Introduction
Liver fibrosis is characterized by the excessive
deposition of extracellular matrix (ECM) in the hepatic parenchyma
and occurs following complex interactions between matrix-producing
hepatic stellate cells (HSCs) and liver-resident and infiltrating
cells (1). Fibrosis is an
intrinsic response to chronic injury, maintaining organ integrity
when extensive necrosis or apoptosis occurs. With protracted
damage, fibrosis can progress toward excessive scarring and organ
failure, as in liver cirrhosis. Although hepatic fibrosis in humans
can be caused by various stimuli (congenital, metabolic,
inflammatory, parasitic, vascular, toxins or drugs), the molecular
mechanisms underlying the development of fibrosis are basically the
same (2). Following liver injury,
a defined program of molecular changes occurs that is tightly
orchestrated at the cellular and molecular level (3). Importantly, fibrosis is no longer
considered static, but the result of a continuous remodeling
process. This process is characterized mainly by the activation of
HSCs which acquire a myofibroblast (MFB) phenotype and are able to
express and deposit large quantities of ECM components within the
liver (4,5). If the injury is temporary, these
changes are transient and liver fibrosis may not occur. If the
injury is sustained, however, chronic inflammation and the
accumulation of the ECM persist, leading to the progressive
substitution of the normal liver parenchyma by scar tissue. In the
pathogenesis of chronic liver disease, the ECM homeostasis is
further disturbed by an unbalanced activity of matrix
metalloproteinases (MMPs) and their tissue inhibitors (TIMPs).
Experimental studies conducted on isolated primary hepatic cells
and experimental animal models have led to the identification of
several pathogenetic mediators, namely signaling pathways that are
involved in the fibrogenic response. The aberrant activity of
transforming growth factor (TGF)-β1 or members of the
platelet-derived growth factor family are the most prominent
drivers of the activation and transdifferentiation of HSCs into
MFBs.
To date, the antifibrotic treatment of fibrosis
represents an unconquered area for drug development, with enormous
potential but also high risks. Preclinical research has yielded
numerous targets for antifibrotic agents, some of which have
entered early-phase clinical studies, but progress has been
hampered due to the relative lack of sensitive and specific
biomarkers to measure the progression or reversal of fibrosis
(6,7). The aim of the present study was to
identify candidate genes and putative biomarkers for the diagnosis
and treatment of hepatic fibrosis.
Materials and methods
Initial screening of microarray
datasets
The microarray datasets used in this study were
obtained from NCBI Gene Expression Omnibus (GEO, http://www.ncbi.nlm.nih.gov/geo/) (8,9)
and ArrayExpress (10). MESH
terms ʽliver fibrosis', ʽmyofibroblasts' and ʽhepatic stellate
cells' were used to identify potential datasets of interest, and
GSE34949 and E-MTAB-2081 were selected for the analysis. Both
datasets included whole-genome transcriptional data from rodent
HSCs in the quiescent and activated state. For GSE34949, total RNA
was extracted from primary C57BL/6 mouse HSCs on days 0 and 7 of
culture. For the E-MTAB-2081 dataset, RNA samples were obtained
from quiescent and activated HSCs derived from Sprague-Dawley rats.
For each dataset, we performed moderate t-statistics to generate a
list of upregulated genes. We used a threshold value of p<0.05
and a fold change of >2. In order to identify a robust
fibrogenic gene signature, the web-based application, Venn Diagram
Generator (http://www.bioinformatics.lu/venn.php), was used to
select the differentially expressed genes (DEGs) commonly shared
between the two datasets.
Functional analysis of genes of the
fibrogenic gene signature
Functional analysis was performed on the selected
genes using the Database for Annotation, Visualization and
Integrated Discovery (DAVID) Bioinformatics Database (http://david.abcc.ncifcrf.gov/), as previously
described (11). The biological
significance of these genes was queried by performing enrichment
analysis for Gene Ontology (GO) categories, including biological
processes (BPs) and molecular functions (MFs), for domain
architectures [Simple Modular Architecture Research Tool (SMART)]
and for the identification of significant biological pathways. The
Kyoto Encyclopedia of Genes and Genomes (KEGG), a collection of
manually curated pathways and molecular networks, was used as
reference. In order to construct a series of interconnected
networks, the selected genes were subjected to k-means clustering
using the STRING database (http://string-db.org/). STRING provides a global view
of all the available interaction data by creating networks, which
represent the current knowledge on the functional interconnections
among genes. All associations are provided with a confidence score
that represents an estimation on how likely a given association
describes a functional linkage between two genes. Clustering was
performed for five clusters and disconnected nodes were excluded.
The web server, REVIGO (http://revigo.irb.hr), was used to summarize the lists
of GO terms in order to find representative subsets of the terms
that rely on semantic similarity.
Results
Identification of fibrogenic genes
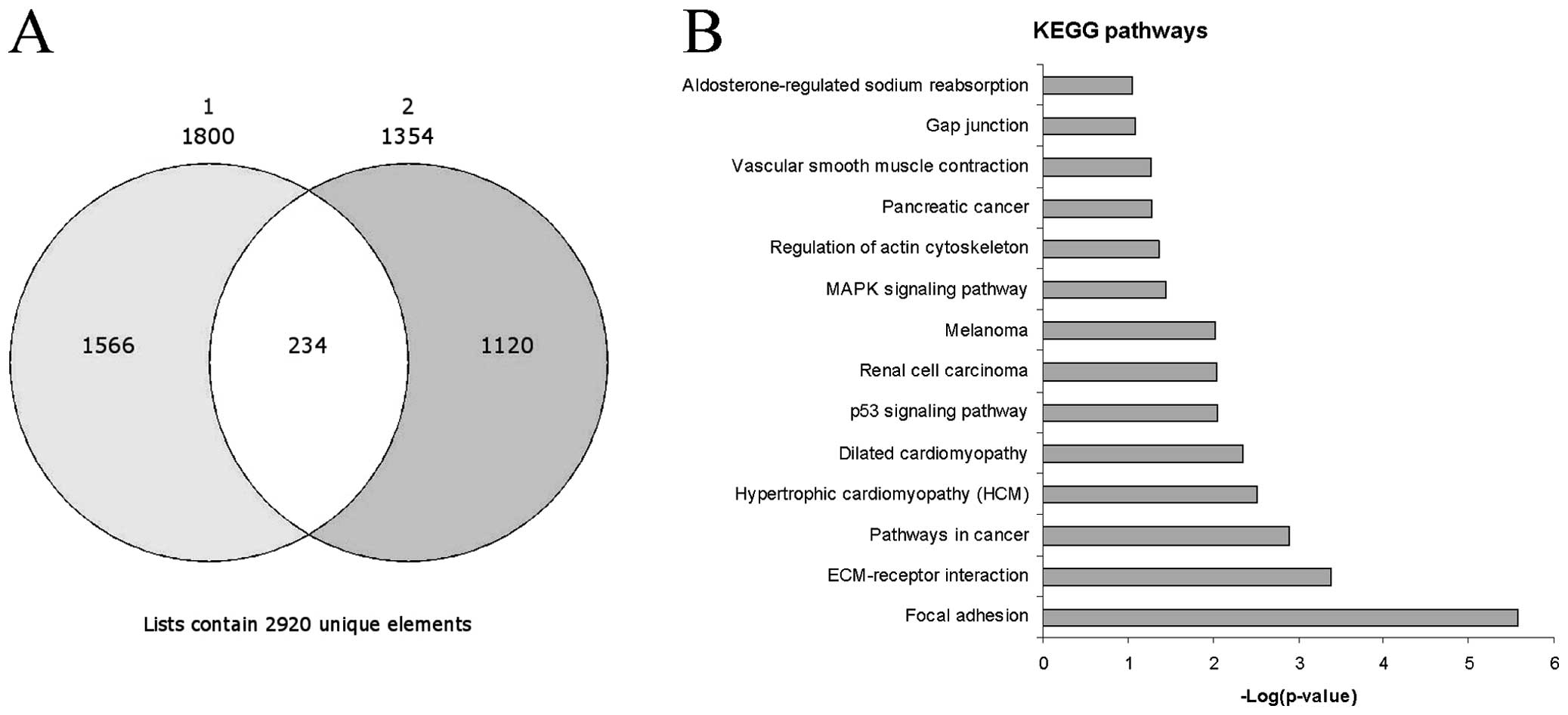
We identified 1,800 genes which were upregulated in
the activated vs. the resting HSCs in the GSE34949 dataset and
1,354 genes in the E-MTAB-2081 dataset. Among the significantly
upregulated genes, 254 genes were common between the two datasets
and were selected for further analysis (Fig. 1A). As expected, some genes had
well-established associations with fibrosis. These included ECM
components [collagen, type I, alpha 1 (COL1A1), collagen, type I,
alpha 2 (COL1A2), collagen, type III, alpha 1 (COL3A1), collagen,
type V, alpha 2 (COL5A2), collagen, type VIII, alpha 1 (COL8A1),
collagen, type XII, alpha 1 (COL12A1), fibronectin 1 (FN1) and
tenascin C (TNC)], metalloproteinases (MMP11, MMP12, MMP19 and
MMP23) and growth factors [TGFB2, TGFB3, fibroblast growth factor
(FGF)18 and FGF2].
Functional analysis of commonly
upregulated genes
We utilized DAVID to identify the major functions,
networks and pathways relevant to the 254 significantly upregulated
genes.
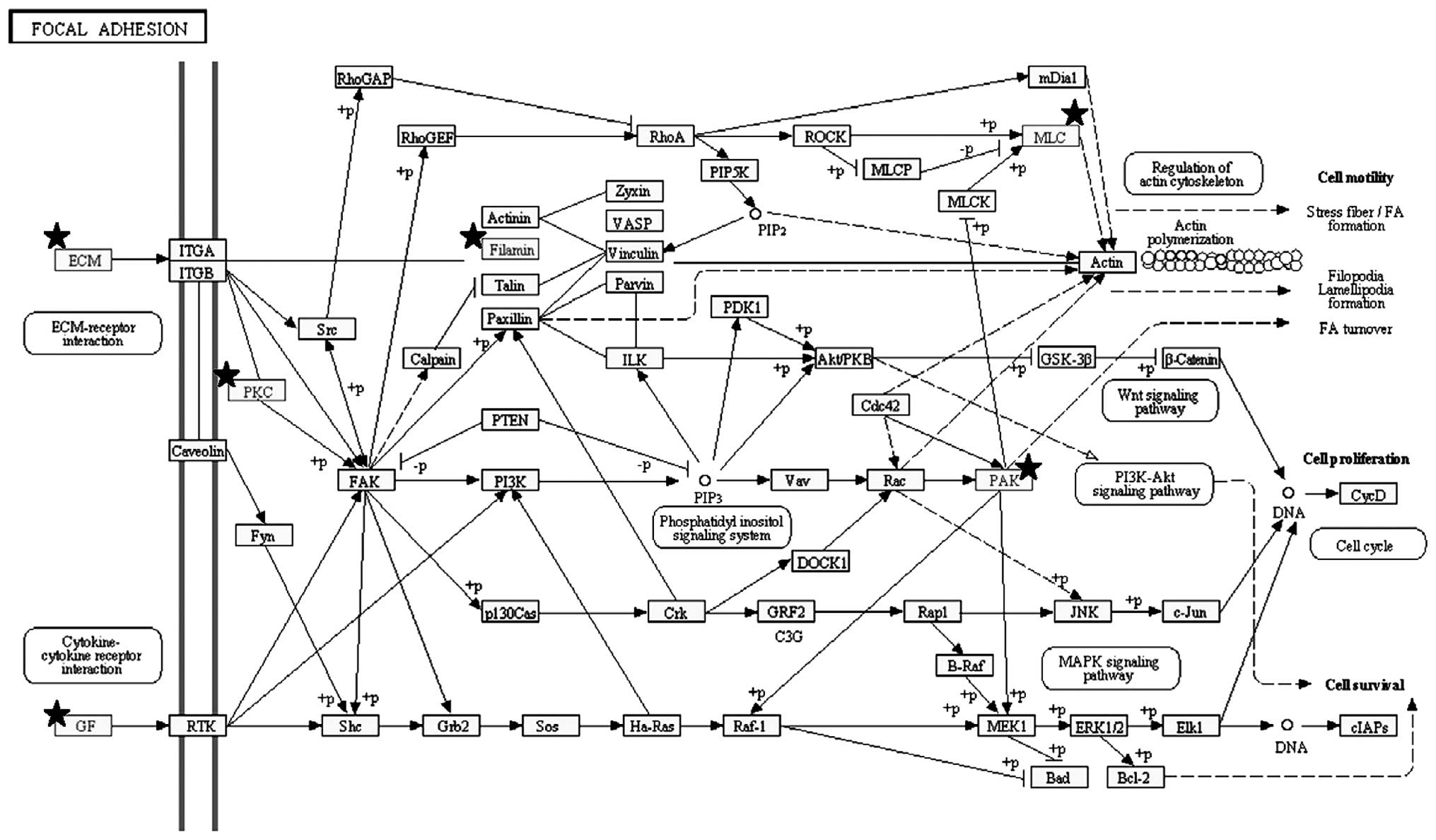
Pathway analysis revealed that the top three
significant pathways enriched by the 254 genes were ʽfocal
adhesion' (Figs. 1B and 3), ʽECM-receptor interaction' and
ʽpathways in cancer'. These pathways shared several genes, i.e.,
the FGFs and ECM components already indicated. Other notable genes
were represented by protein kinase C, alpha (PRKCA), insulin-like
growth factor (IGF1), filamin C, gamma (FLNC), myosin, light chain
9, regulatory (MYL9), p21 protein (Cdc42/Rac)-activated kinase 3
(PAK3), platelet derived growth factor C (PDGFC) and c-fos-induced
growth factor/vascular endothelial growth factor D (FIGF) in the
ʽfocal adhesion' pathway, syndecan 2 (SDC2) in the ʽECM-receptor
interaction' pathway and PRKCA, cyclin-dependent kinase inhibitor
2A (CDKN2A), cyclin-dependent kinase inhibitor 2B (CDKN2B),
aryl-hydrocarbon receptor nuclear translocator 2 (ARNT2), IGF1,
frizzled class receptor 2 (FZD2) and FIGF in the ʽpathways in
cancer'. Among the other top enriched KEGG pathways, the presence
of those genes related to hypertrofic and dilated cardiomyopathy
was indicative of the myofibroblastic trans-differentiation of
activated HSCs (Fig. 1B). In
addition, the involvement of the p53, renal cell carcinoma,
melanoma and mitogen-activated protein kinase (MAPK) signaling
pathways seemed to correlate with liver fibrosis to liver cirrhosis
and the evolution toward hepatocellular carcinoma (Fig. 1B), and as previously demonstrated
(12).
The use of SMART led to the identification of the
ʽcalcium-binding EGF-like domain', ʽEGF', ʽfibrillar collagens
C-terminal domain', ʽzinc-depen dent metalloprotease' and ʽvon
Willebrand factor, type C' as the principal protein domains
overrepresented by the commonly upregulated genes during
fibrogenesis (Fig. 2A and B).
A characteristic of HSCs is the ability to undergo
cell cycle progression and cell mitosis (13,14). Indeed, biological processes
strongly associated with the upregulated genes were found to be
related to the regulation of cell proliferation, cartilage
development, regulation of cell growth and division (Fig. 2B).
The most significant molecular functions (Fig. 2C) associated with the commonly
upregulated genes were ʽcalcium ion binding', ʽgrowth factor
binding', ʽextracellular matrix structural constituent' and ʽgrowth
factor activity' (Fig. 2C).
Gene network analysis of upregulated
genes
The sole analysis of the gene expression profile
provides an incomplete picture of the biological mechanisms
involved in a process, such as liver fibrosis, since it does not
include the interrelations and communication paths among genes. The
construction of a gene network is the logical continuation of gene
expression profiling. Describing a biological system using a
network helps to identify characteristics that could potentially
lead to the development of novel treatment strategies, by a better
understanding of the process. In a gene network, genes form the
network nodes and relationships serve as network edges.
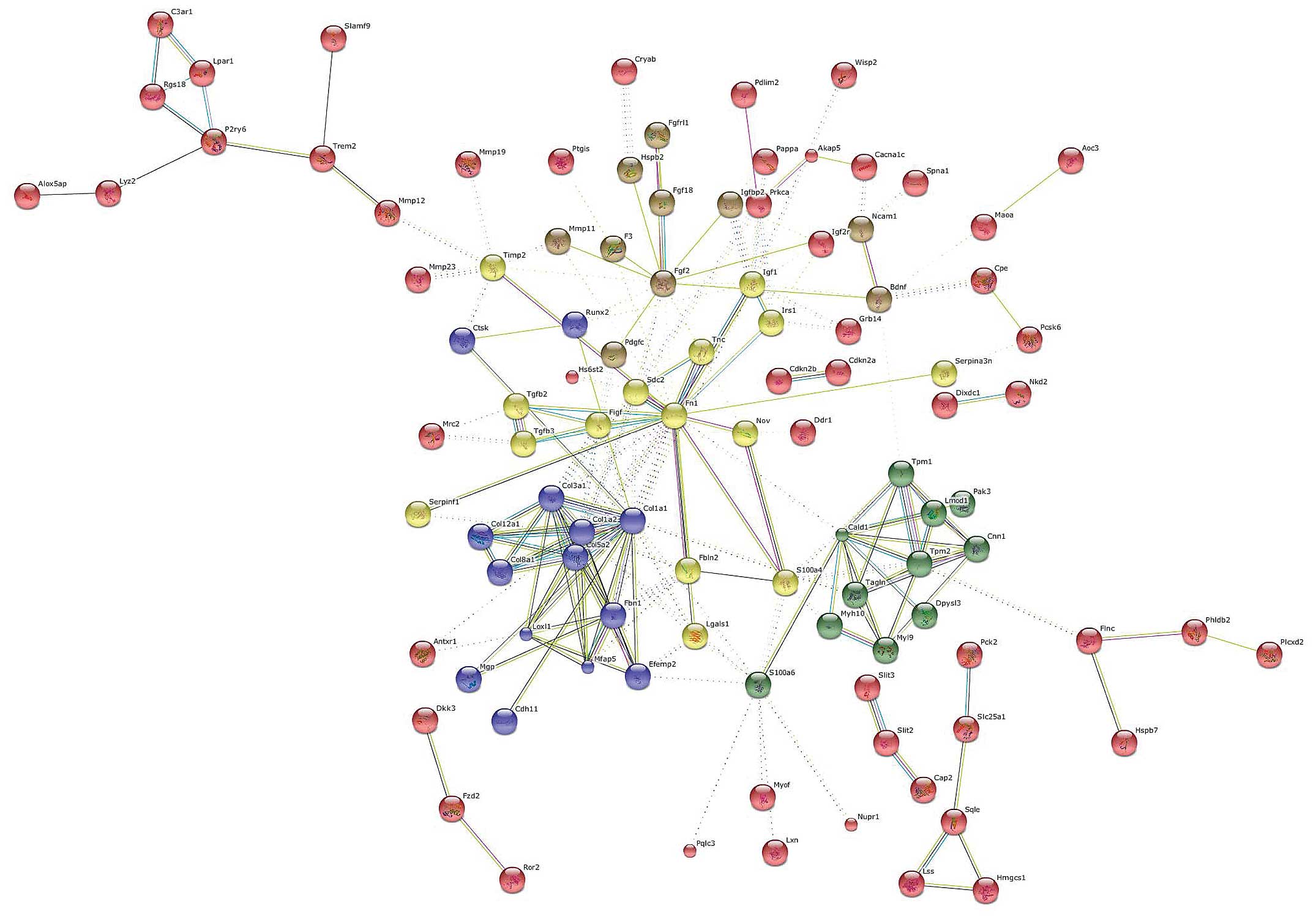
Genes were clustered using the k-means algorithm for
a number of five clusters (Fig.
4). The central hub was occupied by FN1 which interconnected
all of the five clusters. Other important nodes were represented by
caldesmon 1 (CALD1), COL1A1 and FGF2.
CALD1 is a protein expressed in smooth muscle cells,
which combines with calmodulin, tropomyosin and actin thus
regulates cellular contraction (15). Although it has been suggested to
be a biomarker for smooth muscle tumors, its role in liver fibrosis
remains unexplored.
Another important gene, at the interface of three
different clusters, was represented by S100A4. S100A4, also known
as fibroblast-specific protein 1 (FSP1), belongs to the S100 gene
family, a multi-gene family of highly conserved
Ca2+-binding proteins. S100A4 is known to be upregulated
in pulmonary fibrosis (16),
renal fibrosis (17) and in
portal tract fibrosis following bile duct damage (18). Data on endometrial cancer have
indicasted that S100A4 is induced by TGF-β1 and that it is
necessary for the effects of TGF-β1 on cancer cell migration and
invasion (19). S100A4 could thus
play a central role in the induction and/or maintenance of liver
fibrosis in chronic hepatic disease. The development of drugs
specifically targeting S100A4 may thus be equally effective as
anti-TGF-β treatment, but with less potential adverse effects,
given the more downstream target.
Another gene that was identified as a relevant hub
in the network analysis was Runt-related transcription factor 2
(Runx2). The Runx family of transcription factors is composed of
three distinct genes, Runx1, Runx2 and Runx3, which play a key part
in normal development and cancer (20). In particular, Runx2 is involved in
osteoblast differentiation and bone formation and it has been
reported that Runx2 is a downstream target of the TGF/Smad4
pathway. In addition, the MAPK pathway plays an essential role in
the phosphorylation and activation of Runx2 (21). Hattori et al (22) demonstrated that the inhibition of
p38 is able to ameliorate liver cirrhosis through the
down-regulation of Runx2. Taken together, these data suggest the
central involvement of this gene in the progression of hepatic
fibrosis.
Analysis of downregulated genes
In order to define the complete mechanisms
underlying the development of liver fibrosis, we identified
commonly downregulated genes between the GSE34949 and E-MTAB-2081
datasets. We found 1,200 and 1,122 genes downregulated in these
databases, respectively, with 132 genes being shared between the
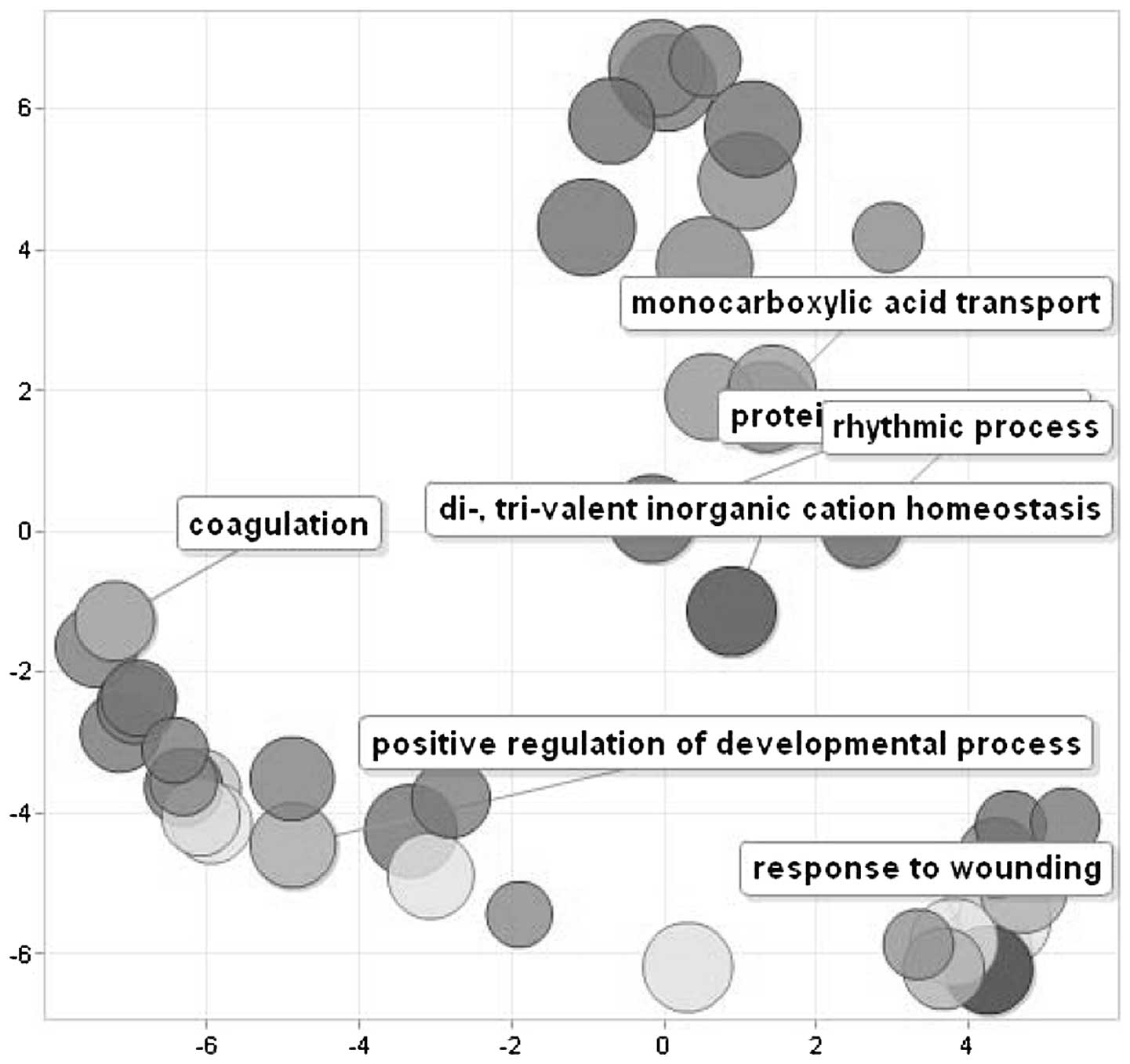
two datasets. GO analysis revealed five major biological processes
enriched by these genes, represented by ʽresponse to wounding',
ʽpositive regulation of developmental process', ʽcoagulation',
ʽcarboxilic acid catabolism' and ʽpolysaccharide metabolism'.
Fig. 5 shows a scatterplot of the
cluster representatives obtained by applying multidimensional
scaling to a matrix of the semantic similarities of the GO terms.
Along the same lines, KEGG pathway analysis identified as the most
enriched pathways, the ʽcomplement and coagulation cascades' [with
kininogen 1 (KNG1), fibrinogen gamma chain (FGG), fibrinogen alpha
chain (FGA), fibrinogen beta chain (FGB), coagulation factor II
(thrombin) (F2), serpin peptidase inhibitor, clade C
(antithrombin), member 1 (SERPINC1) and coagulation factor IX (F9)
being involved], the ʽNOD-like receptor signaling pathway' [which
included MAPK12, interleukin (IL)1B, nuclear factor of kappa light
polypeptide gene enhancer in B-cells inhibitor, alpha (NFKBIA) and
MAPK11], ʽcytokine-cytokine receptor interaction' [including
pro-platelet basic protein (chemokine (C-X-C motif) ligand 7)
(PPBP), IL1B, nerve growth factor receptor (NGFR), interleukin 13
receptor, alpha 1 (IL13RA1), IL10, IL1A and kinase insert domain
receptor (KDR)] and the ʽRIG-I-like receptor signaling pathway'
[with MAPK12, NFKBIA, MAPK11 and DEXH (Asp-Glu-X-His) box
polypeptide 58 (DHX58)]. These data are concordant with those
presented in the study by Ji et al (23) who found that downregulated genes
were primarily related to immune response and lipid metabolism. Of
note, GO analysis revealed that the major molecular functions
involved were the endopeptidase inhibitor, the vitamin transporter,
protein bridging and DNA-directed DNA polymerase activity.
Discussion
HSCs represent the major cell type responsible for
the development of liver fibrosis. Following liver injury, HSCs
become activated and transdifferentiate into MFBs that lead to
intrahepatic ECM accumulation. Depicting the molecular events that
occur during HSC activation is key for a comprehensive
understanding of the biological role of HSCs in liver fibrosis and
for the development of effective strategies for the diagnosis and
treatment of this disorder.
Meta-analysis of multiple microarray datasets yields
more reliable and comprehensive results than using a single dataset
of gene expression profiles. Moreover, the possibility of including
data from different mammal species increases even more the
predictability power of the data extracted from a single dataset
(24).
In the present study, we performed a meta-analysis
of two datasets from two different rodent species and identified
254 and 132 genes that were significantly upregulated and
downregulated in each dataset, respectively, in activated HSCs.
Together these genes draw a robust and reliable picture of the
molecular mechanisms involved in the development of hepatic
fibrosis. The majority of the genes identified in this
meta-analysis corresponded to factors already described in the
literature by independent studies (3,5,14,18,23). This observation strongly supports
the reliability of the data presented herein. These data allow
researchers to identify novel pharmacological targets to prevent
and treat hepatic fibrogenesis, and to monitor its evolution
through the screening of relevant molecules involved in the
degeneration of the liver parenchyma. The ideal biomarker for liver
fibrosis should be reliable and readily available and should be
useful in assessing the progression of liver disease. The
identification of a biomarker with such characteristics would
replace the need for biopsies which, being invasive procedures, are
associated with the risk of pain and/or bleeding.
Among the list of candidate genes identified in this
study, there are several highly modulated genes which have not been
previously associated with liver fibrosis. Bioinformatics analysis
of these modulated genes not only enhanced our understanding of the
major characteristics of activated HSCs, but also suggested new
avenues for the development of more effective therapies for liver
fibrosis.
References
|
1
|
Anthony PP, Ishak KG, Nayak NC, Poulsen
HE, Scheuer PJ and Sobin LH: The morphology of cirrhosis.
Recommendations on definition, nomenclature, and classification by
a working group sponsored by the World Health Organization. J Clin
Pathol. 31:395–414. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hernandez-Gea V and Friedman SL:
Pathogenesis of liver fibrosis. Annu Rev Pathol. 6:425–456. 2011.
View Article : Google Scholar
|
|
3
|
Gressner AM and Weiskirchen R: Modern
pathogenetic concepts of liver fibrosis suggest stellate cells and
TGF-β as major players and therapeutic targets. J Cell Mol Med.
10:76–99. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Friedman SL: Hepatic stellate cells:
Protean, multifunctional, and enigmatic cells of the liver. Physiol
Rev. 88:125–172. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Tacke F and Weiskirchen R: Update on
hepatic stellate cells: Pathogenic role in liver fibrosis and novel
isolation techniques. Expert Rev Gastroenterol Hepatol. 6:67–80.
2012. View Article : Google Scholar
|
|
6
|
Popov Y and Schuppan D: Targeting liver
fibrosis: strategies for development and validation of antifibrotic
therapies. Hepatology. 50:1294–1306. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Schuppan D and Kim YO: Evolving therapies
for liver fibrosis. J Clin Invest. 123:1887–1901. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nicoletti A, Fagone P, Donzuso G, Mangano
K, Dibilio V, Caponnetto S, Bendtzen K, Zappia M and Nicoletti F:
Parkinson's disease is associated with increased serum levels of
macrophage migration inhibitory factor. Cytokine. 55:165–167. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Fagone P, Di Rosa M, Palumbo M, De
Gregorio C, Nicoletti F and Malaguarnera L: Modulation of heat
shock proteins during macrophage differentiation. Inflamm Res.
61:1131–1139. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Parkinson H, Sarkans U, Kolesnikov N,
Abeygunawardena N, Burdett T, Dylag M, Emam I, Farne A, Hastings E,
Holloway E, et al: ArrayExpress update - an archive of microarray
and high-throughput sequencing-based functional genomics
experiments. Nucleic Acids Res. 39(Database issue): D1002–D1004.
2011. View Article : Google Scholar :
|
|
11
|
Fagone P, Muthumani K, Mangano K, Magro G,
Meroni PL, Kim JJ, Sardesai NY, Weiner DB and Nicoletti F: VGX-1027
modulates genes involved in lipopolysaccharide-induced Toll-like
receptor 4 activation and in a murine model of systemic lupus
erythematosus. Immunology. 142:594–602. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Amann T, Bataille F, Spruss T, Mühlbauer
M, Gäbele E, Schölmerich J, Kiefer P, Bosserhoff AK and Hellerbrand
C: Activated hepatic stellate cells promote tumorigenicity of
hepatocellular carcinoma. Cancer Sci. 100:646–653. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bissell DM, Wang SS, Jarnagin WR and Roll
FJ: Cell-specific expression of transforming growth factor-beta in
rat liver. Evidence for autocrine regulation of hepatocyte
proliferation. J Clin Invest. 96:447–455. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Coker RK, Laurent GJ, Shahzeidi S, Lympany
PA, du Bois RM, Jeffery PK and McAnulty RJ: Transforming growth
factors-beta 1, -beta 2, and -beta 3 stimulate fibroblast
procollagen pro duction in vitro but are differentially expressed
during bleomycin-induced lung fibrosis. Am J Pathol. 150:981–991.
1997.PubMed/NCBI
|
|
15
|
Watanabe K, Tajino T, Sekiguchi M and
Suzuki T: h-Caldesmon as a specific marker for smooth muscle
tumors. Comparison with other smooth muscle markers in bone tumors.
Am J Clin Pathol. 113:663–668. 2000.PubMed/NCBI
|
|
16
|
Lawson WE, Polosukhin VV, Zoia O,
Stathopoulos GT, Han W, Plieth D, Loyd JE, Neilson EG and Blackwell
TS: Characterization of fibroblast-specific protein 1 in pulmonary
fibrosis. Am J Respir Crit Care Med. 171:899–907. 2005. View Article : Google Scholar
|
|
17
|
Robertson H, Ali S, McDonnell BJ, Burt AD
and Kirby JA: Chronic renal allograft dysfunction: The role of T
cell-mediated tubular epithelial to mesenchymal cell transition. J
Am Soc Nephrol. 15:390–397. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Rygiel KA, Robertson H, Marshall HL,
Pekalski M, Zhao L, Booth TA, Jones DE, Burt AD and Kirby JA:
Epithelial-mesenchymal transition contributes to portal tract
fibrogenesis during human chronic liver disease. Lab Invest.
88:112–123. 2008. View Article : Google Scholar
|
|
19
|
Xie R, Schlumbrecht MP, Shipley GL, Xie S,
Bassett RL Jr and Broaddus RR: S100A4 mediates endometrial cancer
invasion and is a target of TGF-beta1 signaling. Lab Invest.
89:937–947. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Ito Y: Oncogenic potential of the RUNX
gene family: ʽoverview'. Oncogene. 23:4198–4208. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Xiao G, Jiang D, Thomas P, Benson MD, Guan
K, Karsenty G and Franceschi RT: MAPK pathways activate and
phosphorylate the osteoblast-specific transcription factor, Cbfa1.
J Biol Chem. 275:4453–4459. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hattori S, Dhar DK, Hara N, Tonomoto Y,
Onoda T, Ono T, Yamanoi A, Tachibana M, Tsuchiya M and Nagasue N:
FR-167653, a selective p38 MAPK inhibitor, exerts salutary effect
on liver cirrhosis through downregulation of Runx2. Lab Invest.
87:591–601. 2007.PubMed/NCBI
|
|
23
|
Ji J, Yu F, Ji Q, Li Z, Wang K, Zhang J,
Lu J, Chen L, E Q, Zeng Y and Ji Y: Comparative proteomic analysis
of rat hepatic stellate cell activation: A comprehensive view and
suppressed immune response. Hepatology. 56:332–349. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Rasche A, Al-Hasani H and Herwig R:
Meta-analysis approach identifies candidate genes and associated
molecular networks for type-2 diabetes mellitus. BMC Genomics.
9:3102008. View Article : Google Scholar : PubMed/NCBI
|