Introduction
Brugada syndrome (BrS) is a genetic heart disorder
predominantly exhibiting an autosomal dominant pattern of
inheritance and is thought to be responsible for 20% of sudden
fatalities in people <50 years of age with apparently
structurally normal hearts (1).
It is characterized by a dynamic coved-type ST-segment elevation in
the right precordial leads (V1–V3) of the electrocardiogram (ECG),
and clinically by an increased risk of ventricular tachycardia (VT)
or ventricular fibrillation (VF) and sudden fatality (2). The phenotype and penetrance of the
disease appear to be associated with factors that alter the balance
of outward and inward currents at the end of phase I of the
epicardial ventricular action potential (3). In BrS, it is believed that the
arrhythmic substrate is the result of increased heterogeneity of
the currents involved in the phase I repolarization of the action
potential (AP) in the right ventricle (4). Electrophysiological evidence
indicates that the basis for arrhythmogenicity is the loss of the
AP dome in certain epicardial sites (5). This leads to increased epicardial
dispersion of repolarization, enabling local re-excitation via
phase II reentry (6). Multiple
mutations of genes, SCN5A, SCN1B, SCN3B,
GPD1L, CACNA1C, CACNB2, KCNE3 and
KCND3, have been shown to decrease cardiac Na+
and Ca2+ channel activity or increase fast transient
outward K+ channel activity, and they are linked to BrS
(7).
The voltage-gated cardiac fast transient outward
K+ current (Ito,f) plays a predominant role
in determining the initial repolarization of AP. It is
well-documented that the human cardiac Ito,f channel is
composed of the α subunit/KV4.3 and β
subunit/K+ channel-interacting protein 2 (8). The expression of the
KV4.3 channel in normal or failing human and canine
hearts exhibits a transmural gradient in the left ventricle
(9), which is responsible for the
transmural electrophysiological heterogeneity (10). Accordingly, an early
repolarization abnormality as a result of decreased expression and
dysfunction of the KV4.3 channel in a variety of heart
diseases, such as myocardial infarction and heart failure,
contributes to the pathogenesis of life-threatening cardiac
arrhythmias (11). Recently, two
missense mutations have been identified in the
KV4.3-encoded gene, KCND3, from patients with
BrS. The two individual mutants have been shown to produce a gain
of Ito,f function in HEK-293 cells when co-expressing
with KChIP2 (12). Simulations
using a Luo-Rudy II action potential mode demonstrated the stable
loss of the AP dome as a result of the increase of Ito,f
maximal conductance associated with heterozygous expression of
either the L450F or G600R mutant of KV4.3 in the
presence of KChIP2 (12).
However, the individual role of the mutants and KChIP2 in the
gain-of-function of the KV4.3 channel is unclear. The
present study demonstrated that two mutations are sufficient to
increase the KV4.3 current by increasing membrane
expression, but KChIP2 also participates with the mutation-related
BrS by increasing KV4.3 protein expression and
regulating channel kinetics.
Material and methods
Plasmids and reagents
cDNA plasmids-encoding KV4.3 and KChIP2,
respectively, were kindly provided by Dr Jeanne M. Nerbonne at
Washington University School of Medicine in St. Louis (MO, USA).
Alignment by NCBI Blast software (http://blast.ncbi.nlm.nih.gov/Blast.cgi) showed that
>90% of the amino acids of rat (NCBI protein, NP_001257891.1,
10-Aug-2014) and human (NCBI protein, NP_004971.2, 25-May-2014)
KV4.3 are identical (Fig.
1B). Two targeted mutations in rat KCND3 were generated
using the QuickChange II XL site-directed mutagenesis kit (Agilent,
Santa Clara, CA, USA) according to the manufacturer’s instructions.
Rat KCND3, with two individual mutations corresponding to
BrS in humans, was confirmed by sequencing of the constructs using
the Big T3 terminator kit (Applied Biosystems, Foster City, CA,
USA) (Fig. 1A) and they encode
KV4.3-G581R and KV4.3-L450F, respectively.
The following primers were used for polymerase chain reaction
(PCR): rKV4.3-G581R, 5′AAAGCAGACGATCGACTGAGACCAA-3′
[nucleotides (nt) 1821–1855]; and rKV4.3-L450F,
5′GCGCAATGGACT CTTCAATGAAGCTCTGG-3′ (nt 1427–1455). The underlined
letters (TC and T) refer to the base pair changes corresponding to
the rat KV4.3 mutation G580R and L450F,
respectively.
Cell culture and transfection
HEK-293 cells were cultured in Dulbecco’s modified
Eagle’s medium supplemented with 10% fetal bovine serum
(Sigma-Aldrich, St. Louis, MO, USA), 100 U/ml penicillin and 100
µg/ml streptomycin (Life Technologies, Grand Island, NY,
USA). The cells were maintained at 37°C in a 5% CO2
incubator, and passaged every 3 days at confluence. Prior to
transfection, cells were seeded in 6-well plates at a density of
5×105 cells/ml. After 24 h, a total of 1.0 µg
plasmid DNA was diluted in transfection medium using Lipofectamine
2000 reagent (Invitrogen, Grand Island, NY, USA) according to the
manufacturer’s instructions.
Western blot analysis
HEK-293 cells were lysed on ice with ice-cold Lysis
buffer (Cell Signaling Technology, Boston, MA, USA) following
rinsing with phosphate-buffered saline (PBS). Cell lysates were
subsequently collected and prepared by passing through a 25-G
needle connected to a 1-ml syringe 10 times before centrifugation
at 17,000 × g for 10 min at 4°C. Supernatants were harvested and
the protein concentration was determined using the Bradford method
and a spectrophotometer. Equal amounts of protein were loaded onto
a polyacrymide gel and run at 80–120 V for 2 h, and were
subsequently transferred onto a polyvinyl fluoride membrane
overnight. The membranes were washed in PBS three times for 5 min
each, and blocked with 5% skimmed powdered milk for 1 h.
Subsequently the membranes were incubated with a monoclonal
antibody against KV4.3 (mouse anti-rat, 1:1,000; Cat.
no. 75-017), KChIP2b (mouse anti-rat, 1:2,000; Cat. no. 75-004)
(from NeuroMab, Davis, CA, USA) or glyceraldehyde-3-phosphate
dehydrogenase (GAPDH) (mouse anti-rabbit, 1:4,000; Cat. no. CB1001;
Millipore, Billerica, MA, USA) at 4°C overnight and washed 3 times
prior to incubation with horseradish peroxidase (HRP)-conjugated
goat anti-mouse secondary antibodies (polyclonal, 1:3,000; Cat. no.
AP181P; Millipore) for 1 h at room temperature. ECL
chemiluminescent reagent (Perkin Elmer, Waltham, MA, USA) was the
chemiluminescent substrate used for chemiluminescence-based
immunodetection of HRP.
Reverse transcription (RT)-PCR
Total RNA was extracted from HEK-293 cells using the
RNeasy Miniprep Plus kit (Qiagen, Valencia, CA, USA) according to
the manufacturer’s instructions after transfection for 24 h with
plasmids encoding KV4.3-wild-type (WT), or individual
mutants with plasmids encoding KChIP2. RNA concentration was
determined using a Bradford spectrometer at OD 260. Total RNA was
stored in −80°C for further experiments. Reverse transcription was
carried out with the High Capacity Reverse Transcription kit
(Applied Biosystems) according to the manufacturer’s instructions.
Subsequent PCR amplification was performed using the Bio-Rad
MyCycler Thermal Cycler (Bio-Rad, Hercules, CA, USA). The following
primers were used for amplification and detection of
KV4.3: Forward, 5′-TTTGTCACACTCCGGGTCTTCCGT-3′, and
reverse, 5′-TCATTGAGGAGCCCATTGCGCTTG-3′. GAPDH was
determined using the following primer pairs: Forward,
5′-ACGGATTTGGTCGTATTGGG-3′, and reverse,
5′-CGCTCCTGGAAGATGGTGAT-3′. The PCR cycling conditions were: 94°C
for 3 min; 27 cycles of 94°C for 30 sec, 55°C for 30 sec and 72°C
for 30 sec; and 1 cycle of additional extension at 72°C for 7 min;
and subsequently held at 4°C. The final concentration of all the
reagents were: 1X Taq buffer, 0.2 mM of each dNTP, 0.2 µM of
each primer and 2 units of Taq polymerase. The expression of
KV4.3 mRNA was normalized to the GAPDH level. The
density of the KV4.3 mRNA and protein bands between
groups was quantified using ImageJ (NIH, Bethesda, MA, USA).
Whole-cell KV4.3
recording
Outward K+ currents in the HEK-293 cells
were recorded in a voltage-clamp mode at room temperature (24°C).
Experiments were conducted using a Axopatch 200B amplifier attached
to a Dell desktop computer equipped with a DigiData 1322 series
analog/digital interface and pClamp 10.0 software (all from Axon,
Sunnyvale, CA, USA). Electrodes were pulled using a PC-10 vertical
pipette puller (Narishige, East Meadow, NY, USA) and had a pipette
resistance between 1.5 and 3.0 MΩ subsequent to filling with a
recording pipette solution containing: 135 mM KCl, 1 mM
MgCl2, 10 mM HEPES and 5 mM glucose (pH 7.2). The bath
solution for the recording contained: 136 mM NaCl, 4 mM KCl, 1 mM
CaCl2, 2 mM MgCl2, 10 mM HEPES and 10 mM
glucose (pH 7.4). Only the data acquired from cells with an input
resistance >0.7 GΩ were analyzed. Current densities were
obtained from peak amplitudes normalized to cell capacitances. The
voltage-dependent inactivation and recovery from inactivation were
measured using the protocols shown in the Figs. 2 and 3. The voltage dependence of steady-state
inactivation of the KV4.3-WT, KV4.3-G581R and
KV4.3-L450F-encoded K+ currents in the
presence of KChIP2 evoked from each conditioning potential were
measured and normalized to the current evoked from −70 mV (in the
same cell). Each sweep was applied with 10 sec intervals. Data were
obtained at different sampling frequencies and the current signals
were filtered simultaneously at 5 kHz prior to digitization and
storage.
Examination under immunofluorescence
confocal micros- copy
HEK-293 cells were plated in 35-mm dishes overnight
before transfection with plasmids containing cDNAs. Twenty-four
hours after transfection, cells were fixed using 4%
paraformaldehyde, washed 3 times with PBS and permeablized with
0.1% Triton X-100 (Sigma-Aldrich). After being blocked for 1 h with
10% normal goat serum (Invitrogen), the cells were washed and
incubated with mouse anti-KV4.3 monoclonal antibody
(1:200; NeuroMab) overnight. Four more wash steps of 5 min each
were applied prior to incubation with Alexa Fluor 546 goat
anti-mouse IgG (polyclonal, 1:2,000; Life Technologies) for 1 h at
room temperature. Cells were subsequently washed and incubated in
300 nM 4′,6-diamidino-2-phenylindole (Sigma-Aldrich) for 5 min. The
cells were washed 3 times. Subsequently, the cells were examined
and images were captured using an IX-81 laser confocal microscopy
(Olympus, Tokyo, Japan). Images were captured at magnification,
x400 and analyzed using NIH ImageJ software. Cell membrane
localization of WT and mutant KV4.3 was determined by
calculating the ratio of membrane to cytoplasmic fluorescence
intensity as follows: Membrane/cytosolic ratio = (peak membrane
intensity - background)/(mean cytosolic intensity - background).
The intensities were determined by a line scan through each cell.
The cytosolic intensity was calculated as the mean intensity over
at least two membrane thicknesses inside the cell. The background
was determined as the mean intensity at two membrane thicknesses
away from the cell. The membrane and cytosolic intensity were
measured per cell in a blinded fashion, and the mean ratio for each
mutant was normalized by the mean WT ratio for the same day.
Statistical analysis
All the values are presented as the means ± standard
error of the mean. Two-tailed Student’s t-test or one-way analysis
of variance (multiple groups) followed by the Dunnett’s test (for
single comparisons) was used to compare the difference between
various groups. P<0.05 was considered to indicate a
statistically significant difference.
Results
KV4.3-G581R and
KV4.3-L450F cause a gain-of-function of transient
outward K+ currents
In order to determine the effects of individual
mutations on KV4.3/KChIP2-encoded K+
channels, HEK-293 cells were transfected with plasmids encoding
KV4.3-WT, KV4.3-G581R and
KV4.3-L450F with KChIP2 and green fluorescent protein
(GFP). GFP expression allowed visualization of cells for whole-cell
recording. Voltage-gated transient outward K+ currents
were evoked by a 4,500 msec depolarizing pulse between −70 and +60
mV in 10 mV increments from a holding potential of −70 mV. The
transient outward K+ current was significantly increased
in HEK-293 cells expressing KV4.3-G581R or
KV4.3-L450F with KChIP2 (Fig. 1A). In comparison with
KV4.3-WT plus KChIP2, KV4.3-G581R and
KV4.3-L450F plus KChIP2-encoded peak current densities
were significantly (P<0.05) increased from −30 to +60 mV
(Fig. 1B). For example, they were
increased by 33.7 and 79.4%; from 480.7±26.1 pA/pF (n=12) to
642.9±54.1 pA/pF (n=11) and 862.6±98.5 pA/pF (n=12), respectively,
at +40 mV (Fig. 1B).
KV4.3-G581R and
KV4.3-L450F influences the kinetics of transient outward
K+ currents
Analysis of the decay phases of the outward
K+ currents-encoded by KV4.3-WT,
KV4.3-G581R and KV4.3-L450F with KChIP2
revealed that current decay is well-described by the sum of one
exponential and neither time constant exhibits any appreciable
voltage dependence (Fig. 1C).
Each individual mutation significantly (P<0.05) slowed
KV4.3-L450F + KChIP2 or KV4.3-G581R +
KChIP2-encoded channel inactivation from 0 to +60 mV compared to
KV4.3-WT (Fig. 1C),
confirmed by mean ± standard error (SE) time constants of
inactivation (τinactivation) at 188.8±18.3 msec (L450F)
and 155.9±10.0 msec (G581R) compared to 128.5±5.6 msec (WT).
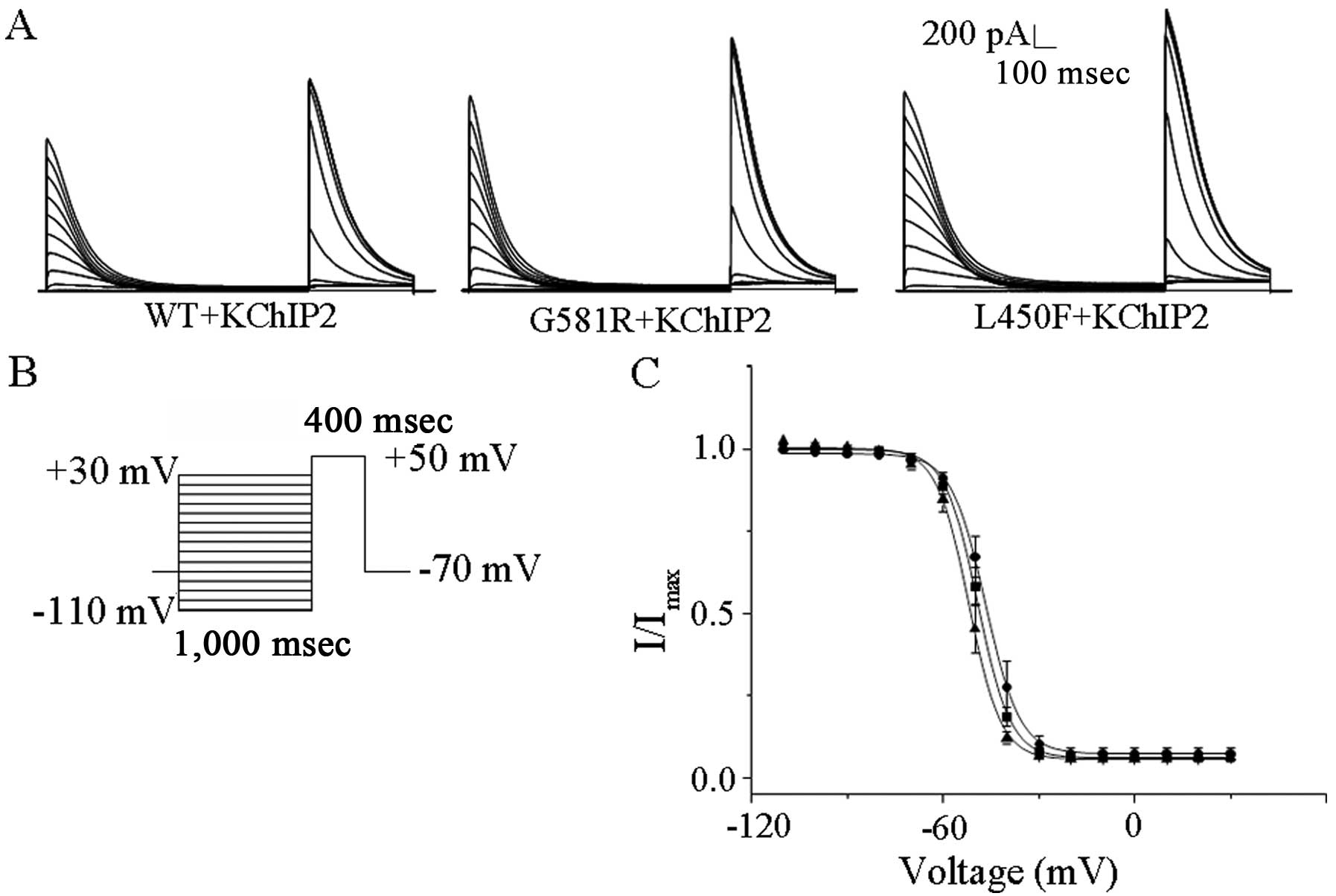
The voltage dependences of steady state inactivation
of KV4.3-WT, KV4.3-G581R and
KV4.3-L450F with KChIP2 were examined during 400 msec
depolarization to +50 mV after 1 sec conditioning prepulses to
potentials between −110 and +30 mV; the protocol (Fig. 2B) is shown below the current
records in Fig. 2A. The steady
state inactivation data for transient outward currents were
well-described by a single Boltzmann equation. The individual
mutations did not affect the values of V1/2 and K (data
not shown), indicating that they did not significantly affect the
steady-state voltage-dependent inactivation of
KV4.3/KChIP2-encoded channels (Fig. 2C).
To examine the effects of two individual mutations
on the time dependency of recovery from steady state inactivation
of KV4.3 alone and with KChIP2-encoded channels, HEK-293
cells expressing KV4.3-WT, KV4.3-G581R or
KV4.3-L450F with KChIP2 were first depolarized to +40 mV
for 400 msec to inactivate the currents, subsequently
hyperpolarized to −70 mV for varying times ranging from 2 to 7,500
msec, and finally stepped to +40 mV to activate the currents and
assess the extent of recovery; the protocol (Fig. 3B) is illustrated below the
K+ currents (Fig. 3A).
Analysis of the normalized current amplitudes as a function of the
recovery time (interpulse interval) revealed that the time courses
of recovery of KV4.3-WT, KV4.3-G581R or
KV4.3-L450F with KChIP2-encoded K+ currents
at −70 mV are well-described by single exponentials. The mean ± SE
time constants of recovery (τrecovery) of
KV4.3-WT, KV4.3-G581R or
KV4.3-L450F with KChIP2-encoded K+ currents
are 64.0±5.8 msec (n=6), 84.0±9.0 msec (n=7) and 71.2±5.9 msec,
respectively, and there were no statistical differences among
them.
KV4.3-G581R and
KV4.3-L450F increases the expression of KV4.3
protein
The mutations slow the
KV4.3/KChIP2-encoded channel inactivation, which
contributes to the increase of a gain of channel function, but
their influences on channel protein expression were not determined.
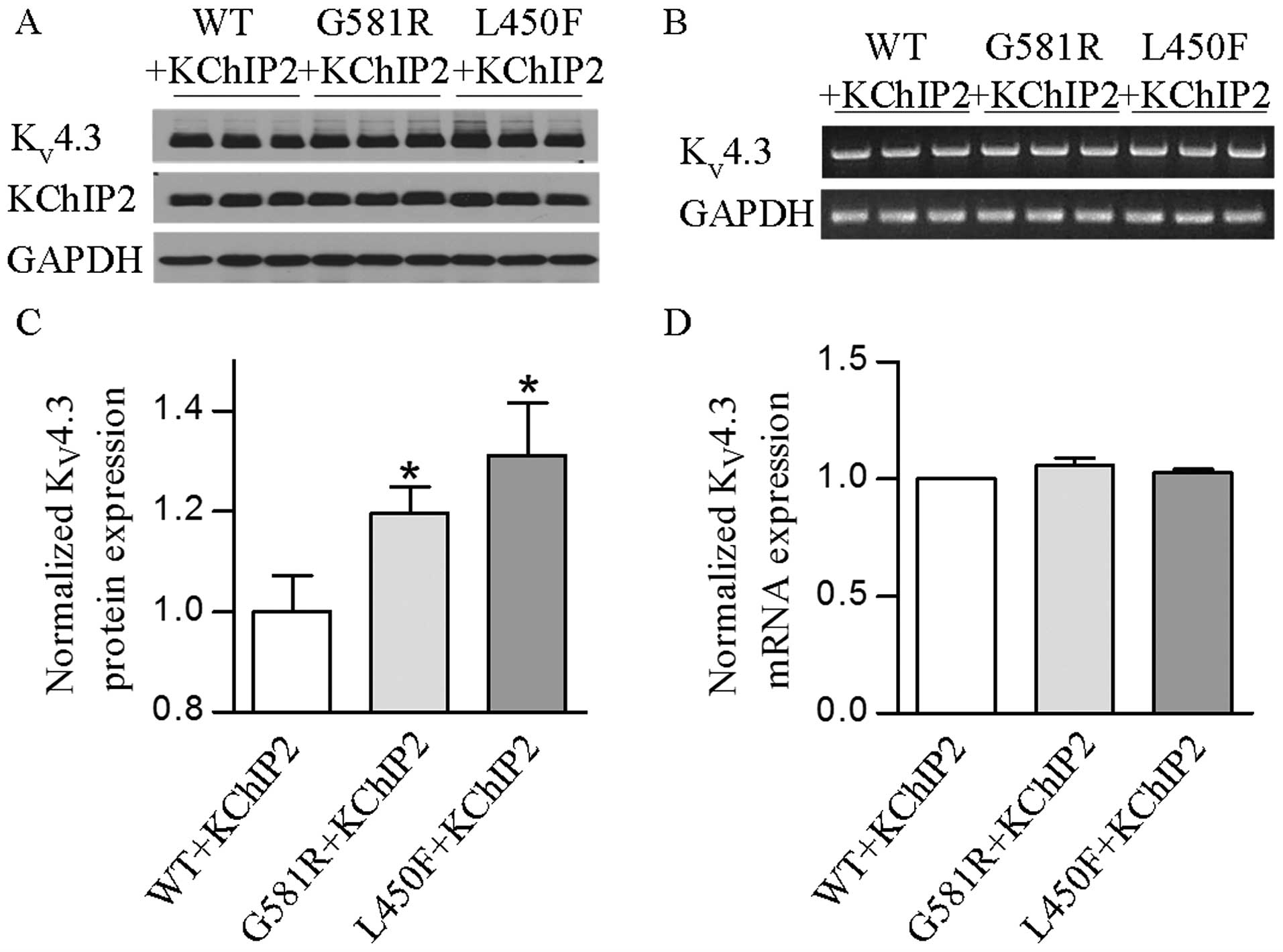
Western blotting was performed on protein extracts from HEK-293
cells for analysis of KV4.3-WT, KV4.3-G581R
and KV4.3-L450F with KChIP2 expression. The results
showed that two individual mutations significantly (P<0.05 or
P<0.01) increased the KV4.3 channel protein in the
presence of KChIP2 (Fig. 4A and
C). These findings indicate that increases of KV4.3
protein expression by two individual mutations also play an
important role in the gain of KV4.3- and
KV4.3/KChIP2-encoded K+ channels.
KV4.3-G581R and
KV4.3-L450F affects the localization of the
KV4.3 protein
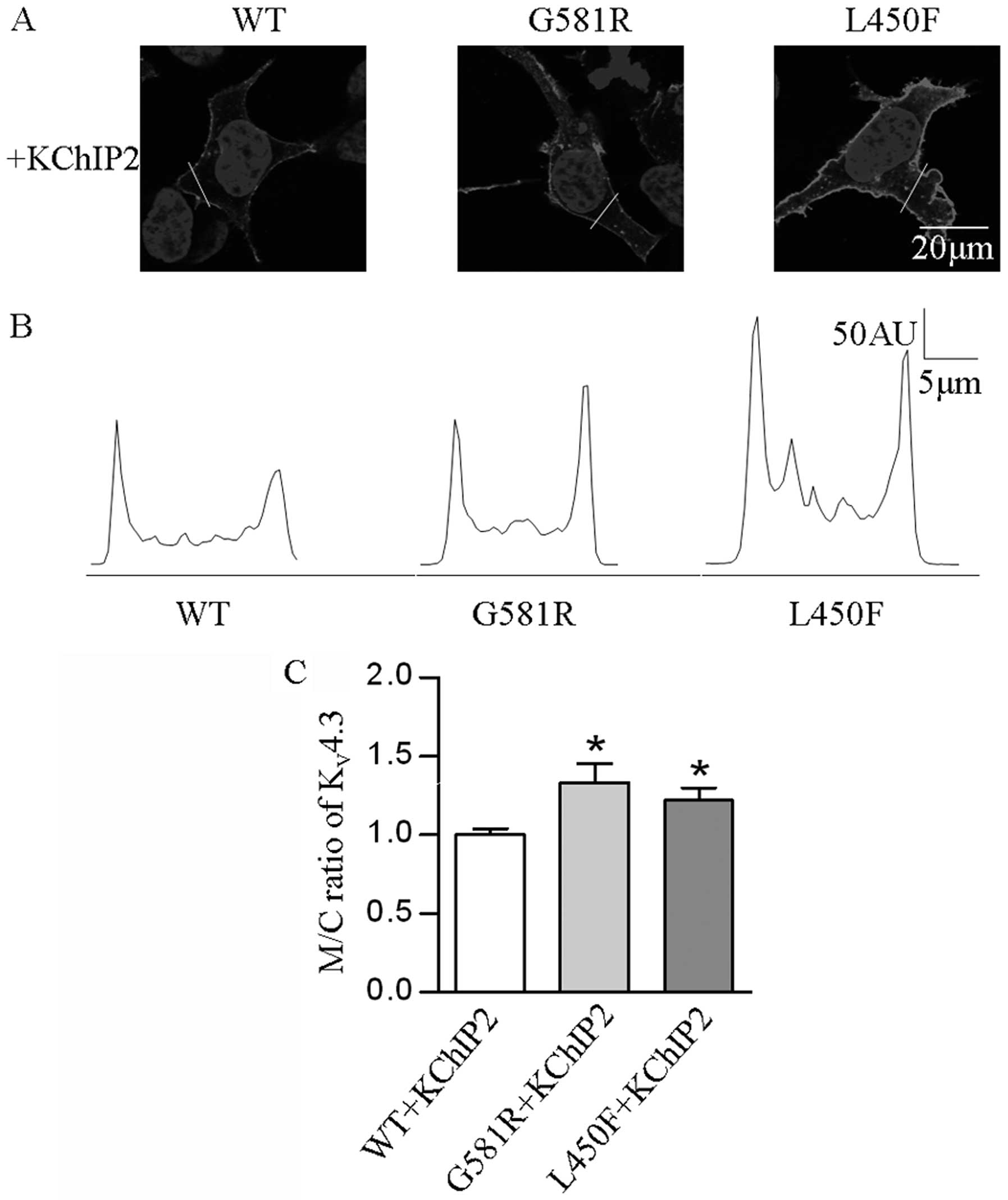
In order to determine if the two mutations affect
the localization of the KV4.3 channel protein in HEK-293
cells expressing KV4.3-WT, KV4.3-G581R and
KV4.3-L450F with KChIP2, confocal immunofluorescence
microscopy was employed. The results revealed a significantly
elevated (P<0.05) M/C ratio of KV4.3 channel in cells
expressing the individual mutants and KChIP2, as compared with
KV4.3-WT and KChIP2 (Fig.
5).
KV4.3-G581R and
KV4.3-L450F have no impact on KV4.3 mRNA
levels
Whether the mutation-induced increase of total and
cell surface KV4.3 protein occurred at its mRNA level
and/or post-translational trafficking remains unclear. Thus, this
was further investigated to determine if KV4.3-G581R and
KV4.3-L450F can affect KV4.3 channel
expression at the transcriptional level. Semi-quantitative PCR
revealed that there was no significant change of the
KV4.3 mRNA level in cells expressing each mutant plus
KChIP2 when compared with KV4.3-WT plus KChIP2 (Fig. 4B and D). These findings indicate
that the two mutations and KChIP2 do not play a regulatory role in
the mRNA processing of the KV4.3 channel.
Discussion
Taken together, our studies revealed that two
BrS-associated KCND3 mutations cause an increase of
Ito,f currents by altering channel kinetics and
increasing membrane protein expression. In comparison with previous
studies (1), the effects of the
mutations on the membrane protein expression were further studied.
The impacts of the rat mutations on the
KV4.3/KChIP2-encoded channel kinetics are slightly
different from those in humans. Regardless, increased transient
outward K+ channel functions by individual mutations are
the same. The present findings provide more insights into the
mechanisms of two KCND3 mutations leading to BrS.
BrS is a genetic heart disease that is characterized
by a specific ECG pattern and an increased risk of sudden cardiac
fatality (13). The typical BrS
electrocardiography findings present ST-segment elevation in leads
V1 to V3 and right bundle branch block. Na+-channel
blockers, such as ajmaline and flecainide (4,14),
have been shown to reveal certain concealed EGC patterns in
patients (7,15). Loss of the AP dome in the right
ventricular epicardium is thought to be caused by ST-segment
elevation in BrS. Abnormalities in electrical heterogeneity of the
right ventricular epicardium is associated with the development of
closely coupled premature ventricular contractions via a phase 2
reentrant mechanism that precipitates VT/VF (16).
The electrophysiological properties of ion channels
are determined by multiple factors, such as an interactive subunit
(8), temperature (17), pH value, ischemia, hypoxia,
chemicals and cytokines (18,19). The preponderance of evidence shows
that the genetic loss or gain-of-function of cardiac ion channels
due to gene mutations may disrupt normal electrical activity and
trigger cardiac arrhythmias, and even sudden cardiac fatality
(1–3). Thus far, 9 BrS-associated gene
mutations have been identified and include SCN5A,
GPD1L, CACNA1C, CACNB2, SCN1B,
KCNE3, SCN3B, HCN4 and KCND3 (1). Mutations in KCNE3 were shown
to induce the gain-of-function of the KV4.3 channel,
which can underlie the development of BrS (20). Recently, Giudicessi et al
(12) established a link between
KCND3 gene mutations and BrS, and demonstrated that two
BrS-associated mutations (KV4.3-G600R and
KV4.3-L450F) lead to the gain-of-function of
Ito, characterized by increasing Ito current
density and the loss of the AP dome, which was suggested as a
pathogenic substrate for BrS. However, the influences of the
individual mutations on the expression and function of the
KV4.3 channel, and the contribution of KChIP2 to the
mutation-induced gain-of-function of the KV4.3 channel
are unclear. Thus, whether the de novo KCND3 mutations are
sufficient to affect the expression and function of the
KV4.3 channel were investigated. Our studies revealed
that either of these two BrS-associated mutations in the
KCND3 gene produce an increase of transient outward
K+ current by increasing KV4.3 protein
expression, particularly in the cell surface. Additionally,
co-expression with KChIP2 will promote an increase of peak current
density, alter the channel kinetics and facilitate augmented
protein expression of KV4.3 mutants. In addition, there
were no significant changes in the mRNA level of the
KV4.3 channel in the cells expressing individual mutants
in conjunction with KChIP2. This suggests that the increased
expression of the KV4.3 channel protein by the mutations
may occur at the post-translational level as a result of increased
protein stability or decreased protein degradation.
In conclusion, two novel BrS-related mutations in
the KCND3 gene were found to cause a gain-of-function of
Ito by altering KV4.3 channel kinetics and
membrane protein expression. These findings provide a comprehensive
insight into the mechanism underlying BrS in two
mutation-associated types. Further investigations are required to
elucidate how the mutations enable KV4.3 to become more
stabilized or what other post-translational modifications are
involved in the increased membrane expression of the
KV4.3 channel.
Acknowledgments
Professor Haodong Xu was supported by a grant from
the National Institute of Health (no. K08HL088127), and Dr Faqian
Li was supported by a grant from the American Heart Association and
the Lawrence J. and Florence A. DeGeorge Charitable Trust (no.
10GRNT4460014).
References
|
1
|
Hedley PL, Jorgensen P, Schlamowitz S, et
al: The genetic basis of Brugada syndrome: A mutation update. Hum
Mutat. 30:1256–1266. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Janse MJ and Wilde AA: Molecular
mechanisms of arrhythmias. Rev Port Cardiol. 17(Suppl 2):
II41–II46. 1998.PubMed/NCBI
|
|
3
|
Antzelevitch C: Molecular biology and
cellular mechanisms of Brugada and long QT syndromes in infants and
young children. J Electrocardiol. 34(Suppl): S177–S181. 2001.
View Article : Google Scholar
|
|
4
|
Gussak I, Antzelevitch C, Bjerregaard P,
Towbin JA and Chaitman BR: The Brugada syndrome: Clinical,
electrophysiologic and genetic aspects. J Am Coll Cardiol. 33:5–15.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fish JM and Antzelevitch C: Cellular and
ionic basis for the sex-related difference in the manifestation of
the Brugada syndrome and progressive conduction disease phenotypes.
J Electrocardiol. 36(Suppl): S173–S179. 2003. View Article : Google Scholar
|
|
6
|
Antzelevitch C, Yan GX and Shimizu W:
Transmural dispersion of repolarization and arrhythmogenicity: The
Brugada syndrome versus the long QT syndrome. J Electrocardiol.
32(Suppl): S158–S165. 1999. View Article : Google Scholar
|
|
7
|
Brugada R, Campuzano O, Brugada P, Brugada
J and Hong K: Syndrome Brugada. Gene Reviews®
[Internet]. Pagon RA, Adam MP, Ardinger HH, et al: University of
Washington; Seattle: 1993–2015
|
|
8
|
Deschênes I and Tomaselli GF: Modulation
of KV4.3 current by accessory subunits. FEBS Lett.
528:183–188. 2002. View Article : Google Scholar
|
|
9
|
Kääb S, Dixon J, Duc J, et al: Molecular
basis of transient outward potassium current downregulation in
human heart failure: A decrease in Kv4.3 mRNA correlates
with a reduction in current density. Circulation. 98:1383–1393.
1998. View Article : Google Scholar
|
|
10
|
Antzelevitch C: Molecular basis for the
transmural distribution of the transient outward current. J
Physiol. 533:12001. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Niwa N and Nerbonne JM: Molecular
determinants of cardiac transient outward potassium current (I(to))
expression and regulation. J Mol Cell Cardiol. 48:12–25. 2010.
View Article : Google Scholar
|
|
12
|
Giudicessi JR, Ye D, Tester DJ, et al:
Transient outward current (I(to)) gain-of-function mutations in the
KCND3-encoded KV4.3 potassium channel and Brugada
syndrome. Heart Rhythm. 8:1024–1032. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Behr ER, Dalageorgou C, Christiansen M, et
al: Sudden arrhythmic death syndrome: familial evaluation
identifies inheritable heart disease in the majority of families.
Eur Heart J. 29:1670–1680. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Brugada J, Brugada P and Brugada R: The
syndrome of right bundle branch block ST segment elevation in V1 to
V3 and sudden death - the Brugada syndrome. Europace. 1:156–166.
1999. View Article : Google Scholar
|
|
15
|
Brugada R, Brugada J, Antzelevitch C, et
al: Sodium channel blockers identify risk for sudden death in
patients with ST-segment elevation and right bundle branch block
but structurally normal hearts. Circulation. 101:510–515. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kaufman ES: Mechanisms and clinical
management of inherited channelopathies: long QT syndrome, Brugada
syndrome, catecholaminergic polymorphic ventricular tachycardia,
and short QT syndrome. Heart Rhythm. 6:S51–55. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Singleton CB, Valenzuela SM, Walker BD, et
al: Blockade by N-3 polyunsaturated fatty acid of the
KV4.3 current stably expressed in Chinese hamster ovary
cells. Br J Pharmacol. 127:941–948. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Singarayar S, Singleton C, Tie H, et al:
Effects of components of ischemia on the KV4.3 current
stably expressed in Chinese hamster ovary cells. J Mol Cell
Cardiol. 34:197–207. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu W, Deng J, Xu J, et al: High-mobility
group box 1 (HMGB1) downregulates cardiac transient outward
potassium current (Ito) through downregulation of Kv4.2
and Kv4.3 channel transcripts and proteins. J Mol Cell Cardiol.
49:438–448. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Delpon E, Cordeiro JM, Núñez L, et al:
Functional effects of KCNE3 mutation and its role in the
development of Brugada syndrome. Circ Arrhythm Electrophysiol.
1:209–218. 2008. View Article : Google Scholar
|