Introduction
Dilated cardiomyopathy (DCM), clinically
characterized by ventricular chamber enlargement and an impaired
contraction of the left ventricle or both ventricles, represents
the most prevalent type of heart muscle disease, with an estimated
prevalence of 1 in 250 individuals (1). It is the most common indication for
heart transplantation and a major cause of chronic congestive heart
failure and sudden cardiac death (2–4).
DCM has diverse etiologies with both genetic defects and
environmental risk factors implicated in the pathogenesis of DCM
(1–3,5).
In approximately half of the cases, DCM occurs in the absence of an
identifiable cause, such as coronary artery disease, viral
myocarditis, cardiac valve disease, hypertension, toxic exposure,
alcohol abuse, nutritional deficiency, autoimmune abnormality or
metabolic disorder, and this type of DCM is defined as idiopathic
or primary DCM. However, in 25–50% of cases, the familial
transmission of idiopathic DCM is observed in an autosomal
dominant, recessive, X-linked, or mitochondrial pattern with
variable expressivity and penetrance, hence termed familial DCM,
which is in contrast to sporadic DCM which is not assoicated with a
family history (5,6). Aggregating evidence has indicated
that genetic pathogenic factors play a crucial role in the
pathogenesis of idiopathic DCM, and genetic mutations in >50
genes have been causally linked to idiopathic or familial DCM
(1–3,7–12).
Among these established DCM-associated genes, the majority encode
sarcomeric, cytoskeletal, nucleoskeletal and calcium-handling
proteins (2). Nevertheless, DCM
is of remarkable genetic heterogeneity and the genetic basis
underlying DCM in a significant proportion of patients remains
unclear.
Previous studies have underlined the pivotal roles
of several core cardiac transcription factors in cardiac
development and structural remodeling, including the GATA zinc
finger-containing transcription factor, NK homeodomain
transcription factor and T-box transcription factor families
(13–17), and mutations in GATA binding
protein (GATA)4, GATA5, GATA6, NK2 homeobox (NKX2)-5, NKX2-6 and
T-Box 5 (TBX5) have been associated with various congenital heart
diseases and arrhythmias, including cardiac septal defect,
tetralogy of Fallot, double outlets of the right ventricle and
atrial fibrillation (18–49). Recently, mutations in GATA4,
GATA5, GATA6, NKX2-5 and TBX5 have been associated with familial
DCM (50–56). Additionally, mutated GATA4 has
also been shown to be involved in sporadic DCM (57). However, the prevalence and
spectrum of TBX5 mutations in patients with sporadic DCM remain to
be investigated.
Subjects and methods
Study subjects
A cohort of 146 unrelated patients with sporadic DCM
was recruited from the Han Chinese population. The controls
consisted of 200 unrelated healthy individuals, who were matched to
the patients with DCM in ethnicity and gender. All study
participants underwent a comprehensive clinical evaluation,
including a report of individual and familial histories, medical
records, physical examination, two-dimensional transthoracic
echocardiography with color flow Doppler, X-ray, standard 12-lead
electrocardiogram and exercise tolerance testing. Cardiac
catheterization, angiography, or cardiac magnetic resonance imaging
was performed only if there was a strong clinical indication. The
diagnosis of idiopathic DCM was made as previously described: a
left ventricular end-diastolic diameter of >27 mm/m2
and an ejection fraction of <40% or fractional shortening
<25% in the absence of abnormal loading conditions, coronary
artery disease, congenital heart lesions and other systemic
diseases (50–53,58,59). The patients with co-existent
conditions that may result in ventricular systolic dysfunction,
such as coronary heart disease, hypertensive heart disease,
valvular heart disease and viral myocarditis, were excluded from
the study. The clinical studies were performed by investigators
blinded to the genotypic results. This study was performed in
conformity with the principles of the Declaration of Helsinki. The
study protocol was reviewed and approved by the Ethics Committee of
Shanghai Eighth People’s Hospital, Shanghai, China. Written
informed consent was obtained from all participants prior to the
enrollment in the study.
Mutational screening of TBX5
A peripheral venous blood sample was prepared from
each study participant, and the genomic DNA was extracted using a
Wizard Genomic DNA Purification kit (Promega Corp., Madison, WI,
USA). The coding exons and flanking introns of the TBX5 gene
were sequenced in the 146 unrelated patients with sporadic DCM. A
total of 200 unrelated healthy control individuals were also
genotyped for TBX5. The primer pairs used to amplify the
coding regions and splicing junctions of TBX5 by polymerase
chain reaction (PCR) were designed as previously described
(56). PCR was carried out using
HotStar Taq DNA Polymerase (Qiagen, Hilden, Germany) on a Veriti
Thermal Cycler (Applied Biosystems, Foster, CA, USA). Both strands
of each PCR product were sequenced using a BigDye®
Terminator v3.1 Cycle Sequencing kit under an ABI PRISM 3130xl DNA
Analyzer (both from Applied Biosystems). The identified TBX5
variant was validated by re-sequencing an independent PCR-generated
amplicon from the same individual, and was queried in the single
nucleotide polymorphism (SNP) database at the National Center for
Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/), the human gene mutation
database (HGMD; http://www.hgmd.org/) and the 1000
Genomes Project (1000 GP) database (http://www.1000genomes.org/) to confirm its
novelty.
Alignment of multiple TBX5 protein
sequences across species
The conservation of an affected amino acid was shown
by using the online MUSCLE program on the NCBI website (http://www.ncbi.nlm.nih.gov/homologene?cmd=Retrieve&dopt=MultipleAlignment&list_uids=160).
Analysis of TBX5 variation in silico
The pathogenic potential of a novel TBX5
sequence variation was predicted by MutationTaster (an online
program at: http://www.mutationtaster.org), which automatically
yielded a probability for the variation to be either a deleterious
mutation or a benign polymorphism, and PolyPhen-2 (an online
program at http://genetics.bwh.harvard.edu/pph2/).
Plasmids and site-targeted
mutagenesis
The recombinant expression plasmid, TBX5-pcDNA3.1,
was constructed as previously described (56). The identified mutation was
introduced into the wild-type TBX5-pcDNA3.1 construct by PCR-based
site-directed mutagenesis using a QuikChange II XL Site-Directed
Mutagenesis kit (Stratagene, La Jolla, CA, USA) and confirmed by
sequencing. The expression vector, GATA4-pSSRa, and the (atrial
natriuretic factor) ANF-luciferase (ANF-luc) reporter, which
contains the 2600-bp 50-flanking region of the ANF gene and
expresses Firefly luciferase, were kindly provided by Dr Ichiro
Shiojima from Chiba University School of Medicine (Chiba,
Japan).
Biological analysis of mutated TBX5
COS-7 cells (a fibroblast-like cell line derived
from monkey kidney tissue) from our cell bank were seeded in
24-well plates and cultured in Dulbecco’s modified Eagle’s medium
supplemented with 10% fetal bovine serum. Cells at approximately
90% confluency were transfected using Lipofectamine™ 2000
transfection reagent (Invitrogen Life Technologies, Carlsbad, CA,
USA). The internal control reporter plasmid pGL4.75 (hRluc/CMV;
Promega Corp.), which expresses the Renilla luciferase, were
used in transient transfection assays to normalize transfection
efficiency. For each transfection, the COS-7 cells were transfected
with 0.4 μg of wild-type or mutant TBX5-pcDNA3.1, 1.0
μg of ANF-luc reporter construct and 0.04 μg of
pGL4.75. For co-transfection experiment, 0.2 μg of wild-type
TBX5-pcDNA3.1 and 0.2 μg of mutant TBX5-pcDNA3.1 were used
in the presence of 0.4 μg of ANF-luc and 0.04 μg of
pGL4.75. For the synergistic activation experiment, 0.4 μg
of wild-type GATA4-pSSRa was added. Firefly and Renilla
luciferase activities were measured with the Dual-Glo Luciferase
Assay system (Promega Corp.) 48 h after transfection. The activity
of the ANF promoter was expressed as the fold activation of
Firefly luciferase relative to Renilla luciferase. Three
independent experiments were conducted at minimum for each
transfection.
Statistical analysis
Continuous variables are presented as the means ± SD
and the Student’s unpaired t-test was used to compare the
continuous variables between 2 groups. Non-continuous and
categorical variables are presented as frequencies or percentages
and were compared using Pearson’s χ2 test or Fisher’s
exact test where appropriate. All statistical analyses were
two-sided and P-values <0.05 were considered to indicate a
statistically significant difference.
Results
Baseline clinical characteristics of the
study population
In this case-control study, 146 unrelated patients
with sporadic DCM were clinically evaluated in contrast to 200
unrelated healthy control individuals. The patients presented with
the typical phenotype of DCM as previously described by Elliott
et al (58). The control
individuals had normal echocardiographic parameters without any
evidence of heart disease. All the study participants stated to
have no family history of DCM, and none of them had known risk
factors or comorbidities for DCM, such as coronary artery disease,
hypertension, viral myocarditis, valvular heart disease, congenital
heart disease, drug abuse or exposure to toxicants. The baseline
clinical characteristics of the study subjects are summarized in
Table I.
 | Table IBaseline clinical characteristics of
the study subjects and control individuals. |
Table I
Baseline clinical characteristics of
the study subjects and control individuals.
| Variables | Patients
(n=146) | Controls
(n=200) | P-value |
|---|
| Age (years) | 54±10 | 55±9 | 0.3309 |
| Male gender
(%) | 72 (49) | 98 (49) | 0.9538 |
| SBP (mmHg) | 119±13 | 126±12 | <0.0001 |
| DBP (mmHg) | 80±8 | 83±7 | 0.0002 |
| HR (bpm) | 87±14 | 75±12 | <0.0001 |
| LVEDD (mm) | 69±7 | 46±6 | <0.0001 |
| LVESD (mm) | 59±7 | 36±6 | <0.0001 |
| LVEF (%) | 38±8 | 63±7 | <0.0001 |
| LVFS (%) | 19±6 | 31±5 | <0.0001 |
| NYHA function class
(%) | | | |
| I | 17 (12) | NA | NA |
| II | 45 (31) | NA | NA |
| III | 62 (43) | NA | NA |
| IV | 21 (14) | NA | NA |
Identification of a novel TBX5
mutation
By sequence analysis of TBX5, a heterozygous
variation was identified in 1 of the 146 unrelated patients with
sporadic DCM, with a mutational prevalence of approximately 0.68%.
Specifically, a transversion of guanine into adenine at nucleotide
position 143 (c.427G>A), predicting the substitution of
threonine for alanine at amino acid 143 (p.A143T), was identified
in a 47-year-old female with DCM. She stated to have not positive
family history of DCM, and had no overt congenital cardiovascular
abnormalities or forelimb malformations. The sequence
electropherograms showing the detected heterozygous TBX5
variation in contrast to its corresponding control sequence are
presented in Fig. 1. The
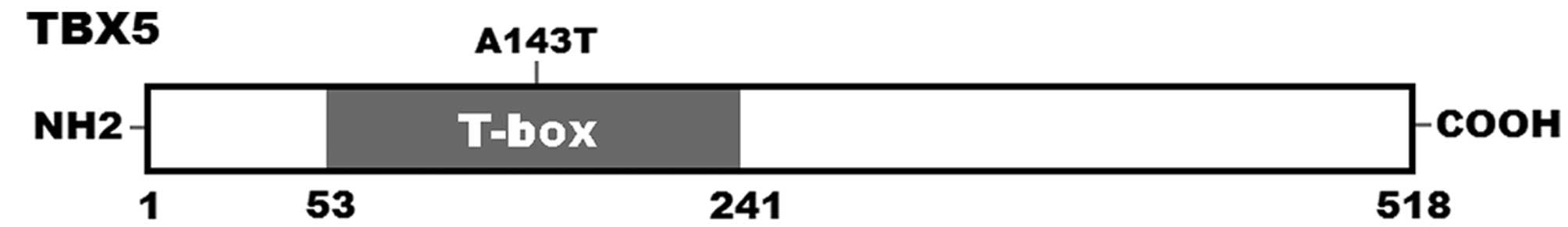
schematic diagram of TBX5 protein showing the T-box structural
domain and the location of the mutation identified in this study is
illustrated in Fig. 2. The
mutation was not observed in the 400 control chromosomes and it was
not found n the SNP, HGM or 1000 GP databases, which were consulted
again on March 19, 2015, suggesting a novel mutation.
Alignment of multiple TBX5 protein
sequences across species
As shown in Fig.
3, a cross-species alignment of multiple TBX5 protein sequences
displayed that the alanine at amino acid position 143 of human TBX5
was completely conserved evolutionarily among species, implying
that the amino acid is functionally important.
Causative potential of a TBX5
variation
The TBX5 sequence variation of c.427G>A
was predicted to be pathogenic by MutationTaster, with a P-value of
0.9999999932. No SNPs in the altered region were reported in the
MutationTaster database. The amino acid substitution of p.A143T was
also predicted to be possibly damaging by another online program,
PolyPhen-2, with a score of 0.994 (sensitivity, 0.46; specificity,
0.96). These results indicate that the TBX5 mutation contributes to
DCM in this mutation carrier.
Reduced transcriptional activity of the
mutant TBX5
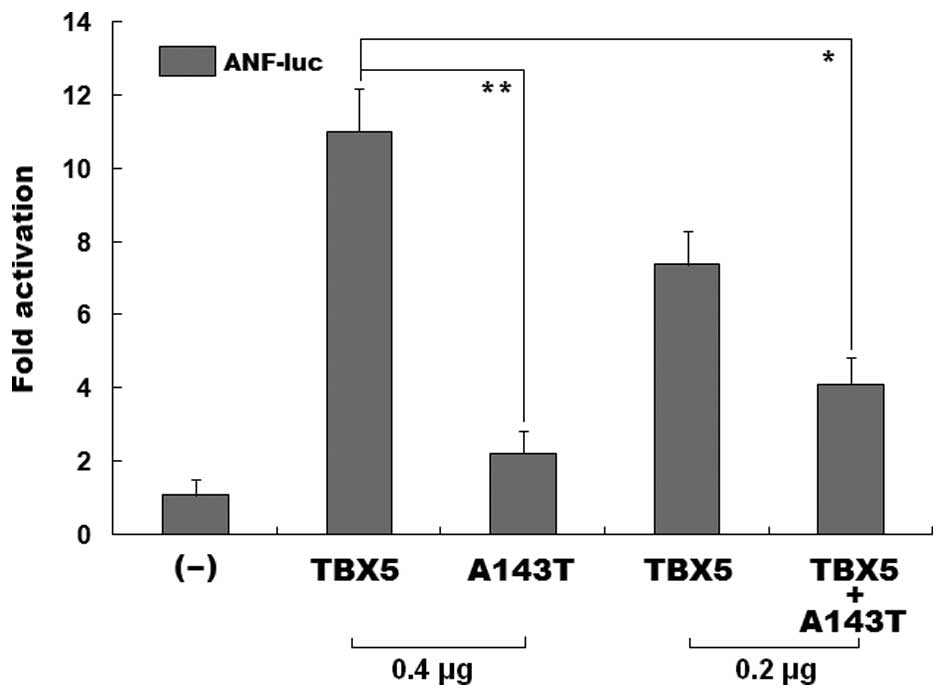
As shown in Fig.
4, the same amount (0.4 μg) of wild-type TBX5 and
its mutant counterpart, A143T, activated the ANF promoter by
approximately 11- and 2-fold, respectively. When wild-type
TBX5 was co-expressed with the same amount (0.2 μg)
of the TBX5 mutant, A143T, the induced activation of the
ANF promoter was approximately 4-fold; while 0.2 μg
of wild-type TBX5 alone activated the ANF promoter by
approximately 7-fold. These functional data reveal that the TBX5
mutant, A143T, had a significantly reduced transcriptional activity
and a dominant-negative effect on its wild-type counterpart.
Diminished synergistic transcriptional
activity of the TBX5 mutant
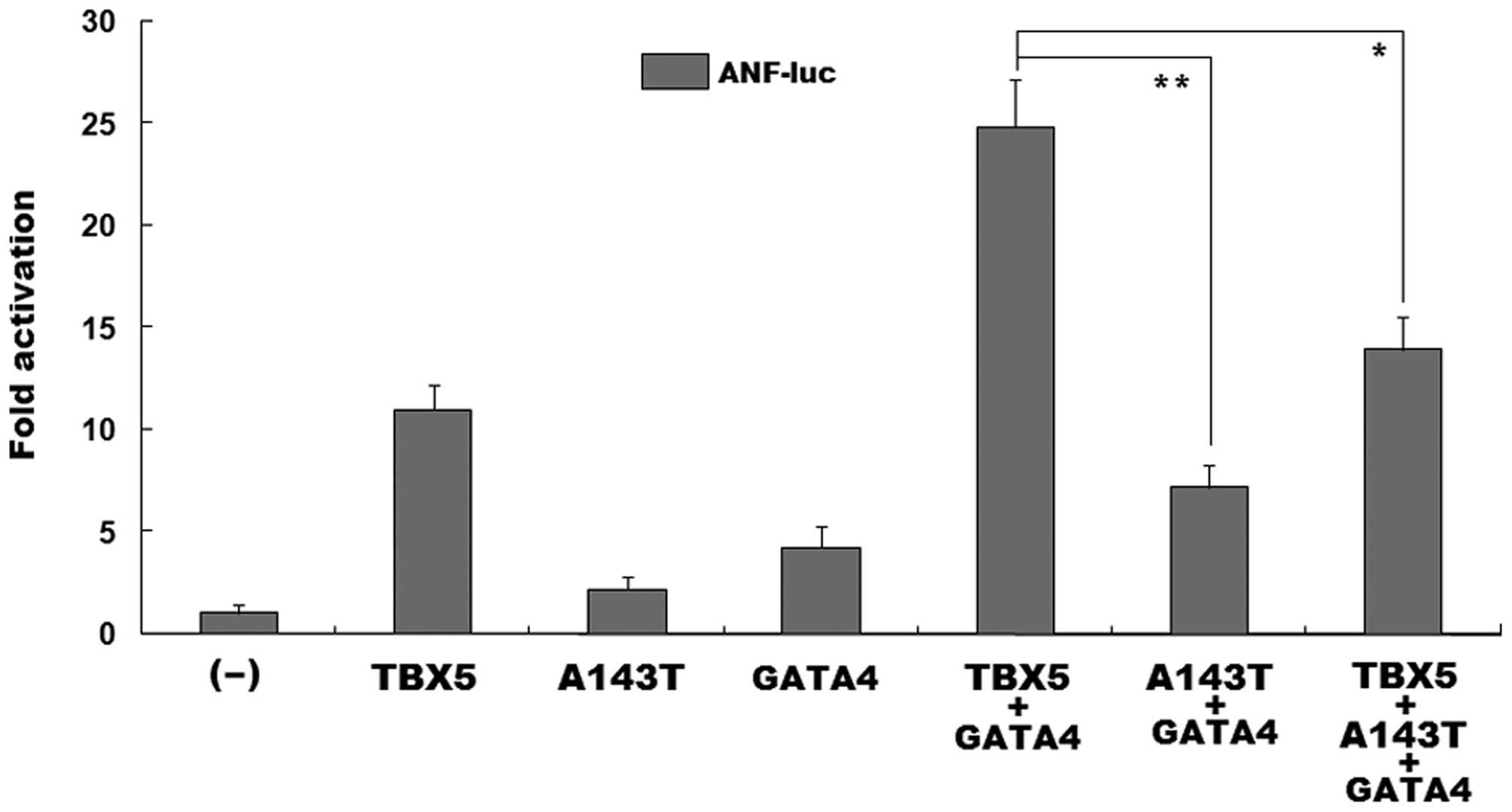
As shown in Fig.
5, in the presence of 0.4 μg of GATA4, the same
amount (0.4 μg) of wild-type TBX5 and the TBX5
mutant, A143T, activated the ANF promoter by approximately
25- and 7-fold, respectively. When wild-type TBX5 was
co-expressed with the same amount (0.2 μg) of the
TBX5 mutant, A143T, the induced activation of the ANF
promoter was approximately 14-fold. These functional data reveal
that the TBX5 mutant, A143T, had a significantly diminished
synergistic transcriptional activity with GATA4.
Discussion
In this study, a novel heterozygous mutation in
TBX5, p.A143T, was identified in a patient with sporadic DCM. The
mutation was absent in the 400 referral chromosomes, and altered
the amino acid that was completely conserved evolutionarily.
Functional analyses revealed that the TBX5 mutant, A143T, was
associated with a significantly decreased transcriptional activity
and had a dominant-negative effect on its wild-type counterpart.
Furthermore, the mutation diminished the synergistic activation
between TBX5 and GATA4. Therefore, it is likely that mutant TBX5
predisposes the mutation carrier to DCM. To the best of our
knowledge, this is the first clinical study that demonstrates the
association of TBX5 loss-of-function mutation with an enhanced
susceptibility to sporadic DCM.
To date, 17 members of the T-box containing
transcription factor family have been discovered in mammals, of
which 6 members, including TBX1, TBX18 and TBX20 of the TBX1
subfamily, and TBX2, TBX3 and TBX5 of the TBX2 subfamily, are
expressed in the heart (16). In
humans, the TBX5 gene, which maps to chromosome 12q24.1 and
contains 8 coding exons, encodes a protein of 518 amino acids. The
TBX5 protein has an important structural domain termed T-box, which
is highly conserved throughout members of the T-box family and
across various species. The T-box motif is essential for the
specific binding to target DNA and the protein-protein
interactions, and previous studies have substantiated that TBX5
regulates the expression of multiple important target genes
expressed in the heart during embryogenesis, including ANF,
CX40 and serum response factor (SRF), alone or in
synergy with trascriptionally cooperative partners, such as GATA4
and NKX2-5 (16,18,38,60,61). In the present study, the TBX5
mutation identified in a patient with sporadic DCM is located in
the T-box domain, and functional analyses unveiled that the
mutation led to a significantly reduced transcriptional activity of
TBX5, alone or synergistically with GATA4, and exerted a
dominant-negative effect on wild-type TBX5. These findings suggest
that haploinsufficiency or a dominant-negative effect resulted from
the TBX5 mutation may be an alternative pathological mechanism of
the pathogenesis of sporadic DCM.
The association of TBX5 loss-of-function
mutation with an increased vulnerability to familial DCM has been
reported previously. Zhang et al (56) sequenced the coding regions and
splice junction sites of TBX5 in a cohort of 190 unrelated
patients with idiopathic DCM, and found a novel heterozygous
mutation (p.S154A) in an index patient with a positive family
history, with a mutational prevalence of approximately 0.53%.
Genetic analysis of the proband’s pedigree revealed that the
mutation co-segregated with DCM transmitted in an autosomal
dominant pattern with complete penetrance. Biological assays
revealed that the mutation significantly decreased the
transcription activating function of TBX5, as well as its synergism
with partners GATA4 and NKX2-5 at the ANF promoter.
Similarly, in the present study, a novel TBX5 loss-of-function
mutation, p.A143T, was linked to sporadic DCM. These observational
results suggest that genetically compromised TBX5 contributes to
the pathogenesis of DCM in a subset of patients.
The discovery that functionally impaired TBX5
predisposes to DCM may be partially attributed to the abnormal
development and structural remodeling of the heart. In vertebrates
and humans, TBX5 is amply expressed in the embryonic and adult
hearts, and is required for normal cardiac development and
structural remodeling, including cellular specification,
proliferation, differentiation and migration, tissue patterning and
morphogenesis (62). In mice,
TBX5 is highly expressed in the cardiac crescent, linear heart
tube, common atrium, left ventricle, left-side ventricular septum,
trabeculae of the right ventricle, and the atrial aspect of the
atrioventricular valves (63).
Mice with a homozygous deletion of two Tbx5 alleles died by
E10.5, due to failure of cardiac looping, hypoplasia of sinuatria
and left ventricle; while mice with a heterozygous deletion of one
Tbx5 allele presented with atrial septal defects,
ventricular septal defects, endocardial cushion defects,
hypoplastic left heart, aberrant trabeculation, and
atrioventricular block, similar with what were observed in patients
with Holt-Oram syndrome (64).
Moreover, in mice TBX5 and GATA4 are co-expressed and interact
physically in the developing atria and ventricles, and mice with
doubly heterozygous deletion of Tbx5 and Gata4
suffered embryonic lethality, thin atrial and ventricular
myocardium with reduced cell proliferation and atrioventricular
septation defects (65). Taken
together, these results from experimental animals support that TBX5
loss-of-function mutation is involved in DCM in humans.
It has been substantiated that TBX5 physically
interacts with multiple core cardiac transcriptional factors,
including GATA4, NKX2-5, MEF2c and TBX20, to form a transcriptional
complex resulting in the synergistic activation of target genes
required for proper cardiac development, including ANF,
CX40, SRF, MHY6 and ID2 (16), and loss-of-function mutations in
several transcriptional cooperative partners of TBX5, including
NKX2-5, GATA4, GATA5, GATA6 and TBX20, have been reported to be
responsible for DCM in humans (50–57,66). Therefore, genetically defective
TBX5 may contribute to DCM by decreasing the expression of
some target genes important for cardiac development and structural
remodeling.
Notably, TBX5 mutations have previously been related
to Holt-Oram syndrome, an autosomal dominant disease characteristic
of congenital cardiac defects and anterior upper limb deformities.
Cardiac phenotypes constitute a wide spectrum of cardiovascular
developmental anomalies, including atrial septal defect,
ventricular septal defect, tetralogy of Fallot, atrioventricular
septal defect, patent ductus arteriosus, mitral valve defect and
aberrant pulmonary vein, as well as cardiac conduction block and
atrial fibrillation (37,48,67). There are three variations of
Holt-Oram syndrome which have been encountered in clinical
practice: patients may have only cardiovascular abnormalities (4%),
only forelimb malformations (27%), or both (69%), and these cardiac
and skeletal deformations vary widely ranging from mild to severe,
even within families (48). In
the present study, in addition to DCM, the mutation carrier had no
apparent cardiac or skeletal anomaly. Different genetic
backgrounds, epigenetic modifiers and functional characteristics of
gene mutations (loss-of-function, dominant-negative, or
gain-of-function effect) as well as its temporal and spatial
effects during cardiac development (germline or somatic) are
potential explanations for the pronounced variability in clinical
presentation (46).
In conclusion, to the best of our knowledge, this
study is the first to present an association of a TBX5
loss-of-function mutation with an increased susceptibility to
sporadic DCM, providing novel insight into the molecular mechanisms
of the pathogenesis DCM, and suggesting potential implications for
prenatal prophylaxis and personalized treatment of the most common
type of primary cardiomyopathy.
Acknowledgments
The authors are thankful to the participants for
their participation in the study. This study was supported in part
by grants from the National Natural Science Fund of China (81270161
and 81470372), the key basic research program of Shanghai, China
(14JC1405500) and the Natural Science Fund of Shanghai, China
(15ZR1438100).
References
|
1
|
McNally EM, Golbus JR and Puckelwartz MJ:
Genetic mutations and mechanisms in dilated cardiomyopathy. J Clin
Invest. 123:19–26. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hershberger RE, Hedges DJ and Morales A:
Dilated cardiomyopathy: the complexity of a diverse genetic
architecture. Nat Rev Cardiol. 10:531–547. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Garcia-Pavia P, Cobo-Marcos M,
Guzzo-Merello G, Gomez-Bueno M, Bornstein B, Lara-Pezzi E, Segovia
J and Alonso-Pulpon L: Genetics in dilated cardiomyopathy.
Biomarkers Med. 7:517–533. 2013. View Article : Google Scholar
|
|
4
|
Koutalas E, Kanoupakis E and Vardas P:
Sudden cardiac death in non-ischemic dilated cardiomyopathy: a
critical appraisal of existing and potential risk stratification
tools. Int J Cardiol. 167:335–341. 2013. View Article : Google Scholar
|
|
5
|
Yoshikawa T: Contribution of acquired
factors to the pathogenesis of dilated cardiomyopathy. -The cause
of dilated cardiomyopathy: genetic or acquired? (acquired-side)-.
Circ J. 75:1766–1773. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kamisago M, Sharma SD, DePalma SR, Solomon
S, Sharma P, McDonough B, Smoot L, Mullen MP, Woolf PK, Wigle ED,
et al: Mutations in sarcomere protein genes as a cause of dilated
cardiomyopathy. N Engl J Med. 343:1688–1696. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Arndt AK, Schafer S, Drenckhahn JD, Sabeh
MK, Plovie ER, Caliebe A, Klopocki E, Musso G, Werdich AA, Kalwa H,
et al: Fine mapping of the 1p36 deletion syndrome identifies
mutation of PRDM16 as a cause of cardiomyopathy. Am J Hum Genet.
93:67–77. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pankuweit S, Ruppert V, Jónsdóttir T,
Müller HH and Meyer T: German Competence Network of Heart Failure:
The HLA class II allele DQB1 0309 is associated with dilated
cardiomyopathy. Gene. 531:180–183. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wahbi K, Béhin A, Bécane HM, Leturcq F,
Cossée M, Laforêt P, Stojkovic T, Carlier P, Toussaint M, Gaxotte
V, et al: Dilated cardiomyopathy in patients with mutations in
anoctamin 5. Int J Cardiol. 168:76–79. 2013. View Article : Google Scholar
|
|
10
|
Ruppert V, Meyer T, Richter A, Maisch B
and Pankuweit S: German Competence Network of Heart Failure:
Identification of a missense mutation in the melusin-encoding
ITGB1BP2 gene in a patient with dilated cardiomyopathy. Gene.
512:206–210. 2013. View Article : Google Scholar
|
|
11
|
Agrawal PB, Pierson CR, Joshi M, Liu X,
Ravenscroft G, Moghadaszadeh B, Talabere T, Viola M, Swanson LC,
Haliloğlu G, et al: SPEG interacts with myotubularin, and its
deficiency causes centronuclear myopathy with dilated
cardiomyopathy. Am J Hum Genet. 95:218–226. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Matsa LS, Sagurthi SR, Ananthapur V, Nalla
S and Nallari P: Endothelin 1 gene as a modifier in dilated
cardiomyopathy. Gene. 548:256–262. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Pikkarainen S, Tokola H, Kerkelä R and
Ruskoaho H: GATA transcription factors in the developing and adult
heart. Cardiovasc Res. 63:196–207. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cai H, Katoh-Kurasawa M, Muramoto T,
Santhanam B, Long Y, Li L, Ueda M, Iglesias PA, Shaulsky G and
Devreotes PN: Nucleocytoplasmic shuttling of a GATA transcription
factor functions as a development timer. Science. 343:12495312014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Akazawa H and Komuro I: Cardiac
transcription factor Csx/Nkx2-5: Its role in cardiac development
and diseases. Pharmacol Ther. 107:252–268. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Greulich F, Rudat C and Kispert A:
Mechanisms of T-box gene function in the developing heart.
Cardiovasc Res. 91:212–222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Oka T, Xu J and Molkentin JD:
Re-employment of developmental transcription factors in adult heart
disease. Semin Cell Dev Biol. 18:117–131. 2007. View Article : Google Scholar
|
|
18
|
Garg V, Kathiriya IS, Barnes R,
Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS,
Hirayama-Yamada K, Joo K, et al: GATA4 mutations cause human
congenital heart defects and reveal an interaction with TBX5.
Nature. 424:443–447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Rajagopal SK, Ma Q, Obler D, Shen J,
Manichaikul A, Tomita-Mitchell A, Boardman K, Briggs C, Garg V,
Srivastava D, et al: Spectrum of heart disease associated with
murine and human GATA4 mutation. J Mol Cell Cardiol. 43:677–685.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang YQ, Wang J, Liu XY, Chen XZ, Zhang W
and Wang XZ: Mutation spectrum of GATA4 associated with congenital
atrial septal defects. Arch Med Sci. 9:976–983. 2013. View Article : Google Scholar
|
|
21
|
Yang YQ, Gharibeh L, Li RG, Xin YF, Wang
J, Liu ZM, Qiu XB, Xu YJ, Xu L, Qu XK, et al: GATA4
loss-of-function mutations underlie familial tetralogy of Fallot.
Hum Mutat. 34:1662–1671. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Xiang R, Fan LL, Huang H, Cao BB, Li XP,
Peng DQ and Xia K: A novel mutation of GATA4 (K319E) is responsible
for familial atrial septal defect and pulmonary valve stenosis.
Gene. 534:320–323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Jiang JQ, Li RG, Wang J, Liu XY, Xu YJ,
Fang WY, Chen XZ, Zhang W, Wang XZ and Yang YQ: Prevalence and
spectrum of GATA5 mutations associated with congenital heart
disease. Int J Cardiol. 165:570–573. 2013. View Article : Google Scholar
|
|
24
|
Wei D, Bao H, Zhou N, Zheng GF, Liu XY and
Yang YQ: GATA5 loss-of-function mutation responsible for the
congenital ventriculoseptal defect. Pediatr Cardiol. 34:504–511.
2013. View Article : Google Scholar
|
|
25
|
Wei D, Bao H, Liu XY, Zhou N, Wang Q, Li
RG, Xu YJ and Yang YQ: GATA5 loss-of-function mutations underlie
tetralogy of fallot. Int J Med Sci. 10:34–42. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu
L, Liu H, Li RG, Xu YJ, Wang Q, et al: GATA5 loss-of-function
mutations associated with congenital bicuspid aortic valve. Int J
Mol Med. 33:1219–1226. 2014.PubMed/NCBI
|
|
27
|
Huang RT, Xue S, Xu YJ, Zhou M and Yang
YQ: Somatic GATA5 mutations in sporadic tetralogy of Fallot. Int J
Mol Med. 33:1227–1235. 2014.PubMed/NCBI
|
|
28
|
Zheng GF, Wei D, Zhao H, Zhou N, Yang YQ
and Liu XY: A novel GATA6 mutation associated with congenital
ventricular septal defect. Int J Mol Med. 29:1065–1071.
2012.PubMed/NCBI
|
|
29
|
Huang RT, Xue S, Xu YJ and Yang YQ:
Somatic mutations in the GATA6 gene underlie sporadic tetralogy of
Fallot. Int J Mol Med. 31:51–58. 2013.
|
|
30
|
Schott JJ, Benson DW, Basson CT, Pease W,
Silberbach GM, Moak JP, Maron BJ, Seidman CE and Seidman JG:
Congenital heart disease caused by mutations in the transcription
factor NKX2-5. Science. 281:108–111. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Huang W, Meng H, Qiao Y, Pang S, Chen D
and Yan B: Two novel and functional DNA sequence variants within an
upstream enhancer of the human NKX2-5 gene in ventricular septal
defects. Gene. 524:152–155. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Qu XK, Qiu XB, Yuan F, Wang J, Zhao CM,
Liu XY, Zhang XL, Li RG, Xu YJ, Hou XM, et al: A novel NKX2.5
loss-of-function mutation associated with congenital bicuspid
aortic valve. Am J Cardiol. 114:1891–1895. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Heathcote K, Braybrook C, Abushaban L, Guy
M, Khetyar ME, Patton MA, Carter ND, Scambler PJ and Syrris P:
Common arterial trunk associated with a homeodomain mutation of
NKX2.6. Hum Mol Genet. 14:585–593. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ta-Shma A, El-lahham N, Edvardson S,
Stepensky P, Nir A, Perles Z, Gavri S, Golender J,
Yaakobi-Simhayoff N, Shaag A, et al: Conotruncal malformations and
absent thymus due to a deleterious NKX2-6 mutation. J Med Genet.
51:268–270. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhao L, Ni SH, Liu XY, Wei D, Yuan F, Xu
L, Xin-Li Li RG, Qu XK, Xu YJ, et al: Prevalence and spectrum of
Nkx2.6 mutations in patients with congenital heart disease. Eur J
Med Genet. 57:579–586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Wang J, Mao JH, Ding KK, Xu WJ, Liu XY,
Qiu XB, Li RG, Qu XK, Xu YJ, Huang RT, et al: A novel NKX2.6
mutation associated with congenital ventricular septal defect.
Pediatr Cardiol. 36:646–656. 2015. View Article : Google Scholar
|
|
37
|
Baban A, Postma AV, Marini M, Trocchio G,
Santilli A, Pelegrini M, Sirleto P, Lerone M, Albanese SB, Barnett
P, et al: Identification of TBX5 mutations in a series of 94
patients with Tetralogy of Fallot. Am J Med Genet A.
164A:3100–3107. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
McCulley DJ and Black BL: Transcription
factor pathways and congenital heart disease. Curr Top Dev Biol.
100:253–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Andersen TA, Troelsen KL and Larsen LA: Of
mice and men: molecular genetics of congenital heart disease. Cell
Mol Life Sci. 71:1327–1352. 2014. View Article : Google Scholar :
|
|
40
|
Wang J, Sun YM and Yang YQ: Mutation
spectrum of the GATA4 gene in patients with idiopathic atrial
fibrillation. Mol Biol Rep. 39:8127–8135. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang XH, Huang CX, Wang Q, Li RG, Xu YJ,
Liu X, Fang WY and Yang YQ: A novel GATA5 loss-of-function mutation
underlies lone atrial fibrillation. Int J Mol Med. 31:43–50.
2013.
|
|
42
|
Li J, Liu WD, Yang ZL and Yang YQ: Novel
GATA6 loss-of-function mutation responsible for familial atrial
fibrillation. Int J Mol Med. 30:783–790. 2012.PubMed/NCBI
|
|
43
|
Xie WH, Chang C, Xu YJ, Li RG, Qu XK, Fang
WY, Liu X and Yang YQ: Prevalence and spectrum of Nkx2.5 mutations
associated with idiopathic atrial fibrillation. Clinics (Sao
Paulo). 68:777–784. 2013. View Article : Google Scholar
|
|
44
|
Huang RT, Xue S, Xu YJ, Zhou M and Yang
YQ: A novel NKX2.5 loss-of-function mutation responsible for
familial atrial fibrillation. Int J Mol Med. 31:1119–1126.
2013.PubMed/NCBI
|
|
45
|
Perera JL, Johnson NM, Judge DP and
Crosson JE: Novel and highly lethal NKX2.5 missense mutation in a
family with sudden death and ventricular arrhythmia. Pediatr
Cardiol. 35:1206–1212. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Yu H, Xu JH, Song HM, Zhao L, Xu WJ, Wang
J, Li RG, Xu L, Jiang WF, Qiu XB, et al: Mutational spectrum of the
NKX2-5 gene in patients with lone atrial fibrillation. Int J Med
Sci. 11:554–563. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang J, Zhang DF, Sun YM, Li RG, Qiu XB,
Qu XK, Liu X, Fang WY and Yang YQ: NKX2-6 mutation predisposes to
familial atrial fibrillation. Int J Mol Med. 34:1581–1590.
2014.PubMed/NCBI
|
|
48
|
Postma AV, van de Meerakker JB, Mathijssen
IB, Barnett P, Christoffels VM, Ilgun A, Lam J, Wilde AA, Lekanne
Deprez RH and Moorman AF: A gain-of-function TBX5 mutation is
associated with atypical Holt-Oram syndrome and paroxysmal atrial
fibrillation. Circ Res. 102:1433–1442. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hong K and Xiong Q: Genetic basis of
atrial fibrillation. Curr Opin Cardiol. 29:220–226. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li RG, Li L, Qiu XB, Yuan F, Xu L, Li X,
Xu YJ, Jiang WF, Jiang JQ, Liu X, et al: GATA4 loss-of-function
mutation underlies familial dilated cardiomyopathy. Biochem Biophys
Res Commun. 439:591–596. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Zhao L, Xu JH, Xu WJ, Yu H, Wang Q, Zheng
HZ, Jiang WF, Jiang JF and Yang YQ: A novel GATA4 loss-of-function
mutation responsible for familial dilated cardiomyopathy. Int J Mol
Med. 33:654–660. 2014.
|
|
52
|
Zhang XL, Dai N, Tang K, Chen YQ, Chen W,
Wang J, Zhao CM, Yuan F, Qiu XB, Qu XK, et al: GATA5
loss-of-function mutation in familial dilated cardiomyopathy. Int J
Mol Med. 35:763–770. 2015.
|
|
53
|
Xu L, Zhao L, Yuan F, Jiang WF, Liu H, Li
RG, Xu YJ, Zhang M, Fang WY, Qu XK, et al: GATA6 loss-of-function
mutations contribute to familial dilated cardiomyopathy. Int J Mol
Med. 34:1315–1322. 2014.PubMed/NCBI
|
|
54
|
Costa MW, Guo G, Wolstein O, Vale M,
Castro ML, Wang L, Otway R, Riek P, Cochrane N, Furtado M, et al:
Functional characterization of a novel mutation in NKX2-5
associated with congenital heart disease and adult-onset
cardiomyopathy. Circ Cardiovasc Genet. 6:238–247. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Yuan F, Qiu XB, Li RG, Qu XK, Wang J, Xu
YJ, Liu X, Fang WY, Yang YQ and Liao DN: A novel NKX2-5
loss-of-function mutation predisposes to familial dilated
cardiomyopathy and arrhythmias. Int J Mol Med. 35:478–486.
2015.
|
|
56
|
Zhang XL, Qiu XB, Yuan F, Wang J, Zhao CM,
Li RG, Xu L, Xu YJ, Shi HY, Hou XM, et al: TBX5 loss-of-function
mutation contributes to familial dilated cardiomyopathy. Biochem
Biophys Res Commun. 459:166–171. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Li J, Liu WD, Yang ZL, Yuan F, Xu L, Li RG
and Yang YQ: Prevalence and spectrum of GATA4 mutations associated
with sporadic dilated cardiomyopathy. Gene. 548:174–181. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Elliott P, O’Mahony C, Syrris P, Evans A,
Rivera Sorensen C, Sheppard MN, Carr-White G, Pantazis A and
McKenna WJ: Prevalence of desmosomal protein gene mutations in
patients with dilated cardiomyopathy. Circ Cardiovasc Genet.
3:314–322. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Richardson P, McKenna W, Bristow M, Maisch
B, Mautner B, O’Connell J, Olsen E, Thiene G, Goodwin J, Gyarfas I,
et al: Report of the 1995 World Health Organization/International
Society and Federation of Cardiology Task Force on the Definition
and Classification of cardiomyopathies. Circulation. 93:841–842.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Hiroi Y, Kudoh S, Monzen K, Ikeda Y,
Yazaki Y, Nagai R and Komuro I: Tbx5 associates with Nkx2-5 and
synergistically promotes cardiomyocyte differentiation. Nat Genet.
28:276–280. 2001. View
Article : Google Scholar : PubMed/NCBI
|
|
61
|
Stennard FA, Costa MW, Elliott DA, Rankin
S, Haast SJ, Lai D, McDonald LP, Niederreither K, Dolle P, Bruneau
BG, et al: Cardiac T-box factor Tbx20 directly interacts with
Nkx2-5, GATA4, and GATA5 in regulation of gene expression in the
developing heart. Dev Biol. 262:206–224. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Plageman TF Jr and Yutzey KE: T-box genes
and heart development: putting the ‘T’ in heart. Dev Dyn.
232:11–20. 2005. View Article : Google Scholar
|
|
63
|
Bruneau BG, Logan M, Davis N, Levi T,
Tabin CJ, Seidman JG and Seidman CE: Chamber-specific cardiac
expression of Tbx5 and heart defects in Holt-Oram syndrome. Dev
Biol. 211:100–108. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
64
|
Bruneau BG, Nemer G, Schmitt JP, Charron
F, Robitaille L, Caron S, Conner DA, Gessler M, Nemer M, Seidman
CE, et al: A murine model of Holt-Oram syndrome defines roles of
the T-box transcription factor Tbx5 in cardiogenesis and disease.
Cell. 106:709–721. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Misra C, Chang SW, Basu M, Huang N and
Garg V: Disruption of myocardial Gata4 and Tbx5 results in defects
in cardiomyocyte proliferation and atrioventricular septation. Hum
Mol Genet. 23:5025–5035. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Kirk EP, Sunde M, Costa MW, Rankin SA,
Wolstein O, Castro ML, Butler TL, Hyun C, Guo G, Otway R, et al:
Mutations in cardiac T-box factor gene TBX20 are associated with
diverse cardiac pathologies, including defects of septation and
valvulogenesis and cardiomyopathy. Am J Hum Genet. 81:280–291.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
67
|
Böhm J, Heinritz W, Craig A, Vujic M,
Ekman-Joelsson BM, Kohlhase J and Froster U: Functional analysis of
the novel TBX5 c.1333delC mutation resulting in an extended TBX5
protein. BMC Med Genet. 9:882008. View Article : Google Scholar : PubMed/NCBI
|