil migration due to high GRK2 expression (45). Since TLR ligations to their
ligands induces the activation of MAP kinases, it is therefore
conceivable that due to the activation of MAP kinases, the
migration of neutrophils may be attenuated, which is in agreement
with our findings that the MFG-E8-mediated upregulation of MAP
kinase phosphorylation attenuated neutrophil migration. MFG-E8
contains EGF domains at its N-terminal domain. Since several
proteins which have an EGF domain in their backbone, e.g., EGF,
heparin-binding EGF-like growth factor (HB-EGF), Notch, and growth
arrest-specific 6 (Gas6) are known to upregulate MAP kinases to
execute their relevant functions (46–49), the upregulation of MAP kinase
phosphorylation by rhMFG-E8 may be comparable.
Developmental endothelial locus-1 (Del-1), a
probable paralogue protein of MFG-E8, which has a sequence and
domain structure similar to MFG-E8, also shows identical
biochemical functions of divalent binding activity to cell membrane
molecules, such as MFG-E8 (50,51). The RGD motif in the second
EGF-like domain is conserved between MFG-E8 and Del-1, both of
which show binding to cells expressing αvβ3
and αvβ5 integrins. A recent study utilizing
Del-1 indicated a significant inhibition of neutrophil migration
through the blocking of the interaction of leukocyte functional
antigen-1 (LFA-1) iin neutrophils and intercellular adhesion
molecule-1 (ICAM-1) in endothelial cells, hence inhibiting
neutrophil migration (52). In
this study, we revealed a mechanism involving MAP kinases and the
GRK2-mediated downregulation of CXCR2 by stimulation with MFG-E8
for the inhibition of neutrophil migration; this provides a novel
direction for the modulation of the intracellular signaling
cascade. Since MFG-E8 and Del-1 are homologous, the findings
regarding Del-1 may be implemented so as to reveal the complete
mechanisms through which MFG-E8 attenuates neutrophil migration. In
this regard, since MFG-E8 has RGD in its backbone, emphasis
therefore be placed on whether MFG-E8 blocks the interaction of
extracellular matrix proteins to their integrin receptor to
attenuate their binding for the initial attachment of neutrophils
to endothelial cells as the first step of neutrophil migration.
Therefore, deducing the role of MFG-E8 to the steps of roling and
adherence may prove to be of considerable interest.
In conclusion, in this study, we identified a novel
link between MAP kinases and GRK2, playing a negative regulatory
role on CXCR2 surface expression (Fig. 6C). Our data may lead to
translational studies being carried out for the identification of
potential drug candidates which can modulate neutrophil migration,
leading to the remission of several inflammatory diseases in which
controlling exaggerated neutrophil infiltration is a major
challenge.
Acknowledgments
This study was supported in part by a National
Institutes of Health Grant R01 GM 057468 (to P.W.). This study was
presented partially as an abstract at the 46th Annual Meeting of
the Society for Leukocyte Biology in Newport, Rhode Island on
October 20–22, 2013. P.W. is an inventor of pending Patent
Cooperation Treaty applications, which covers the fundamental
concept of using MFG-E8 for the treatment of sepsis and ischemia
injury. P.W. is co-founder of TheraSource LLC. TheraSource LLC
holds the exclusive option to license the technology from the
Feinstein Institute for Medical Research.
Abbreviations:
|
MFG-E8
|
milk fat globule-epidermal growth
factor-factor 8
|
|
NF-κB
|
nuclear factor-κB
|
|
MAP kinase
|
mitogen-activated protein kinase
|
|
ROS
|
reactive oxygen species
|
|
ALI
|
acute lung injury
|
|
BALF
|
bronchoalveolar lavage fluid
|
|
ARDS
|
acute respiratory distress
syndrome
|
|
GRK2
|
G protein-coupled receptor kinase
2
|
|
GPCR
|
G protein-coupled receptor
|
|
PMNs
|
polymorphonuclear leukocytes
|
References
|
1
|
Nathan C: Neutrophils and immunity:
Challenges and opportunities. Nat Rev Immunol. 6:173–182. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rot A and von Andrian UH: Chemokines in
innate and adaptive host defense: basic chemokinese grammar for
immune cells. Annu Rev Immunol. 22:891–928. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Etzioni A and Tonetti M: Leukocyte
adhesion deficiency II-from A to almost Z. Immunol Rev.
178:138–147. 2000. View Article : Google Scholar
|
|
4
|
Webb PR, Wang KQ, Scheel-Toellner D,
Pongracz J, Salmon M and Lord JM: Regulation of neutrophil
apoptosis: a role for protein kinase C and
phosphatidylinositol-3-kinase. Apoptosis. 5:451–458. 2000.
View Article : Google Scholar
|
|
5
|
Ayala A, Chung CS, Lomas JL, Song GY,
Doughty LA, Gregory SH, Cioffi WG, LeBlanc BW, Reichner J, Simms HH
and Grutkoski PS: Shock-induced neutrophil mediated priming for
acute lung injury in mice: divergent effects of TLR-4 and
TLR-4/FasL deficiency. Am J Pathol. 161:2283–2294. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abraham E: Neutrophils and acute lung
injury. Crit Care Med. 31(Suppl): S195–S199. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Aziz M, Jacob A, Yang W-L, Matsuda A and
Wang P: Current trends in inflammatory and immunomodulatory
mediators in sepsis. J Leukoc Biol. 93:329–342. 2013. View Article : Google Scholar :
|
|
8
|
Aziz M, Matsuda A, Yang W-L, Jacob A and
Wang P: Milk fat globule-epidermal growth factor-factor 8
attenuates neutrophil infiltration in acute lung injury via
modulation of CXCR2. J Immunol. 189:393–402. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Cui T, Miksa M, Wu R, Komura H, Zhou M,
Dong W, Wang Z, Higuchi S, Chaung W, Blau SA, et al: Milk fat
globule epidermal growth factor 8 attenuates acute lung injury in
mice after intestinal ischemia and reperfusion. Am J Respir Crit
Care Med. 181:238–246. 2010. View Article : Google Scholar :
|
|
10
|
Wagner JG and Roth RA: Neutrophil
migration mechanisms, with an emphasis on the pulmonary
vasculature. Pharmacol Rev. 52:349–374. 2000.PubMed/NCBI
|
|
11
|
Lee WL and Downey GP: Neutrophil
activation and acute lung injury. Curr Opin Crit Care. 7:1–7. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zarbock A and Ley K: Mechanisms and
consequences of neutrophil interaction with the endothelium. Am J
Pathol. 172:1–7. 2008. View Article : Google Scholar :
|
|
13
|
Donnelly SC, Strieter RM, Kunkel SL, Walz
A, Robertson CR, Carter DC, Grant IS, Pollok AJ and Haslett C:
Interleukin-8 and development of adult respiratory distress
syndrome in at-risk patient groups. Lancet. 341:643–647. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Goodman RB, Strieter RM, Martin DP,
Steinberg KP, Milberg JA, Maunder RJ, Kunkel SL, Walz A, Hudson LD
and Martin TR: Inflammatory cytokines in patients with persistence
of the acute respiratory distress syndrome. Am J Respir Crit Care
Med. 154:602–611. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Olson TS and Ley K: Chemokines and
chemokine receptors in leukocyte trafficking. Am J Physiol Regul
Integr Comp Physiol. 283:R7–R28. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reutershan J, Morris MA, Burcin TL, Smith
DF, Chang D, Saprito MS and Ley K: Critical role of endothelial
CXCR2 in LPS-induced neutrophil migration into the lung. J Clin
Invest. 116:695–702. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Murphy PM: Neutrophil receptors for
interleukin-8 and related CXC chemokines. Semin Hematol.
34:311–318. 1997.PubMed/NCBI
|
|
18
|
Johnston RA, Mizgerd JP and Shore SA:
CXCR2 is essential for maximal neutrophil recruitment and
methacholine responsiveness after ozone exposure. Am J Physiol Lung
Cell Mol Physiol. 288:L61–L67. 2005. View Article : Google Scholar
|
|
19
|
Vroon A, Heijnen CJ and Kavelaars A: GRKs
and arrestins: Regulators of migration and inflammation. J Leukoc
Biol. 80:1214–1221. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chuang TT, Iacovelli L, Sallese M and De
Blasi A: G protein- coupled receptors: Heterologous regulation of
homologous desensitization and its implications. Trends Pharmacol
Sci. 17:416–421. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Aragay AM, Ruiz-Gómez A, Penela P, Sarnago
S, Elorza A, Jiménez-Sainz MC and Mayor F Jr: G protein-coupled
receptor kinase 2 (GRK2): mechanisms of regulation and
physiological functions. FEBS Lett. 430:37–40. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Penn RB, Pronin AN and Benovic JL:
Regulation of G protein-coupled receptor kinases. Trends Cardiovasc
Med. 10:81–89. 2000. View Article : Google Scholar
|
|
23
|
Penela P, Ribas C and Mayor F Jr:
Mechanisms of regulation of the expression and function of G
protein-coupled receptor kinases. Cell Signal. 15:973–981. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pitcher JA, Tesmer JJ, Freeman JL, Capel
WD, Stone WC and Lefkowitz RJ: Feedback inhibition of G
protein-coupled receptor kinase 2 (GRK2) activity by extracellular
signal-regulated kinases. J Biol Chem. 274:34531–34534. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Liu X, Ma B, Malik AB, Tang H, Yang T, Sun
B, Wang G, Minshall RD, Li Y, Zhao Y, et al: Bidirectional
regulation of neutrophil migration by mitogen-activated protein
kinases. Nat Immunol. 13:457–464. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hanayama R, Tanaka M, Miwa K, Shinohara A,
Iwamatsu A and Nagata S: Identification of a factor that links
apoptotic cells to phagocytes. Nature. 417:182–187. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hanayama R, Tanaka M, Miyasaka K, Aozasa
K, Koike M, Uchiyama Y and Nagata S: Autoimmune disease and
impaired uptake of apoptotic cells in MFG-E8-deficient mice.
Science. 304:1147–1150. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ensslin MA and Shur BD: Identification of
mouse sperm SED1, a bimotif EGF repeat and discoidin-domain protein
involved in sperm-egg binding. Cell. 114:405–417. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hanayama R and Nagata S: Impaired
involution of mammary glands in the absence of milk fat globule EGF
factor 8. Proc Natl Acad Sci USA. 102:16886–16891. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ensslin MA and Shur BD: The EGF repeat and
discoidin domain protein, SED1/MFG-E8, is required for mammary
gland branching morphogenesis. Proc Natl Acad Sci USA.
104:2715–2720. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Aziz M, Jacob A, Matsuda A, Wu R, Zhou M,
Dong W, Yang W-L and Wang P: Pre-treatment of recombinant mouse
MFG-E8 downregulates LPS-induced TNF-α production in macrophages
via STAT3-mediated SOCS3 activation. PLoS One. 6:e276852011.
View Article : Google Scholar
|
|
32
|
Brissette MJ, Lepage S, Lamonde AS, Sirois
I, Groleau J, Laurin LP and Cailhier JF: MFG-E8 released by
apoptotic endothelial cells triggers anti-inflammatory macrophage
reprogramming. PLoS One. 7:e363682012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Matsuda A, Jacob A, Wu R, Zhou M, Nicastro
JM, Coppa GF and Wang P: Milk fat globule-EGF factor VIII in sepsis
and ischemia-reperfusion injury. Mol Med. 17:126–133. 2011.
View Article : Google Scholar :
|
|
34
|
Nuzzi PA, Lokuta MA and Huttenlocher A:
Analysis of neutrophil chemotaxis. Methods Mol Biol. 370:23–36.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Qiang X, Li J, Wu R, Ji Y, Chaung W, Dong
W and Wang P: Expression and characterization of recombinant human
milk fat globule-EGF factor VIII. Int J Mol Med. 28:1071–1076.
2011.PubMed/NCBI
|
|
36
|
Schneider CA, Rasband WS and Eliceiri KW:
NIH Image to ImageJ: 25 years of image analysis. Nat Methods.
9:671–675. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Belperio JA, Keane MP, Burdick MD, Londhe
V, Xue YY, Li K, Phillips RJ and Strieter RM: Critical role for
CXCR2 and CXCR2 ligands during the pathogenesis of
ventilator-induced lung injury. J Clin Invest. 110:1703–1716. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Matsuda A, Wu R, Jacob A, Komura H, Zhou
M, Wang Z, Aziz MM and Wang P: Protective effect of milk fat
globule-epidermal growth factor-factor VIII after renal
ischemia-reperfusion injury in mice. Crit Care Med. 39:2039–2047.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Brooks PC, Clark RA and Cheresh DA:
Requirement of vascular integrin alpha v beta 3 for angiogenesis.
Science. 264:569–571. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cheyuo C, Jacob A, Wu R, Zhou M, Qi L,
Dong W, Ji Y, Chaung WW, Wang H, Nicastro J, et al: Recombinant
human MFG-E8 attenuates cerebral ischemic injury: Its role in
anti-inflammation and anti-apoptosis. Neuropharmacology.
62:890–900. 2012. View Article : Google Scholar :
|
|
41
|
Aziz MM, Ishihara S, Mishima Y, Oshima N,
Moriyama I, Yuki T, Kadowaki Y, Rumi MA, Amano Y and Kinoshita Y:
MFG-E8 attenuates intestinal inflammation in murine experimental
colitis by modulating osteopontin-dependent alphavbeta3 integrin
signaling. J Immunol. 182:7222–7232. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Bu HF, Zuo XL, Wang X, Ensslin MA, Koti V,
Hsueh W, Raymond AS, Shur BD and Tan XD: Milk fat globule-EGF
factor 8/lactadherin plays a crucial role in maintenance and repair
of murine intestinal epithelium. J Clin Invest. 117:3673–3683.
2007.PubMed/NCBI
|
|
43
|
Jinushi M, Nakazaki Y, Carrasco DR,
Draganov D, Souders N, Johnson M, Mihm MC and Dranoff G: Milk fat
globule EGF-8 promotes melanoma progression through coordinated Akt
and twist signaling in the tumor microenvironment. Cancer Res.
68:8889–8898. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Raymond A, Ensslin MA and Shur BD:
SED1/MFG-E8: A bi-motif protein that orchestrates diverse cellular
interactions. J Cell Biochem. 106:957–966. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Alves-Filho JC, Sônego F, Souto FO,
Freitas A, Verri WA Jr, Auxiliadora-Martins M, Basile-Filho A,
McKenzie AN, Xu D, Cunha FQ, et al: Interleukin-33 attenuates
sepsis by enhancing neutrophil influx to the site of infection. Nat
Med. 16:708–712. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Oda K, Matsuoka Y, Funahashi A and Kitano
H: A comprehensive pathway map of epidermal growth factor receptor
signaling. Mol Syst Biol. 1:00102005. View Article : Google Scholar
|
|
47
|
Reynolds CM, Eguchi S, Frank GD and Motley
ED: Signaling mechanisms of heparin-binding epidermal growth
factor-like growth factor in vascular smooth muscle cells.
Hypertension. 39:525–529. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liu ZJ, Xiao M, Balint K, Smalley KS,
Brafford P, Qiu R, Pinnix CC, Li X and Herlyn M: Notch1 signaling
promotes primary melanoma progression by activating
mitogen-activated protein kinase/phosphatidylinositol 3-kinase-Akt
pathways and up-regulating N-cadherin expression. Cancer Res.
66:4182–4190. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Goruppi S, Ruaro E and Schneider C: Gas6,
the ligand of Axl tyrosine kinase receptor, has mitogenic and
survival activities for serum starved NIH3T3 fibroblasts. Oncogene.
12:471–480. 1996.PubMed/NCBI
|
|
50
|
Hidai C, Zupancic T, Penta K, Mikhail A,
Kawana M, Quertermous EE, Aoka Y, Fukagawa M, Matsui Y, Platika D,
et al: Cloning and characterization of developmental endothelial
locus-1: an embryonic endothelial cell protein that binds the
alphavbeta3 integrin receptor. Genes Dev. 12:21–33. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Hanayama R, Tanaka M, Miwa K and Nagata S:
Expression of developmental endothelial locus-1 in a subset of
macrophages for engulfment of apoptotic cells. J Immunol.
172:3876–3882. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Choi EY, Chavakis E, Czabanka MA, Langer
HF, Fraemohs L, Economopoulou M, Kundu RK, Orlandi A, Zheng YY,
Prieto DA, et al: Del-1, an endogenous leukocyte-endothelial
adhesion inhibitor, limits inflammatory cell recruitment. Science.
322:1101–1104. 2008. View Article : Google Scholar : PubMed/NCBI
|