Introduction
Aldosterone is a mineralocorticoid, which regulates
electrolyte transport through epithelial mineralocorticoid
receptors (1). There has been
increased attention on the role of aldosterone in the renal injury.
For several years, aldosterone was considered to execute its
function only at distal fragments of renal tubules, whereas,
subsequent investigation has revealed functional targets additional
to the distal renal tubule, including proximal tubular cells,
mesangial cells and podocytes (2). Several experimental data have
demonstrated that aldosterone is involved in the progression of
renal injury by triggering the production of reactive oxygen
species (ROS), resulting in the apoptosis of tubular cells
(3,4). Specifically, aldosterone has been
reported to promote the expression of nicotinamide adenine
dinucleotide phosphate oxidase in renal tissues, and generate ROS
to induce glomerular injury (5).
This can be inferred from evidence that aldosterone promotes
podocyte injury via the generation of ROS (6). In normal conditions, the balance of
ROS generation is maintained by the antioxidant defense system,
including superoxide dismutase (SOD) and catalase. However, when
the balance is broken, DNA and proteins are oxidized by excessive
ROS, accompanied by damage to organelles (7). In this case, autophagy is triggered
to degrade the oxidized DNA, proteins and damaged organelles.
Autophagy is highly conserved between yeasts and
humans, and is involved in the degradation of long-lived proteins
and damaged organelles (8). It
has been described as single- or double-membraned vesicles that
contain cytoplasm, including organelles, at various stages of
digestion (9). Autophagy in
mammals can be stimulated by stress, including starvation and
pathology, or by pharmacological agents, including rapamycin
(10). It is also triggered by
ROS, resulting from incomplete reduction of ROS (11). In addition, the induced autophagy
can be inhibited by antioxidant drugs, including N-acetylcysteine
(NAC), catalase, glutathione, melatonin and vitamin E (9).
In mammalian cells, several autophagy genes are
involved in the formation of autophagosomes, including
microtubule-associated protein 1 light chain 3 (MAP1-LC3; LC3),
phosphatidylinositol 3-kinase and beclin-1 (12). LC3-I is the cytosolic form of LC3
and, during autophagy, LC3-I is modified to the membrane-bound form
(LC3-II). Thus, the level of LC3-II is considered an early marker
for the formation of autophagosomes (13). Vps34 is part of the
autophagy-regulating macromolecular complex (PI3K complex),
consisting of Beclin-1/Atg6, Atg14/barkor and p150/Vps15. The
activity of Vps34 is increased by interaction with Beclin-1,
suggesting that increasing the expression of Beclin-1 may enhance
autophagy (14–17). Cells treated with pharmacological
agents, including 3-methyladenine (3-MA) and chloroquine can
inhibit autophagy (18).
Dysfunctional autophagy has been reported in several human
diseases, suggesting that decreased dysfunctional autophagy may
relieve these diseases, however, whether autophagy can be induced
by aldosterone in renal tubular cells and, if so, whether
aldosterone-induced autophagy can be relieved by certain drugs,
remains to be elucidated.
Ginsenoside-Rg1 (Rg1) is the major pharmacological
active component of ginseng, which is a widely used traditional
Chinese medicine (19). It has
extensive pharmacological activities, including antioxidant,
anti-inflammatory and anticancer properties (20,21). Previous investigation has
demonstrated that ginsenoside Rg1 enhances antioxidative protection
and intracellular calcium homeostasis in a cardiomyocyte
hypoxia/reoxygenation model (22), and can also inhibit autophagy in
cardiomyocytes exposed to hypoxia/reoxygenation (23). Our previous study reported the
effects of Rg1 in the protection of mouse podocytes from
aldosterone-induced injury by inhibiting ROS generation (24). Based on the properties of Rg1 on
relieving autophagy in several types of cell, it was hypothesized
that Rg1 may also promote antioxidative protection in renal tubular
cells to inhibit autophagy.
The present study was perforned to investigate the
potential for aldosterone to induce autophagy in NRK-52E cells and
to examine the effects of ginsenoside-Rg1 on relieving
aldosterone-induced autophagy. In addition, the molecular mechanism
underlying aldosterone-induced autophagy and Rg1-mediated therapy
was investigated.
Materials and methods
Cell culture
The NRK-52E rat renal tubular cell line was
purchased from the Type Culture Collection of the Chinese Academy
of Sciences (Shanghai, China). The cells were cultured in
Dulbecco’s modified Eagle’s medium (DMEM; GE Healthcare Life
Sciences, Logan, UT, USA) supplemented with 5% fetal bovine serum
(FBS; GE Healthcare Life Sciences), 100 U/ml penicillin and 100
mg/ml streptomycin at 37°C in 5% CO2 air, conditioned at
100% humidity.
Drug treatment
The NRK-52E cells were seeded in a 6-well plate at a
density of 0.5 million cells/well followed by drug treatment the
following day. The NRK-52E cells were incubated with aldosterone
(10−8 M), NAC (50 mM), chloroquine (CL; 10 µM),
3-Methyladenine (3-MA; l4 mM) and rapamycin (1 ng/ml),
respectively. All the above drugs were purchased from Sigma-Aldrich
(St. Louis, MO, USA). To inhibit autophagy, the cells were
pre-incubated with ginsenoside Rg1 (Rg1; 80 ng/ml) for 2 h (at
37°C), following which aldosterone was added to the culture for up
to 12 h. Rg1 (>98% pure) was purchased from DiDa Kexiang
Biological Co., Ltd. (Guizhou, China). Cells treated with fresh
medium only were used as the controls.
Transmission electron microscopy
(TEM)
For the analysis of autophagosomes using
transmission electron microscopy (TEM), following treatment, the
cells were washed briefly with phosphate-buffer saline (PBS) and
fixed with 3% glutaraldehyde (Sangon Biotech, Shanghai, China)
followed by post-fixation with 1% OsO4 (Sangon Biotech).
The sample was then dehydrated in acetone and embedded in Epon 812
(Nissin EM, Tokyo, Japan). Ultra-thin sections were prepared and
stained with 2.0% uranyl acetate/lead citrate, which were further
visualized using a Hitachi H-600IV electron microscope (Hitachi,
Ltd., Tokyo, Japan).
Western blot analysis
For total protein extraction, the cells were rinsed
with ice-cold PBS containing 5 mM EDTA and lysed with
radioimmunoprecipitation assay lysis buffer (20 µl; Sangon
Biotech) containing 1 mM phenylmethylsulfonyl fluoride (Beyotime
Biotech, Beijing, China) on ice. Following removal of cellular
debris by centrifugation (at 13,000 rpm at 4°C for 15 min), the
protein concentration was determined using a bicinchoninic acid
protein assay kit (Nanjing KeyGen Biotech Co., Ltd., Nanjing,
China). Total protein (25 µg) was resolved on an 10%
SDS-PAGE gel (Solarbio, Beijing, China) and transferred onto a
polyvinylidene difluoride membrane (EMD Millipore, Billerica MA,
USA). Following blocking with 5% milk, the blot was probed with the
indicated primary antibodies at 4°C overnight, followed by washing
with Tris-buffered saline with Tween 20 three times for 5 min each
time. The blots were then incubated with the corresponding
secondary antibody, to recognize the primary antibody, at room
temperature for 1 h. Following washing, as above, the signal was
developed using Immobilon Western Chemiluminescent HRP substrate
(EMD Millipore) and exposed to X-ray film (Kodak, Rochester, NY,
USA). The primary antibodies used in the present study were as
follows: LC3-II (#2775, polyclonal, rabbit anti-mouse, 1:1,000;
Cell Signaling Technology, Inc., Danvers, MA, USA), beclin-1
(#3495S, polyclonal, rabbit anti-mouse, 1:1,000; Cell Signaling
Technology, Inc.), SOD2 (#2299-1, polyclonal, rabbit anti-mouse,
1:2,000; Epitomics, Burlingame, CA, USA) and catalase (#8841,
polyclonal, rabbit anti-mouse, 1:1,000; Cell Signaling Technology,
Inc.) and the secondary antibodies were horseradish
peroxidase-conjugated secondary anti-mouse or anti-rabbit
antibodies (#ZB2301, goat anti-rabbit, 1:5,000; ZSGB-Bio, Beijing,
China).
Immunofluorescence
For immunofluorescence, the NRK-52E cells, grown on
coverslips, were harvested at indicated time-points and fixed with
4% paraformaldehyde (Sangon Biotech) at room temperature following
a brief rinse with PBS. The cells were then washed twice with PBS,
permeablized with 2.5% Triton X-100 (Sangon Biotech), blocked with
10% goat serum in PBS and incubated with primary rabbit polyclonal
anti-LC3B antibody (#2775, polyclonal, rabbit anti-mouse, 1:400;
Cell Signaling Technology, Inc.) overnight at 4°C in a humidified
chamber. Subsequently, the cells were incubated with Alexa
Fluor® 488-conjugated donkey anti-rabbit IgG (H+L)
secondary antibody (#A21206, 1:400; Invitrogen Life Technologies,
Carlsbad, CA, USA) and rhodamine-labeled phalloidin (# R415,
1:1,000; Invitrogen Life Technologies) at room temperature for 1 h.
The nuclei of the cells were visualized using
4′,6-diamino-2-phenylindole (DAPI) staining. Finally,
immunofluorescence images were captured using a Nikon Eclipse 50i
microscope (Nikon) and the number of green fluorescent puncta in 50
cells/group were counted..
Acridine orange staining
The NRK-52E cells were grown on coverslips to 40%
density, and treated with the indicated drug combinations,
described above, for the indicated period of time. The cell medium
was replaced with fresh medium containing acridine orange (0.05
g/ml; Sigma-Aldrich) and Hoechst 33342 (1 µg/ml; Invitrogen
Life Technologies) and incubated for a further 15 min (at 37°C) in
the cell culture incubator. Following brief washing, the cells were
fixed with 4% paraformaldehyde at room temperature for 10 min. The
cells were subjected a to additional washing in PBS for 5 min, and
the corresponding coverslip was mounted to a glass slide. Images of
the cells were captured using a Nikon Eclipse 50i microscope
(Nikon).
Measurement of intracellular levels of
ROS
Intracellular ROS was measured using a
2,7-dichlorofuorescin diacetate (DCFH-DA) assay, which was
performed according to the manufacturer’s instructions (Nanjing
KeyGen Biotech). Briefly, 5 µl DCFH-DA (1 mM) was added to
each well of a 6-well plate; Rosup (1:1,000) was added to the well
as a positive control of ROS, and the cells were cultured for 30
min at 37°C in the dark. Following washing three times with PBS,
the cells were analyzed using a flow cytometer (FACSAria; BD
Biosciences, San Jose, CA, USA) and data were processed using the
CellQuest program (version 3.2.1; BD Biosciences).
Data analysis
Data are presented as the mean ± standard deviation.
Multiple groups of quantitative data were compared using analysis
of variance. Data were analyzed using SPSS software 13.0 (SPSS,
Inc., Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
Aldosterone promotes autophagy in NRK-52E
cells
To investigate the formation of autophagy in
aldosterone-treated NRK-52E cells in the present study, TEM was
used to observe the ultrastructural changes. This observation
revealed that certain morphological changes had appeared, including
the formation of double- and multiple-membrane encapsulated
portions of the cytoplasm and/or organelles (autophagosomes)
(Fig. 1A). Increased numbers of
autophagosomes were observed in the aldosterone-treated NRK-52E
cells, compared with the control cells (Fig. 1B). In order to further confirm
that the autophagy in the NRK-52E cells was induced by aldosterone,
immunofluorescence analysis was performed. The NRK-52E cells were
treated with anti-LC3B antibody, following which the green
fluorescent puncta, which represented autophagosomes, were examined
under fluorescence microscopy (Fig.
1C). Prior to antibody treatment, the cells were pre-incubated
with either buffer (control), aldosterone, rapamycin, rapamycin +
aldosterone, chloroquine, chloroquine + aldosterone, 3-MA or 3-MA +
aldosterone for 2 h. Subsequently, the green fluorescent puncta of
50 cells in each group were counted (Fig. 1D). Western blot analysis
demonstrated that aldosterone promoted the expression of Beclin-1
and the conversion of LC3-I to LC3-II in the NRK-52E cells
(Fig. 1E and F).
 | Figure 1Aldosterone promotes autophagy in
NRK-52E cells. (A) Cells were treated with buffer (control) or 10
nmol/l aldosterone for 12 h, respectively. Under a transmission
electron microscope (original magnification, x10,000), increased
numbers of autophagosomes were observed in the ALDO-treated
cells.(B) Comparison of the number of autophagosomes between the
control and ALDO-treated cells, 20 cells were counted in each group
(n=3). *P<0.05, vs. control. (C) Immunofluorescence
analysis of the expression of LC3-II in NRK-52E cells exposed to
drugs for 12 h. Green fluorescent puncta represent autophagosomes.
Images were captured under fluorescence microscopy (magnification,
x630). (D) Percentage of autophagosomes with green-fluorescent
puncta in each group (50 cells/group; n=3). **P<0.01,
vs. control. *P<0.05 vs. rapamycin.
#P<0.05 vs. CL. (E) Western blot analysis to
determine the protein levels of LC3-I, LC3-II and Beclin-1. (F)
Quantification of protein levels normalized to β-actin.
*P<0.05, vs. control in the LC3-II group.
**P<0.01, vs. control in the Beclin-1 group. Data are
expressed as the mean ± standard deviation. ALDO, aldosterone;
3-MA, 3-Methyladenine; CL, chloroquine; Rapa, rapamycin. |
Aldosterone induces autophagy through the
generation of ROS
The NRK-52E cells were stained with acridine orange
to investigate the formation of aldosterone-induced autophagic
vacuoles (Fig. 2A and B). Under
confocal microscopy, the number of autophagic vacuole was
significantly increased in the cells treated with aldosterone,
rapamycin, and rapamycin + aldosterone. Notably, fewer green
fluorescent puncta were observed following incubation with 3-MA and
NAC, compared with the aldosterone-induced cells. Western blot
analysis demonstrated that the expression of Beclin-1 and LC3-II
were inhibited in the cells treated with NAC (Fig. 2C and E). Western blot analysis was
also performed to detect the expression levels of SOD2 and
catalase. The cells treated with aldosterone promoted the
expression levels of SOD2 and catalase, inversely, the expression
levels of SOD2 and catalase were inhibited in the NAC-pretreated
cells (Fig. 2D and F).
 | Figure 2Aldosterone-induced autophagy is
reduced through inhibition of reactive oxygen species generation.
(A) NRK-52E cells were incubated with acridine orange (0.05
µg/ml) and Hoechst 33342 (1 µg/ml) for 15 min
following drug treatment for 12 h, and were observed using confocal
microscopy. Green fluorescent puncta represent autophagic vacuoles
(magnification, x800). (B) Number of autophagic vacuoles in each
group (50 cells/group; n=3). **P<0.01, vs. control.
*P<0.05, vs. aldosterone. (C) Western blot analysis
to detect the protein levels of Beclin-1, LC3-I and LC3-II in
NRK-52E cells exposed for 12 h to buffer (control), ALDO, NAC or
NAC + ALDO. (D) Western blot analysis to detect the protein levels
of SOD2 and catalase. (E) Quantification of protein levels of
Beclin-1, LC3-I and LC3-II, normalized to β-actin.
*P<0.05, vs. aldosterone in each group. (F)
Quantification of protein levels of SOD2 and catalase, normalized
to β-actin. *P<0.05, vs. aldosterone in each group.
Data are expressed as the mean ± standard deviation. ALDO,
aldosterone; 3-MA, 3-Methyladenine; NAC, N-acetylcysteine; SOD2,
superoxide dismutase 2. |
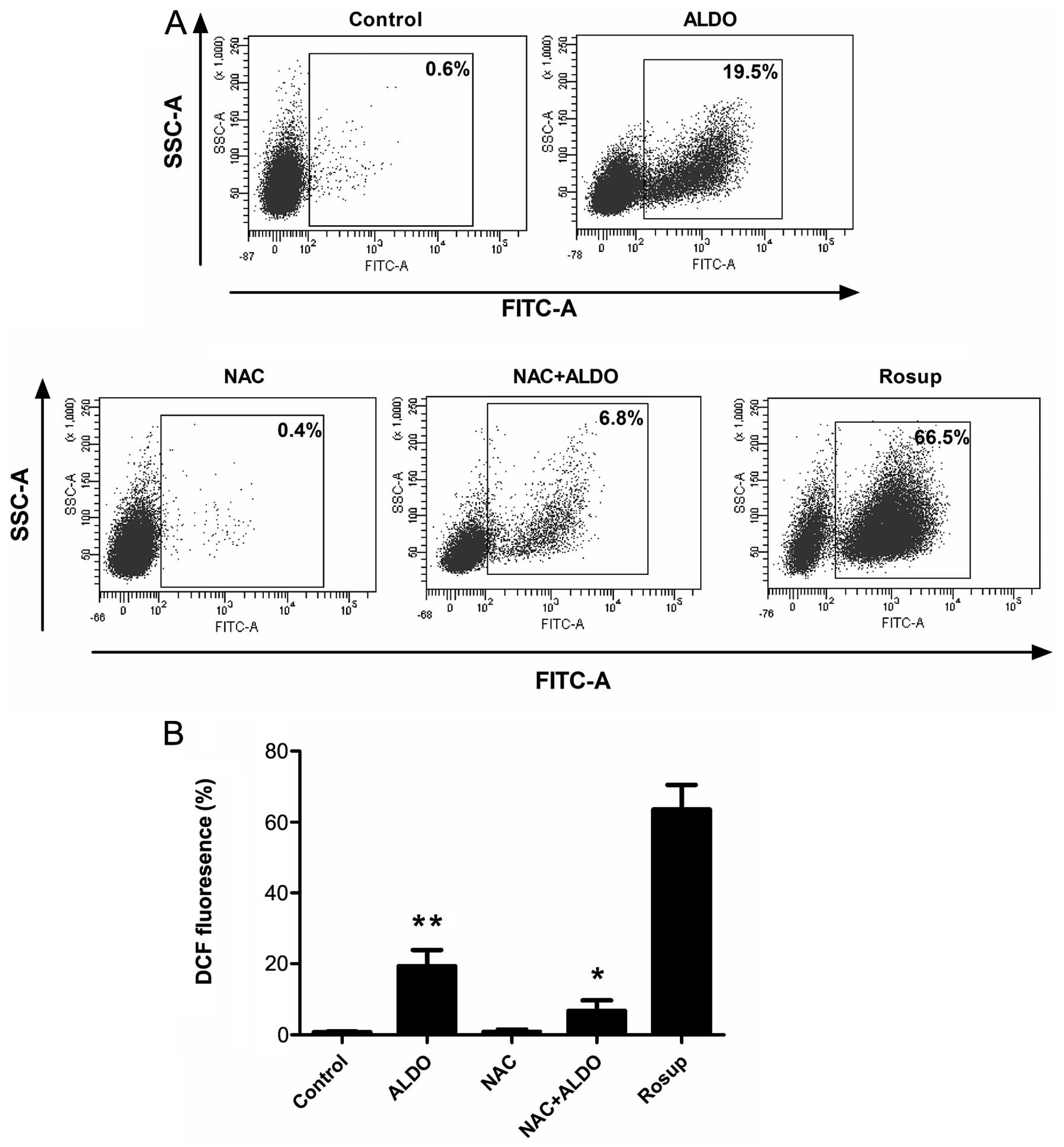
To determine the levels of oxidative stress in the
aldosterone-treated cells, the cells were stained with DCFH-DA,
which is oxidized to DCF by intracellular ROS, and the fluorescence
of the DCF was measured using flow cytometry (Fig. 3). The results revealed that
treatment with NAC markedly reduced the generation of
aldosterone-induced ROS. Rosup was used as a positive control.
Taken together, these findings suggested that cells treated with
NAC repressed the aldosterone-induced autophagy by inhibiting the
generation of ROS.
Effects of Rg1 on aldosterone-induced
autophagy in NRK-52E cells
To investigate the effects of Rg1 on
aldosterone-induced autophagy, the NRK-52E cells were pre-treated
with control (buffer), aldosterone, Rg1 and Rg1 + aldosterone, and
the proteins were isolated from these cells to perform western blot
analysis for catalase, SOD2, LC3B and Beclin-1. The results
demonstrated that cells incubated with aldosterone showed higher
protein levels of SOD2 and catalase, compared with the control
cells, and the cells treated with Rg1 exhibited lower protein
levels of SOD2 and catalase following pre-treatment with
aldosterone, compared with the cells treated with aldosterone only.
This indicated that Rg1 inhibited the generation of ROS. In
addition, the protein levels of LC3-II and Beclin-1 were
significantly increased in the aldosterone-treated cells, compared
with the control, however, when the cells incubated with Rg1
following aldosterone treatment, the protein levels of LC3-II and
Beclin-1 decreased (Fig. 4).
These data suggested that Rg1 relieved aldosterone-induced
autophagy by inhibiting the generation of ROS in the NRK-52E
cells.
Rg1 inhibits aldosterone-induced
autophagy in NRK-52E cells by regulating the AMPK/mTOR signaling
pathway
To investigate the signaling pathway involved in the
inhibition of autophagy by Rg1, the presents study performed
western blot analysis for phosphorylated (p)-AMPK and AMPK
(Fig. 5A). Proteins were isolated
from the cells following incubation with buffer, aldosterone,
aldosterone + Compound C or aldosterone + Rg1 for 12 h. Following
pre-treatment with aldosterone, the expression of p-AMPK in the
NRK-52E cells was significantly increased, compared with the
control, however, there was no significant change in the expression
of AMPK, suggesting that aldosterone may induce the phosphorylation
of AMPK. However, the expression of p-AMPK was reduced to normal
levels following incubation of the aldosterone-treated cells with
Compound C, which is an inhibitor of AMPK. In addition, the
expression of p-AMPK was also reversed in the aldosterone-treated
cells by Rg1, indicating that Rg1 may reduce the
aldosterone-induced phosphorylation of AMPK. The ratio of
p-AMPK/AMPK was calculated to measure the phosphorylation of AMPK
(Fig. 5B). It was previously
demonstrated that activated AMPK induces autophagy in cells by
inhibiting the activation of mTOR and phosphorylation of p70
ribosomal S6 protein kinase (P70S6K), which acts as an important
downstream target protein in the mTOR signaling pathway (25). Therefore, the present study
performed western blot analysis for p-P70S6K and P70S6K (Fig. 5C). Proteins were harvested from
the cells following pre-treatment with buffer, aldosterone, Rg1 and
aldosterone + Rg1 for 24 h. The results demonstrated that the
expression of p-P70S6K was markedly reduced in the
aldosterone-treated cells, compared with the control, and
incubation of the cells with aldosterone and Rg1 reversed the
reduced expression of p-P70S6K to normal, suggested that Rg1
recovered the phosphorylation of P70S6K, which reduced by
aldosterone. The p-P70S6K/P70S6K ratio was calculated to measure
the phosphorylation of P70S6K (Fig.
5D). Taken together, these data suggested that Rg1 inhibited
aldosterone-induced autophagy through regulation of the AMPK/mTOR
signaling pathway in NRK-52E cells.
 | Figure 5Rg1 inhibits aldosterone-induced
autophagy in NRK-52E cells by regulating the AMPK/mTOR signaling
pathway. (A) Western blot analysis to detect the protein levels of
p-AMPK and AMPK. Proteins were isolated from cells following
pre-treatment with buffer (control), ALDO, ALDO + Compound C or
ALDO + Rg1, (B) Value of p-AMPK/AMPK. *P<0.05, vs.
aldosterone. (C) Western blot analysis to detect the protein levels
of p-P70S6K and P70S6K. Proteins were isolated from the cells
following pretreatment with buffer (control), ALDO, Rg1 or ALDO +
Rg1. (D) Value of p-P70S6K/P70S6K. *P<0.05, vs.
aldosterone. Data are expressed as the mean ± standard deviation.
ALDO, aldosterone; SOD2, superoxide dismutase 2; Rg1,
ginsenoside-Rg1; P70S6K, p70 ribosomal S6 protein kinase; p-P70S6K,
phosphorylated P70S6K. |
Discussion
In the present study, NRK-52E cells were exposed to
aldosterone to examine the potential for aldosterone to induce
autophagy in renal tubular epithelial cells, and to investigate the
association between aldosterone-induced autophagy and ROS
generation, as well as the effects of Rg1 on this process.
Initially, increased autophagosomes and elevated conversion of
LC3-I to LC3-II were observed in the aldosterone-treated NRK-52E
cells, suggesting enhanced autophagy in these cells, which was
further validated using acridine orange staining, flow cytometry
and western blot analysis. Furthermore, increased ROS generation
was observed to mediat aldosterone-induced autophagy and this was
reversed by Rg1, possibly through regulation of the AMPK/mTOR
signaling pathway.
Aldosterone is involved in various renal
pathologies, including glomerulosclerosis, proteinuria and fibrosis
(26). A number of previous
studies investigating the role of aldosterone in renal diseases
have revealed the importance of aldosterone in the processes of
inflammation and fibrosis (27,28). Furthermore, these reports also
demonstrated that aldosterone promotes the production of ROS via
MR-dependent mechanisms, and that inhibiting ROS synthesis can
prevent the progression of proteinuria and ameliorate renal injury,
suggesting the importance of oxidative stress in
aldosterone-induced renal injury. However, accumulating evidence
has suggested that ROS is important for the regulation of
autophagy. Thus, the present study hypothesized that aldosterone
promotes renal injury through inducing autophagy via the generation
of ROS. Our previous report demonstrated that aldosterone induces
podocyte autophagy through the ROS signaling pathway (24). In the present study, to further
investigate the importance of aldosterone in renal injury,
aldosterone-induced autophagy was examined in the NRK-52E renal
tubular cell line, which is another important cell in kidney.
To investigate the effects of aldosterone in NRK-52E
cells, the cells were exposed to 10 nmol/l aldosterone, and the
results revealed that the formation of autophagosomes was notably
increased in cells treated with aldosterone, compared with the
control cells. Consistent with previous reports, chloroquine and
rapamycin markedly promoted autophagy in the aldosterone-treated
cells in the present study (23,24). Chloroquine is a lysosomal
inhibitor, which inhibits lysosome-autophagosome fusion, and
rapamycin is an inhibitor of mTOR. To investigate the formation of
autophagosomes, the present study measured the LC3-II, the protein
levels of which in cells represent the dynamic balance between the
production and degradation of autophagosomes (13). The results revealed that the
expression of LC3-II was elevated in aldosterone-treated cells. In
addition, the expression of Beclin-1, which is involved in the
formation of autophagosomes, was markedly increased in these cells.
In the present study, the results of the immunofluorescence
analysis demonstrated that the effects on the cells treated with
aldosterone were the same effects as those exposed to chloroquine
or rapamycin, and further enhanced aldosterone-induced autophagy.
By contrast, 3-MA inhibited the formation of autophagosome in the
aldosterone-treated cells. These results suggested that aldosterone
promoted autophagy in the NRK-52E cells.
To elucidate the role of antioxidation in autophagy
in the present study, the NRK-52E cells were treated with the
antioxidant, NAC, following incubation with aldosterone, which
resulted in the formation of fewer autophagosomes in the NAC +
aldosterone treatment group, compared with the aldosterone
treatment group, assessed by staining with acridine orange. Further
investigation demonstrated that cellular antioxidative enzymes,
including SOD2 and catalase, were increased in aldosterone-treated
cells, indicatinga protective mechanism against ROS-mediated
injury. In addition, the protein levels of SOD2 and catalase were
notably decreased in the cells treated with NAC, which also
inhibited the expression of Beclin-1 and LC3-II. Results of the
flow cytometry also confirmed the increased oxidation in the
aldosterone-treated cells, which was reversed by NAC. Therefore, it
was hypothesized that inhibiting the generation of ROS can reduce
aldosterone-induced autophagy.
Due to the property of antioxidants, ginsenoside-Rg1
was used to inhibit the generation of ROS, thereby attempting to
reduce aldosterone-induced autophagy in the NRK-52E cells. Previous
reports have already demonstrated the role of Rg1 in inhibiting
autophagy in hypoxia/reoxygenation-treated H9c2 cardiomyocytes. The
results of the present study revealed that the cells pretreated
with Rg1 reduced the expression of SOD2, catalase, Beclin-1 and
LC3-II. In addition, Rg1 significantly inhibited
aldosterone-induced autophagy. To investigate the signaling
pathways involved in Rg1 inhibiting autophagy, the expression
levels of AMPK and p-AMPK were examined. AMPK is a major regulator
of energy homeostasis, and adenosine triphosphate depletion can
activate AMPK to decompose fatty acids and induce autophagy
(29). In addition, AMPK can
activate TSC1/2, and inhibit Rheb and mTORC1, promoting autophagy.
P70S6K, is a downstream serine/threonine kinase of mTOR, and
activated mTOR is involved in several cellular functions by
phosphorylating specific factors, including P70S6K (30). In the present study, Rg1 inhibited
the activation of AMPK by reducing the phosphorylation of AMPK and
increasing the expression of p-P70S6K in the NRK-52E cells, which
revealed that the activation of Rg1 may involve in AMPK/mTOR
signaling pathway.
In conclusion, the present study demonstrated that
aldosterone induced autophagy via the generation of ROS in NRK-52E
cells. Rg1 notably relieved oxidative stress in the
aldosterone-pre-treated NRK-52E cells and inhibited the activation
of AMPK to sustain mTOR activity, thereby markedly reducing
aldosterone-induced autophagy. These findings provide novel insight
into the protective effects of Rg1 in renal tubular cells and may
assist in developing novel treatments for kidney diseases.
Acknowledgments
The present study was supported by grants from the
National Natural Science Foundation of China (grant. no. 81170667),
the Youth Science and Technology Support Program of Sichuan
Province (grant. no. 2011JDT0014), the Education Department Program
of Sichuan Provine (grant. no. 15ZA0253) and the Chengdu Medical
College Program (grant. no. CYZ14-011).
References
|
1
|
Crabbe J: Stimulation of active sodium
transport by the isolated toad bladder with aldosterone in vitro. J
Clin Invest. 40:2103–2110. 1961. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nguyen Dinh Cat A and Jaisser F:
Extrarenal effects of aldosterone. Curr Opin Nephrol Hypertens.
21:147–156. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
McGowan AJ, Fernandes RS, Samali A and
Cotter TG: Anti-oxidants and apoptosis. Biochem Soc Trans.
24:229–233. 1996.PubMed/NCBI
|
|
4
|
Patni H, Mathew JT, Luan L, Franki N,
Chander PN and Singhal PC: Aldosterone promotes proximal tubular
cell apoptosis: Role of oxidative stress. Am J Physiol Renal
Physiol. 293:F1065–F1071. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Nishiyama A and Abe Y: Aldosterone and
renal injury. Nippon Yakurigaku Zasshi. 124:101–109. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Shibata S, Nagase M, Yoshida S, Kawachi H
and Fujita T: Podocyte as the target for aldosterone: Roles of
oxidative stress and Sgk1. Hypertension. 49:355–364. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Queisser N, Amann K, Hey V, Habib SL and
Schupp N: Blood pressure has only minor influence on
aldosterone-induced oxidative stress and DNA damage in vivo. Free
Radic Biol Med. 54:17–25. 2013. View Article : Google Scholar
|
|
8
|
Yu L: Recent progress in autophagy. Cell
Res. 24:1–2. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ravikumar B, Sarkar S, Davies JE, Futter
M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M,
Korolchuk VI, Lichtenberg M, Luo S, et al: Regulation of mammalian
autophagy in physiology and pathophysiology. Physiol Rev.
90:1383–1435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mizushima N, Levine B, Cuervo AM and
Klionsky DJ: Autophagy fights disease through cellular
self-digestion. Nature. 451:1069–1075. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen Y and Gibson SB: Is mitochondrial
generation of reactive oxygen species a trigger for autophagy?
Autophagy. 4:246–248. 2008. View Article : Google Scholar
|
|
12
|
Xie Z and Klionsky DJ: Autophagosome
formation: Core machinery and adaptations. Nat Cell Biol.
9:1102–1109. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kabeya Y, Mizushima N, Ueno T, Yamamoto A,
Kirisako T, Noda T, Kominami E, Ohsumi Y and Yoshimori T: LC3, a
mammalian homologue of yeast Apg8p, is localized in autophagosome
membranes after processing. EMBO J. 19:5720–5728. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kihara A, Kabeya Y, Ohsumi Y and Yoshimori
T: Beclin-phosphatidylinositol 3-kinase complex functions at the
trans-Golgi network. EMBO Rep. 2:330–335. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Furuya N, Yu J, Byfield M, Pattingre S and
Levine B: The evolutionarily conserved domain of Beclin 1 is
required for Vps34 binding, autophagy and tumor suppressor
function. Autophagy. 1:46–52. 2005. View Article : Google Scholar
|
|
16
|
Itakura E, Kishi C, Inoue K and Mizushima
N: Beclin 1 forms two distinct phosphatidylinositol 3-kinase
complexes with mammalian Atg14 and UVRAG. Mol Biol Cell.
19:5360–5372. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Sun Q, Fan W, Chen K, Ding X, Chen S and
Zhong Q: Identification of Barkor as a mammalian autophagy-specific
factor for Beclin 1 and class III phosphatidylinositol 3-kinase.
Proc Natl Acad Sci USA. 105:19211–19216. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Butler D, Brown QB, Chin DJ, Batey L,
Karim S, Mutneja MS, Karanian DA and Bahr BA: Cellular responses to
protein accumulation involve autophagy and lysosomal enzyme
activation. Rejuvenation Res. 8:227–237. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Lü JM, Yao Q and Chen C: Ginseng
compounds: An update on their molecular mechanisms and medical
applications. Curr Vasc Pharmacol. 7:293–302. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Liu CX and Xiao PG: Recent advances on
ginseng research in China. J Ethnopharmacol. 36:27–38. 1992.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Mochizuki M, Yoo YC, Matsuzawa K, Sato K,
Saiki I, Tono-oka S, Samukawa K and Azuma I: Inhibitory effect of
tumor metastasis in mice by saponins, ginsenoside-Rb2, 20(R)-and
20(S)-ginsenoside-Rg3, of red ginseng. Biol Pharm Bull.
18:1197–1202. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhu D, Wu L, Li CR, Wang XW, Ma YJ, Zhong
ZY, Zhao HB, Cui J, Xun SF, Huang XL, et al: Ginsenoside Rg1
protects rat cardiomyocyte from hypoxia/reoxygenation oxidative
injury via antioxidant and intracellular calcium homeostasis. J
Cell Biochem. 108:117–124. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zhang ZL, Fan Y and Liu ML: Ginsenoside
Rg1 inhibits autophagy in H9c2 cardiomyocytes exposed to
hypoxia/reoxygenation. Mol Cell Biochem. 365:243–250. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Mao N, Cheng Y, Shi XL, Wang L, Wen J,
Zhang Q, Hu QD and Fan JM: Ginsenoside Rg1 protects mouse podocytes
from aldosterone-induced injury in vitro. Acta Pharmacol Sin.
35:513–522. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kim J, Kundu M, Viollet B and Guan KL:
AMPK and mTOR regulate autophagy through direct phosphorylation of
Ulk1. Nat Cell Biol. 13:132–141. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ibrahim HN, Rosenberg ME and Hostetter TH:
Role of the renin-angiotensin-aldosterone system in the progression
of renal disease: A critical review. Semin Nephrol. 17:431–440.
1997.PubMed/NCBI
|
|
27
|
Namsolleck P and Unger T: Aldosterone
synthase inhibitors in cardiovascular and renal diseases. Nephrol
Dial Transplant. 29(Suppl 1): i62–i68. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Nagase M, Yoshida S, Shibata S, Nagase T,
Gotoda T, Ando K and Fujita T: Enhanced aldosterone signaling in
the early nephropathy of rats with metabolic syndrome: possible
contribution of fat-derived factors. J Am Soc Nephrol.
17:3438–3446. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hardie DG: AMP-activated/SNF1 protein
kinases: conserved guardians of cellular energy. Nat Rev Mol Cell
Biol. 8:774–785. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xu JT, Zhao X, Yaster M and Tao YX:
Expression and distribution of mTOR, p70S6K, 4E-BP1, and their
phosphorylated counterparts in rat dorsal root ganglion and spinal
cord dorsal horn. Brain Res. 1336:46–57. 2010. View Article : Google Scholar : PubMed/NCBI
|