Introduction
The O-linked β-N-acetylglucosamine (O-GlcNAc)
modification (O-GlcNAcylation) of cytoplasmic and nuclear proteins
has been shown to have important regulatory implications on the
pathophysiology of cardiovascular disorders (1,2).
O-GlcNAcylation is catalyzed by the enzyme, O-GlcNAc transferase
(OGT), which is responsible for the addition of O-GlcNAc using
substrate UDP-N-acetylglucosamine (UDP-GlcNAc), which is reversed
by the complementary O-GlcNAcase (OGA) for the hydrolytic cleavage
of O-GlcNAc (3). O-GlcNAcylation
modifies the same or neighbouring serine/threonine sites as
phosphorylation. A complex and extensive cross-talk exists between
O-GlcNAcylation and phosphorylation. OGT and OGA form transient
complexes with kinases and phosphatases (4). Moreover, globally increased
O-GlcNAcylation is known to influence the phosphorylation dynamics
in a site-specific manner, resulting in either a reduction or
augmentation of phosphorylation (5).
The post-translational modification by
O-GlcNAcylation acts as a critical regulatory mechanism for
numerous biological processes involving a range of target proteins,
including kinases, phosphatases, transcriptional factors and signal
transduction molecules (1,4,6).
Studies have demonstrated that the acute augmentation of O-GlcNAc
levels improves the tolerance to stress stimuli (7), confers cardioprotective effects
against ischemia-reperfusion (IR) injury (8), blunts the cardiomyocyte nuclear
factor of activated T-cells (NFAT)-induced changes during
hypertrophy (9) and attenuates
vascular inflammation (10).
Conversely, a sustained increase in O-GlcNAcylation contributes to
the development of vascular and cardiac dysfunction (1,2).
Its ability to regulate reactive oxygen species (ROS) generation,
calcium homeostasis (7,11,12), nuclear factor-κB (NF-κB)
activation, the production of inflammatory cytokines (13,14) and intracellular signaling
pathways, such as mitogen-activated protein kinases (MAPKs) and
NFAT (1,9) may underlie the protective or
negative effects of O-GlcNAcylation on the cardiovascular
system.
Obstructive sleep apnea (OSA) has been identified as
a risk factor for cardiovascular diseases, including myocardial
infarction, hypertension and stroke, resulting from oxidative
stress and augmented inflammatory responses caused by a recurrent
decrease in arterial oxygen [intermittent hypoxia (IH)] (15). Animal subjected to long-term IH
have been shown to exhibit myocardial apoptosis, cardiac
hypertrophy, pathological remodeling and an impairment in global
cardiac function (16,17). Post-translational modifications,
particularly phosphorylation, are important in mediating the
OSA-associated pathological manifestations by functionally
regulating a subset of proteins involved in signaling pathways,
transcriptional factor activation, neurotransmitter synthesis and
cardioprotection in rodents (18). Members of the MAPK family,
primarily extracellular signal-regulated kinase 1/2 (ERK1/2) and
p38 MAPK participate in cardiovascular pathophysiology processes
related to OSA by regulating the systemic inflammation and redox
status (16,19). Given the resemblance between the
periodic reoxygenation in IH and IR injury, during which the
processes of the overproduction of ROS and the enhancement of
inflammatory reactions contribute to organ damage, in study, we
aimed to determine whether protein O-GlcNAc levels are related to
the impairment in left ventricular (LV) function and the associated
pathological hypertrophy in the heart in a rat model prone to the
development of hypertension (20).
Materials and methods
IH procedures
Male Wistar rats (weighing 200–220 g, 8 weeks old)
were provided by the Experimental Animal Center of Tongji Hospital,
Huazhong University of Science and Technology, Wuhan, China. The
rats were housed under specific pathogen-free (SPF) conditions on a
12-h light-dark cycle with free access to regular chow and water.
All protocols were approved by the Animal Care and Use Committee of
Huazhong University of Science and Technology.
The normoxia and IH protocols were performed as
described in our previous study with some modifications (20). The rats were randomly assigned and
exposed to either normoxia (21% O2) or IH (alternating
from 2 min 21% O2 to 2 min 6–8% O2) from 8:00
a.m. to 4:00 p.m. for 4 weeks [4 weeks normoxia (CON); 4 weeks IH
(CIH)]. The desired gas profile was achieved by a computerized
system (BioSpherix OxyCycler A84; BioSpherix Ltd., Lacona, NY, USA)
to control the distribution of pure nitrogen or oxygen into the
chambers. Weekly, the rats were weighed, and several rats from each
group were anesthetized with urethane [1.2 g/kg, by intraperitoneal
(i.p.) injection] for arterial blood pressure measurements and
cardiac catheterization. In parallel, rats were anesthetized and 3
arterial blood samples were drawn from the right carotid artery
prior to exposure to IH (baseline, C0), at the most hypoxic portion
of the cycle (I2) and at the peak of the reoxygeneration (C2),
respectively. Arterial blood gas assessments were performed using
an ABL5 blood gas analyzer (Radiometer, Copenhagen, Denmark).
Cardiac catheterization and remodeling
index
Cardiac hemodynamics measurements were conducted
according to previous studies (20,21). Briefly, the rats were anesthetized
with spontaneous respiration. A polyethylene cannula (PE-50) was
introduced into the right carotid artery and advanced into the left
ventricle. The heart rate (HR), LV end-systolic pressure (LVESP)
and LV end-diastolic pressure (LVEDP) were monitored using a
PowerLab/4SP data acquisition system (ML118; AD Instruments,
Medford, MA, USA). The maximal rates of LV systolic and diastolic
pressure (±dp/dt) were calculated. Thereafter, the rats were
sacrificed by exsanguination. The hearts were removed and placed in
ice-cold phosphate-buffered saline (PBS) solution, blotted and
weighed. The heart weight-to-body weight index (HW/BW) and the left
ventricule plus septum weight-to-body weight index (LVW/BW) were
calculated to measure the degree of LV hypertrophy. The LV
lateral-mid free wall was fixed in 4% formaldehyde and embedded in
paraffin. A portion of the left ventricle was immediately frozen in
liquid nitrogen and stored at −80°C.
Histological analysis
LV free wall sections (5-µm-thick) were
stained with hematoxylin and eosin (H&E) or Masson’s trichrome.
The cardiomyocyte diameter and myocardial interstitial collagen
content were quantitatively analyzed using Image-Pro Plus 7.0
software (IPP; Media Cybernetics, Silver Spring, MD, USA) as
previously described (22,23).
Briefly, for the cardiomyocyte diameter assessment, 100 cells with
a round shape and central nuclei randomly selected from 5 regions
of each slide (×200 magnification) were analyzed. The total
myocardial interstitial collagen area, excluding the perivascular
collagen was quantitatively determined in Masson’s trichromestained
sections (×200 magnification).
Terminal deoxynucleotidyl
transferase-mediated dUTP nick-end labeling (TUNEL) assay
Cardiac apoptosis was determined by TUNEL assay
using the In Situ Cell Death Detection kit (Roche, Mannheim,
Germany) following the instructions provided by the manufacturer.
Briefly, following deparaffinization and rehydration, the sections
were treated with proteinase K (20 µg/ml) for 15 min. The
slides were then immersed in TUNEL reaction mixture at 37°C for 1 h
in a humidified atmosphere in the dark. A total of 10 random fields
per tissue section were captured at ×200 magnification (BX51
microscope; Olympus, Tokyo, Japan) and analyzed using IPP software.
The apoptotic index (%) was calculated as the ratio of the number
of TUNEL-positive nuclei divided by the total number of nuclei.
Immunohistochemistry (IHC)
IHC was carried out on the paraffin-embedded LV
sections (5-µm-thick) using a 3,3′-diaminobenzidine (DAB)
kit (Dako, Copenhagen, Denmark) following the manufacturer’s
instructions. Briefly, the sections were deparaffinized, blocked
with 3% hydrogen peroxide, treated with 3% bovine serum albumin
(BSA) in PBS (pH 7.4), and incubated with the primary antibodies
specific for caspase-3 (1:50), NF-κB p65 (1:100), OGA (1:100) or
OGT (1:100) at 4°C overnight. The slides were washed and incubated
with horseradish peroxidase (HRP)-conjugated secondary antibody
(1:200) for 1 h at room temperature. The slides were then washed,
developed with DAB and counter-stained with hematoxylin. The slides
were visualized and captured at ×200 magnification (BX51;
Olympus).
Preparation of tissue homogenates
The heart samples were homogenized in ice-cold RIPA
buffer containing protease inhibitor cocktail and phosphatase
inhibitor cocktail (both from Roche). The homogenate was then
centrifuged at 12,000 x g for 15 min at 4°C. The protein
concentration in the supernatant was determined using the Enhanced
BCA Protein assay kit (from the Beyotime Institute of
Biotechnology, Jiangsu, China). The supernatant was then stored in
aliquots at −80°C for further analysis.
Measurements of myocardial tumor necrosis
factor-α (TNF-α) and interleukin-6 (IL-6)
The TNF-α and IL-6 content in the supernatant was
measured using quantikine enzyme-linked immunosorbent assay (ELISA)
kits (R&D Systems, Minneapolis, MN, USA) following the
manufacturer’s instructions. The heart supernatant was added to the
respective wells in duplicate followed by incubation with the
detection antibody supplemented with substrate and stop solution.
The optical density (OD) of the colored product was determined
using a spectrophotometer at 450 nm (Microplate Reader, Model 3550;
Bio-Rad Laboratories, Hercules, CA, USA). Cytokine amounts were
normalized to the protein content and expressed as
picograms/milligram protein (pg/mg).
Western blot anlalysis
Equal amounts of protein (40–60 µg) were
separated on 10% sodium dodecyl sulfate-polyacrylamide gel
electrophoresis (SDS-PAGE) and transferred onto polyvinylidene
difluoride membranes (PVDF, 0.45 µm; Millipore, Billerica,
MA, USA). The membranes were blocked in 5% non-fat milk or BSA in
0.05% Tween-20/Tris-buffered saline (TBST) for 1 h at room
temperature and incubated with primary antibodies specific for
O-GlcNAc (1:800), OGT (1:1,000), OGA (1:400), calcium/calmodulin
dependent protein kinase II (CaMKII) (1:800), phosphorylated
(p-)CaMKII (1:800), ERK1/2 (1:1,000), p-ERK1/2 (1:1,000), p38 MAPK
(1:1,000), p-p38 MAPK (1:1,000) and glyceraldehyde 3-phosphate
dehydrogenase (GAPDH) (1:3,000) at 4°C overnight. After washing,
the membranes were incubated with HRP-conjugated secondary
antibodies (1:4,000) for 1 h at room temperature. The bands were
visualized using an enhanced chemiluminescence (ECL) detection kit
(Advansta Corp., Menlo Park, CA, USA) and quantified by AlphaEaseFC
software (Alpha Innotech, San Leandro, CA, USA).
Reagents and antibodies
The antibodies for O-GlcNAc (CTD110.6; #sc-59623;
mouse monoclonal antibody), NF-κB p65 (sc-109; rabbit polyclonal
antibody), CaMKII (M-176; #sc-9035; rabbit polyclonal antibody) and
p-CaMKII (22B1; #sc-32289; mouse monoclonal antibody), as well as
secondary anti-mouse and anti-rabbit IgG were obtained from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Antibodies against
OGA (#14711-1-AP; rabbit polyclonal antibody) and OGT (#11576-2-AP;
rabbit polyclonal antibody) were from Proteintech (Proteintech
Group Inc., Chicago, IL, USA). Antibodies for p38 MAPK (#9212,
rabbit polyclonal antibody), p-p38 MAPK (#4511, rabbit monoclonal
antibody), ERK1/2 (#4695; rabbit monoclonal antibody), p-ERK1/2
(Thr202/Tyr204) (#4376; rabbit monoclonal antibody), caspase-3
(#9662; rabbit polyclonal antibody), and GAPDH (#5174; rabbit
monoclonal antibody) were from Cell Signaling Technology (CST,
Beverly, MA, USA). Most high quality reagents were from
Sigma-Aldrich (St. Louis, MO, USA).
Statistical analysis
Statistical analyses were performed using PASW 18.0
software (SPSS Inc., Chicago, IL, USA). Data are presented as the
means ± standard deviation (SD). Statistical analysis was performed
by one-way analysis of variance (ANOVA), followed by post hoc
comparisons [least significant difference (LSD)] or by a Student’s
t-test where appropriate. A value of P<0.05 was considered to
indicate a statistically significant difference.
Results
Changes in arterial blood gas levels,
blood pressure and HR
Over the course of exposure to IH consisting of 2
min of hypoxia followed by 2 min of re-oxygenation, arterial blood
gases withdrawn during the most hypoxic portion of the cycle showed
a nadir hypoxia within the range of severe OSA, which recovered to
the baseline PaO2 during the air flush (Fig. 1A). Baseline blood pressure
parameters [mean arterial blood pressure (MABP), systolic blood
pressure (SBP), diastolic blood pressure (DBP)] and HR were similar
between the 2 groups. Exposure to IH for 2 weeks evoked a
significant increase in MABP, SBP and DBP, which remained elevated
until the end of the 4-week duration of exposure to IH. However,
exposure to normoxia failed to cause any significant changes in
blood pressure parameters with time. However, neither exposure to
normoxia nor IH markedly altered the rats HR during the same
observation period (Fig. 1B).
Changes in cardiac architecture
The rats in the IH group gained less body weight
than the rats in the normoxia group during the 4-week period of
exposure. The ratio of HW/BW and LVW/BW and the indexes of systemic
hypertension-induced hypertrophy were significantly higher in the
rats in the IH group. The LV tissue sections of the rats in the IH
group stained with H&E showed a gradually abnormal myocardial
architecture, as evidenced by an increase in cardiomyocyte
diameter, cardiomyocyte disarray and structural disorganization. No
signigicant changes in the interstitial collagen deposition were
observed (Fig. 2 and Table I).
 | Table IChanges in body weight, heart weight
and left ventricular function during the 4 weeks of exposure to
IH. |
Table I
Changes in body weight, heart weight
and left ventricular function during the 4 weeks of exposure to
IH.
| | Week 0 | Week 1 | Week 2 | Week 3 | Week 4 |
|---|
| BW (g) | Sham | 211±7 (24) | 261±17 (24)a | 312±23 (24)a | 327±17 (24)a | 368±30 (24)a |
| IH | 208±11 (24) | 243±11 (24)a,b | 269±18 (24)a,b | 289±57 (24)a,b | 335±51 (24)a,b |
| HW/BW | Sham | 2.75±0.14 (6) | 2.77±0.19 (6) | 2.77±0.24 (6) | 2.79±0.28 (6) | 2.73±0.19 (21) |
|
(×103) | IH | 2.73±0.10 (6) | 2.81±0.38 (6) | 2.93±0.28 (6) | 3.00±0.14 (6)a | 3.08±0.21 (20)a,b |
| LV/BW | Sham | 2.17±0.12 (6) | 2.14±0.20 (6) | 2.19±0.19 (6) | 2.19±0.22 (6) | 2.19±0.14 (21) |
|
(×103) | IH | 2.16±0.09 (6) | 2.20±0.34 (6) | 2.29±0.21 (6) | 2.35±0.10 (6)† | 2.45±0.19 (20)a,b |
| LVESP | Sham | 108.2±9.7 (6) | 111.8±11.0
(6) | 114.0±9.5 (6) | 119.3±16.1
(6) | 118.5±21.9
(13) |
| (mmHg) | IH | 111.0±10.6
(6) | 115.6±9.6 (6) | 130.5±9.8 (6)a,b | 147.9±11.8
(7)a,b | 139.6±6.9 (8)a,b |
| LVEDP | Sham | 6.5±3.1 (6) | 6.2±2.8 (6) | 7.2±3.5 (6) | 7.2±3.3 (6) | 6.9±4.0 (13) |
| (mmHg) | IH | 6.7±2.6 (6) | 7.3±3.0 (6) | 7.3±2.6 (6) | 8.4±2.5 (7) | 14.5±2.0 (8)a,b |
| +dP/dt | Sham | 6709±960 (6) | 6850±1042 (6) | 6636±932 (6) | 7065±1070 (6) | 6993±1037 (13) |
| (mmHg/sec) | IH | 6865±1255 (6) | 7089±982 (6) | 7739±868 (6)b | 8255±442 (7)a,b | 5807±729 (8)a,b |
| −dP/dt | Sham | 7645±864 (6) | 7777±1002 (6) | 7827±902 (6) | 7903±962 (6) | 8245±1127 (13) |
| (mmHg/sec) | IH | 7809±867 (6) | 8132±1016 (6) | 8603±961 (6) | 8693±815 (7) | 6233±816 (8)a,b |
Changes in LV systolic and diastolic
function
Of note, compared to the rats in the normoxia group,
the rats exposed to 2 or 3 weeks of IH presented with an augmented
LVESP, +dP/dt and −dP/dt, indicative of improved myocardial
performance as an outcome of exposure to IH, which then declined
from week 4. LVEDP was gradually increased by exposure to IH, with
a significant enhancement at week 4. There were no significant
differences observed in any of these indexes over time for the rats
in the normoxia group (Table
I).
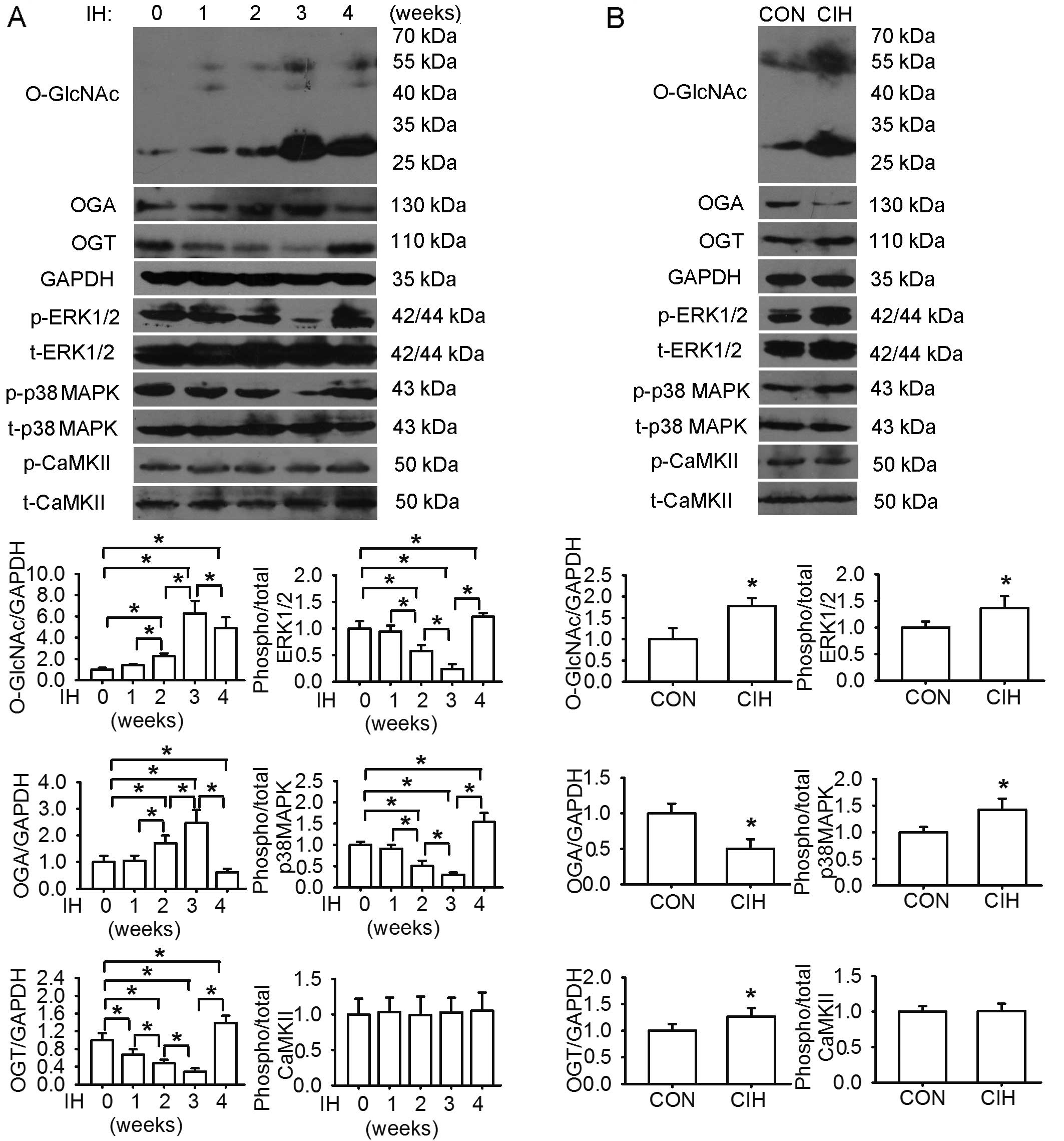
Cardiomyocyte apoptosis and dynamic
changes in protein levels of O-GlcNAc, OGA, OGT, ERK1/2 and p38
MAPK
The protein levels of O-GlcNAc in the LV tissues
steadily increased following exposure to IH, reaching peak levels
at week 3. There was a similar change observed in the OGA levels.
The OGT, ERK1/2 and p38 MAPK phosphorylation levels were affected
in an opposite manner. The levels of phosphorylated CaMKII remained
almost unaltered during the same observation period. In parallel,
compared with exposure to normoxia, 4 weeks of exposure to IH
augmented the O-GlcNAc protein, OGT, phosphorylated ERK1/2 and p38
MAPK levels, accompanied by a decrease in OGA levels and an
increase in cardiomyocyte apoptosis, as well as an increase in the
levels of caspase-3, NF-κB p65, TNF-α and IL-6 in the LV tissues
(Figs. 3 and 4).
Discussion
In the present study, we found that exposure to IH
induced a significant increase in rat blood pressure from weeks 2
to 4 of exposure, associated with a gradually abnormal myocardial
architecture. Compared to the rats in the normoxia group, the rats
exposed to IH for 2 or 3 weeks demonstrated an improved LV systolic
and diastolic function, which then declined from week 4.
Consistently, the protein levels of O-GlcNAc and OGA in the LV
tissues steadily increased following exposure to IH, reaching peak
levels at week 3. However, the OGT levels and the phosphorylation
levels of ERK1/2 and p38 MAPK were affected in an opposite manner.
At week 4, the CIH-induced increase in the protein levels of
O-GlcNAc and the phosphorylation levels of MAPKs was accompanied by
cardiac inflammation, as evidenced by increased cardiomyocyte
apoptosis and the cardiac content of NF-κB, inflammatory cytokines
and caspase-3. Thus, these findings indicate the possible
involvement of protein O-GlcNAc and MAPK signaling in mediating
alterations in LV function and cardiac damage during IH.
During the course of the 4 week of exposure to IH,
the LV systolic and diastolic capability first increased and then
decreased, consistent with previous reports that short-term
exposure to IH may exert cardioprotective effects, whereas
prolonged exposure to IH appears to exert deleterious effects
(24–26). The IH-induced cardiac hypertrophy
may exist at first in a compensatory state to augment LV
contractility and maintain cardiac output, and then progress to a
decompensated state, ultimately evolving to dilated cardiac
hypertrophy with LV dysfunction and remodeling (16,27,28). Our findings are in agreement with
previous results, indicating that IH protects the isolated heart
from reperfusion injury (29) and
induces adaptive responses to improve cardiac function during the
progression of heart failure (26). Naghshin et al (25,26) demonstrated that 4 weeks of
exposure to IH induced beneficial cardiac adaptive responses to
hypoxia in mice. However, other studies have demonstrated a decline
in cardiac function in rats exposed to IH for 4 weeks or longer
(16,17,24,28,30), which is consistent with our
findings. Thus, there may be a very narrow window of around 3 to 4
weeks of exposure to IH in rodent models during which a transition
from physiological adaptations to pathological cardiovascular
outcomes occurs.
Intriguingly, in this study, alterations in LV
function coincided with the dynamic changes in the protein levels
of O-GlcNAc, suggesting a potential mediating role of
O-GlcNAcylation. Paradoxically, the expression level of OGT, which
catalyzes O-GlcNAc formation, was affected in an opposite manner.
However, the expression of OGA, which catalyzes O-GlcNAc removal,
was altered in a manner similar to that of O-GlcNAc. In addition to
the notion that O-GlcNAc, OGA and OGT levels change with age, other
contributing factors, such as changes in enzymatic activity and
UDP-GlcNAc concentrations should also be taken into consideration
(31). Further studies are
warranted to clarify the potential role of these factors in the
regulation of cardiac O-GlcNAc levels under IH conditions. Previous
studies have demonstrated that an acute increase in O-GlcNAc levels
may exert protective effects on the cardiovascular system, whereas
a persistent increase may exert adverse effects on the chronic
disease status (1,32). In accordance with this, in our
study, the protein levels of O-GlcNAc increased rapidly during the
first 3 weeks of exposure to IH, contributing to the maintenance of
LV function in response to an elevated LV afterload as evidenced by
an increase in blood pressure. The sustained increase in O-GlcNAc
levels induced by further exposure to IH may lead to the
pathogenesis of LV dysfunction and cardiac remodeling. The
differential effects of an acute versus a sustained increase in
O-GlcNAc levels on cardiac function are possibly due to the
modifications of different signaling molecules (1).
Although to the best of our knowledge, the
association between O-GlcNAcylation and IH has not yet been
investigated to date, the contributing role of O-GlcNAc
modification in cardiovascular dysfunction is increasingly
recognized (1,2,33).
Studies have revealed that an elevated O-GlcNAc modification is
linked to the adverse effects of diabetes on the heart by
disrupting the cardiomyocyte hypertrophic signaling pathway and
altering mitochondrial and mechanical function. Reducing O-GlcNAc
levels can restore hypertrophic signaling in a rodent model of
diabetes (1,32–34). In addition, an increase in
O-GlcNAc levels may contribute to the development and progression
of pressure-overload or NFAT-associated heart hypertrophy and
infarct-induced heart failure. The properties of O-GlcNAcylation to
modulate signal transduction involved in redox, inflammatory and
apoptotic processes have also been implicated in the dysregulation
of the cardiovascular system (1,2,7,32,33). Accordingly, our finding that CIH
increased both O-GlcNAc protein levels and the cardiac content of
inflammatory cytokines together with an enhanced cardiomyocyte
apoptosis suggests an association between O-GlcNAcyaltion and
inflammatory signaling under IH conditions.
The ERK1/2 and p38 MAPK signaling cascades
participate in the cardiac remodeling process and in the
progression of cardiac dysfunction induced by IH. Béguin et
al (29) demonstrated that
acute IH (AIH) enhanced both ERK1/2 and p38 MAPK phosphorylation
levels in the rat myocardium. Inhibitors specific to ERK1/2 and p38
MAPK abolished the AIH-induced cardioprotective effects against
ischemic insults. Cardiac hypertrophy in rats subjected to CIH may
be attributed to increased p38 MAPK activation (16). The phosphorylation of MAPKs may in
turn activate NF-κB, inducing the overproduction of inflammatory
cytokines, consequently triggering inflammatory responses, the
apoptotic program and the expression of hypertrophy-related genes
(16,19). In this study, we found that
cardiac remodeling occurred following exposure to IH, which was
characterized by cardiac hypertrophy, cardiomyocyte disarray,
structural disorganization and increased cardiomyocyte diameter and
apoptosis. During the course of exposure to IH, the phosphorylation
levels of ERK1/2 and p38 MAPK exhibited dynamic changes, which were
almost opposite to those observed in LV function and protein
O-GlcNAc levels. Moreover, the increase in the phosphorylation
levels of p38 MAPK and ERK1/2 by CIH coincided with LV dysfunction
and enhanced cell apoptosis and the increased protein levels of
NF-κB and inflammatory cytokines. Taken together, these results
indicate that the ERK1/2 and p38 MAPK pathways are possibly
involved in the pathological remodeling and impairment of LV
function under IH conditions, which may be related to O-GlcNAc
modification, since there is an interplay between O-GlcNAcylation
and MAPK activation during oxidative stress (1).
CaMKII, the most abundant CaMK in the heart,
functions as a nodal signaling molecule in the regulation of
cardiac physiology and pathology (35). The activation of CaMKII by
phosphorylation has been implicated in cardiac hypertrophy, dilated
cardiomyopathy and heart failure (36). However, data on the status of
CaMKII activity and the associated changes in CaMKII
phosphorylation under IH conditions are controversial, possibly due
to the variations in experimental protocols in the aspect of the
severity of hypoxia, cycle frequency, exposure duration and cell
and tissue types (37–39). Similarly, in this study, we did
not observe any significant changes in the phosphorylation level of
CaMKII during the course of exposure to IH. CaMKII has 4 isozymes
and the antibody used in this study is raised against a synthetic
peptide corresponding to residue 286 of the CaMKII α-subunit, whose
phosphorylation is known to trigger the autonomous activation of
CaMKII. Recently, the oxidation- and O-GlcNAcylation-dependent
CaMKII activation has also been found to have critical regulatory
implications in cardiac pathologies (40,41). Additional mechanisms for CaMKII
activation include the direct modification by nitric oxide (NO) and
the interaction with protein partners. Thus, the extent of CaMKII
activation in the heart may be partly dependent on phosphorylation
(35). It is possible that the
protein abundance does not linearly correlate with enzyme activity
and the isoform-specific effects may also play a role. Further
studies are warranted to determine the activity of CaMKII and to
elucidate the potential pathways of CaMKII activation contributing
to the regulation of pathological cardiac signaling under IH
conditions.
Due to the limitation that the modulation of
O-GlcNAc levels in vivo in pharmacological or genetical
approaches does not result in the desired outcome (33), we failed to investigate the
potential regulatory mechanism of O-GlcNAcylation in IH-related
cardiac pathophysiology by directly modulating O-GlcNAc levels in
our rat model. However, the dynamic changes in the protein levels
of O-GlcNAc, OGA and OGT coincided with alterations in LV function,
suggesting a possible causal link between O-GlcNAcylation and
IH-induced myocardial pathology. Additionally, the extensive
interplay between O-GlcNAcylation and phosphorylation, both of
which may interfere with redox and inflammatory signaling, points
to a need to clarify their regulatory roles in these
pathophysiological processes in response to IH. The availability of
specific antibodies for O-GlcNAcylated proteins, which can identify
the O-GlcNAc-modified sites, would provide new insight into the
fundamental mechanisms of O-GlcNAc protein in the modulation of
IH-associated cardiac function in future studies. Consequently,
O-GlcNAcylation may emerge as a potential therapeutic target in the
treatment of OSA-induced cardiac diseases.
In conclusion, the findings from the present study
suggested that short-term exposure to IH led to an improvement in
cardiac performance; however, a longer period of exposure to IH
caused detrimental outcomes. Protein O-GlcNAc levels and cellular
signaling involving ERK1/2 and p38 MAPK may act as potential
regulators in IH-associated cardiac remodeling and changes in LV
function.
Acknowledgments
We would like to thank Professor Qinghua Hu
(Department of Pathophysiology, Huazhong University of Science and
Technology) for the use of the CO pod system and the PowerLab data
acquisition system. This study was supported by grants (nos.
81070067 and 81370185) from the National Natural Science Foundation
of China (NSFC).
References
|
1
|
Lima VV, Spitler K, Choi H, Webb RC and
Tostes RC: O-GlcNAcylation and oxidation of proteins: Is signalling
in the cardiovascular system becoming sweeter? Clin Sci (Lond).
123:473–486. 2012. View Article : Google Scholar
|
|
2
|
Dassanayaka S and Jones SP: O-GlcNAc and
the cardiovascular system. Pharmacol Ther. 142:62–71. 2014.
View Article : Google Scholar :
|
|
3
|
Hart GW, Housley MP and Slawson C: Cycling
of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins.
Nature. 446:1017–1022. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hart GW, Slawson C, Ramirez-Correa G and
Lagerlof O: Cross talk between O-GlcNAcylation and phosphorylation:
Roles in signaling, transcription, and chronic disease. Annu Rev
Biochem. 80:825–858. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang Z, Gucek M and Hart GW: Cross-talk
between GlcNAcylation and phosphorylation: Site-specific
phosphorylation dynamics in response to globally elevated O-GlcNAc.
Proc Natl Acad Sci USA. 105:13793–13798. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Vaidyanathan K, Durning S and Wells L:
Functional O-GlcNAc modifications: Implications in molecular
regulation and pathophysiology. Crit Rev Biochem Mol Biol.
49:140–163. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Groves JA, Lee A, Yildirir G and Zachara
NE: Dynamic O-GlcNAcylation and its roles in the cellular stress
response and homeostasis. Cell Stress Chaperones. 18:535–558. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Vibjerg Jensen R, Johnsen J, Buus
Kristiansen S, Zachara NE and Bøtker HE: Ischemic preconditioning
increases myocardial O-GlcNAc glycosylation. Scand Cardiovasc J.
47:168–174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Facundo HT, Brainard RE, Watson LJ, Ngoh
GA, Hamid T, Prabhu SD and Jones SP: O-GlcNAc signaling is
essential for NFAT-mediated transcriptional reprogramming during
cardiomyocyte hypertrophy. Am J Physiol Heart Circ Physiol.
302:H2122–H2130. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hilgers RH, Xing D, Gong K, Chen YF,
Chatham JC and Oparil S: Acute O-GlcNAcylation prevents
inflammation-induced vascular dysfunction. Am J Physiol Heart Circ
Physiol. 303:H513–H522. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ngoh GA, Watson LJ, Facundo HT and Jones
SP: Augmented O-GlcNAc signaling attenuates oxidative stress and
calcium overload in cardiomyocytes. Amino Acids. 40:895–911. 2011.
View Article : Google Scholar :
|
|
12
|
Wu T, Zhou H, Jin Z, Bi S, Yang X, Yi D
and Liu W: Cardio-protection of salidroside from
ischemia/reperfusion injury by increasing N-acetylglucosamine
linkage to cellular proteins. Eur J Pharmacol. 613:93–99. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zou L, Yang S, Champattanachai V, Hu S,
Chaudry IH, Marchase RB and Chatham JC: Glucosamine improves
cardiac function following trauma-hemorrhage by increased protein
O-GlcNAcylation and attenuation of NF-{kappa}B signaling. Am J
Physiol Heart Circ Physiol. 296:H515–H523. 2009. View Article : Google Scholar
|
|
14
|
Yang S, Zou LY, Bounelis P, Chaudry I,
Chatham JC and Marchase RB: Glucosamine administration during
resuscitation improves organ function after trauma hemorrhage.
Shock. 25:600–607. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Fava C, Montagnana M, Favaloro EJ, Guidi
GC and Lippi G: Obstructive sleep apnea syndrome and cardiovascular
diseases. Semin Thromb Hemost. 37:280–297. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chen LM, Kuo WW, Yang JJ, Wang SG, Yeh YL,
Tsai FJ, Ho YJ, Chang MH, Huang CY and Lee SD: Eccentric cardiac
hypertrophy was induced by long-term intermittent hypoxia in rats.
Exp Physiol. 92:409–416. 2007. View Article : Google Scholar
|
|
17
|
Chen L, Zhang J, Gan TX, et al: Left
ventricular dysfunction and associated cellular injury in rats
exposed to chronic intermittent hypoxia. J Appl Physiol (1985).
104:218–223. 2008. View Article : Google Scholar
|
|
18
|
Kumar GK and Prabhakar NR:
Post-translational modification of proteins during intermittent
hypoxia. Respir Physiol Neurobiol. 164:272–276. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ryan S, McNicholas WT and Taylor CT: A
critical role for p38 map kinase in NF-kappaB signaling during
intermittent hypoxia/reoxygenation. Biochem Biophys Res Commun.
355:728–733. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo XL, Deng Y, Shang J, Liu K, Xu YJ and
Liu HG: ERK signaling mediates enhanced angiotensin II-induced rat
aortic constriction following chronic intermittent hypoxia. Chin
Med J (Engl). 126:3251–3258. 2013.
|
|
21
|
Chen L, Einbinder E, Zhang Q, Hasday J,
Balke CW and Scharf SM: Oxidative stress and left ventricular
function with chronic intermittent hypoxia in rats. Am J Respir
Crit Care Med. 172:915–920. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lin J, Lopez EF, Jin Y, Van Remmen H,
Bauch T, Han HC and Lindsey ML: Age-related cardiac muscle
sarcopenia: Combining experimental and mathematical modeling to
identify mechanisms. Exp Gerontol. 43:296–306. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yan L, Zhang JD, Wang B, Lv YJ, Jiang H,
Liu GL, Qiao Y, Ren M and Guo XF: Quercetin inhibits left
ventricular hypertrophy in spontaneously hypertensive rats and
inhibits angiotensin II-induced H9C2 cells hypertrophy by enhancing
PPAR-γ expression and suppressing AP-1 activity. PLoS One.
8:e725482013. View Article : Google Scholar
|
|
24
|
Lee SD, Kuo WW, Wu CH, Lin YM, Lin JA, Lu
MC, Yang AL, Liu JY, Wang SG, Liu CJ, et al: Effects of short- and
long-term hypobaric hypoxia on Bcl2 family in rat heart. Int J
Cardiol. 108:376–384. 2006. View Article : Google Scholar
|
|
25
|
Naghshin J, McGaffin KR, Witham WG, et al:
Chronic intermittent hypoxia increases left ventricular
contractility in C57BL/6J mice. J Appl Physiol (1985). 107:787–793.
2009. View Article : Google Scholar
|
|
26
|
Naghshin J, Rodriguez RH, Davis EM, Romano
LC, McGaffin KR and O’Donnell CP: Chronic intermittent hypoxia
exposure improves left ventricular contractility in transgenic mice
with heart failure. J Appl Physiol (1985). 113:791–798. 2012.
View Article : Google Scholar
|
|
27
|
Han Q, Yeung SC, Ip MS and Mak JC:
Cellular mechanisms in intermittent hypoxia-induced cardiac damage
in vivo. J Physiol Biochem. 70:201–213. 2014. View Article : Google Scholar
|
|
28
|
Yin X, Zheng Y, Liu Q, Cai J and Cai L:
Cardiac response to chronic intermittent hypoxia with a transition
from adaptation to maladaptation: The role of hydrogen peroxide.
Oxid Med Cell Longev. 2012:5695202012.PubMed/NCBI
|
|
29
|
Béguin PC, Belaidi E, Godin-Ribuot D, Lévy
P and Ribuot C: Intermittent hypoxia-induced delayed
cardioprotection is mediated by PKC and triggered by p38 MAP kinase
and Erk1/2. J Mol Cell Cardiol. 42:343–351. 2007. View Article : Google Scholar
|
|
30
|
Ding W and Zhang X, Huang H, Ding N, Zhang
S, Hutchinson SZ and Zhang X: Adiponectin protects rat myocardium
against chronic intermittent hypoxia-induced injury via inhibition
of endoplasmic reticulum stress. PLoS One. 9:e945452014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fülöp N, Feng W, Xing D, He K, Not LG,
Brocks CA, Marchase RB, Miller AP and Chatham JC: Aging leads to
increased levels of protein O-linked N-acetylglucosamine in heart,
aorta, brain and skeletal muscle in Brown-Norway rats.
Biogerontology. 9:139–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Darley-Usmar VM, Ball LE and Chatham JC:
Protein O-linked β-N-acetylglucosamine: A novel effector of
cardiomyocyte metabolism and function. J Mol Cell Cardiol.
52:538–549. 2012. View Article : Google Scholar
|
|
33
|
Zachara NE: The roles of O-linked
β-N-acetylglucosamine in cardiovascular physiology and disease. Am
J Physiol Heart Circ Physiol. 302:H1905–H1918. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Marsh SA, Dell’Italia LJ and Chatham JC:
Activation of the hexosamine biosynthesis pathway and protein
O-GlcNAcylation modulate hypertrophic and cell signaling pathways
in cardiomyocytes from diabetic mice. Amino Acids. 40:819–828.
2011. View Article : Google Scholar :
|
|
35
|
Erickson JR: Mechanisms of CaMKII
Activation in the Heart. Front Pharmacol. 5:592014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang T, Maier LS, Dalton ND, Miyamoto S,
Ross J Jr, Bers DM and Brown JH: The deltaC isoform of CaMKII is
activated in cardiac hypertrophy and induces dilated cardiomyopathy
and heart failure. Circ Res. 92:912–919. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Yu Z, Wang ZH and Yang HT:
Calcium/calmodulin-dependent protein kinase II mediates
cardioprotection of intermittent hypoxia against
ischemic-reperfusion-induced cardiac dysfunction. Am J Physiol
Heart Circ Physiol. 297:H735–H742. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yeung HM, Kravtsov GM, Ng KM, Wong TM and
Fung ML: Chronic intermittent hypoxia alters Ca2+
handling in rat cardiomyocytes by augmented
Na+/Ca2+ exchange and ryanodine receptor
activities in ischemia-reperfusion. Am J Physiol Cell Physiol.
292:C2046–C2056. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yuan G, Nanduri J, Bhasker CR, Semenza GL
and Prabhakar NR: Ca2+/calmodulin kinase-dependent
activation of hypoxia inducible factor 1 transcriptional activity
in cells subjected to intermittent hypoxia. J Biol Chem.
280:4321–4328. 2005. View Article : Google Scholar
|
|
40
|
Erickson JR, Pereira L, Wang L, Han G,
Ferguson A, Dao K, Copeland RJ, Despa F, Hart GW, Ripplinger CM and
Bers DM: Diabetic hyperglycaemia activates CaMKII and arrhythmias
by O-linked glycosylation. Nature. 502:372–376. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Erickson JR, Joiner ML, Guan X, Kutschke
W, Yang J, Oddis CV, Bartlett RK, Lowe JS, O’Donnell SE,
Aykin-Burns N, et al: A dynamic pathway for calcium-independent
activation of CaMKII by methionine oxidation. Cell. 133:462–474.
2008. View Article : Google Scholar : PubMed/NCBI
|