Introduction
Usher syndrome (USH) is an autosomal recessive (AR)
inherited disease belonging to the group of retinitis pigmentosa
(RP) syndromes and is a clinical and genetically heterogeneous
disease (1,2). Patients with USH usually exhibit
progressive visual loss, hearing impairment and vestibule
dysfunction. Clinically, USH is subdivided into three subclasses
based on the severity and progression of the hearing impairment and
whether the vestibule invaded. Type 1 USH is the most severe form,
with the prepubertal onset of progressive RP, profound hearing loss
and vestibular dysfunction. Type 2 USH is the most common type and
is less severe, with moderate to severe congenital deafness and
later-onset RP, but with the absence of vestibular dysfunction.
Type 3 is the least common type, with progressive deafness,
adult-onset RP, hypermetropic astigmatism and a variable impairment
of vestibular function. Currently, 10 genes that are associated
with this disease have been identified, and three loci have been
mapped in human chromosomes (http://www.retinogenetics.org).
Thus far, increasing attention has been paid to the
molecular diagnosis of USH. The Sanger sequencing of the coding
region, a traditional approach, is reliable and provides an easy
strategy to determine the genetic causes of a disease (3). However, Sanger sequencing is not
always affordable due to the large number of coding fragments. A
USH genotyping microarray based on arrayed primer extension
technology was used to simultaneously screen multiple known sites;
however, it was unable to detect new mutations, insertions or
deletions (Indels) (4,5). Custom-designed targeted exome
sequencing is a high-throughput and cost-effective method that
permits the screening of a number of previously targeted coding
regions (6,7). To cover full coding regions in the
human genome, whole-exome sequencing has been developed to
facilitate the discovery of novel disease genes (8).
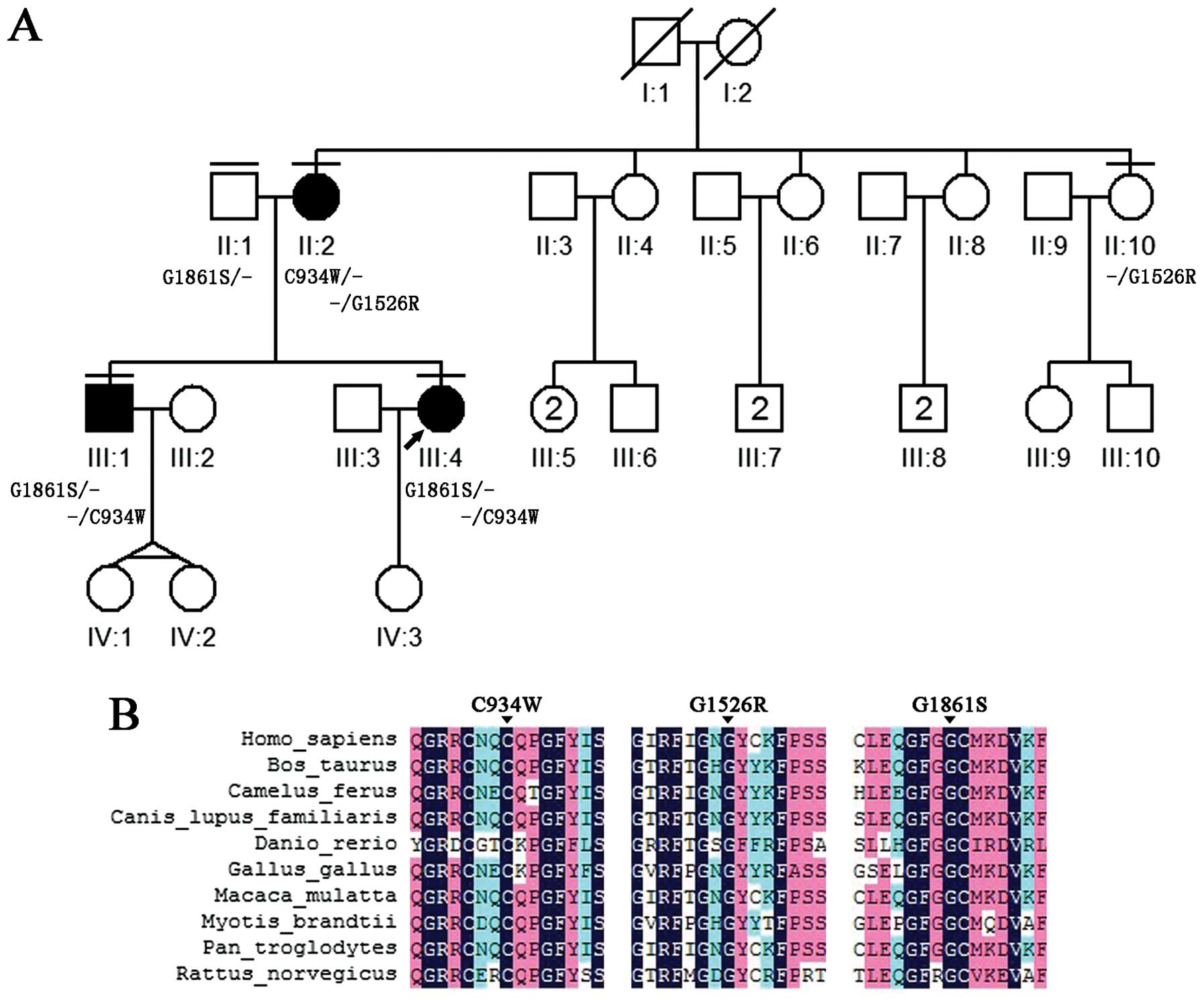
In the present study, a pseudo-dominant pedigree of
USH was identified, which presented as dominant heritance, in
patients over two successive generations. As all of the known genes
are a recessive trait, we speculated that a novel causative gene in
the dominant pattern was mutated in this family. To determine the
genetic predisposition, whole-exome sequencing was applied and one
novel and two known USH2A mutations were identified that
successfully explained the genetic architecture in this family.
Materials and methods
Subject recruitment
The study was carried out in adherence to the tenets
of the Declaration of Helsinki and was approved by the Ethics
Committee of the Eye Hospital of Wenzhou Medical University
(Wenzhou, Zhejiang, China). All the study subjects were fully
informed, and consent was obtained. In the study, five individuals,
including two males and three females, from a Chinese family
exhibited phenotypic features that were consistent with USH and a
pseudo-dominant inheritance pattern from the Division of Ophthalmic
Genetics at the Eye Hospital of Wenzhou Medical University. The
clinical diagnosis of USH was based on typical visual loss due to
RP and progressive hearing impairment. Comprehensive ophthalmic
tests were performed on each patient, including tests of visual
acuity, fundus photography, optical coherence tomography,
electro-retinography (ERG) and perimetry. The primary complaints
from patients were night blindness, visual field restriction and
hearing loss, in addition to typical symptoms, including bone
spicule-like pigmentation, retinal vessel attenuation and waxy disc
pallor in the fundus. A more detailed family history was obtained
via personal interviews with the patients and family members
(Fig. 1). Peripheral blood
samples were collected following informed consent from all five of
the subjects. Three genomic DNA samples, including samples from two
affected individuals (II:2 and III:1) and one unaffected individual
(II:1), were selected for whole-exome sequencing, and two samples
from affected individuals (III:4 and II:10) were tested for
mutation validation using Sanger sequencing.
DNA preparation
Genomic DNA was extracted from leukocytes using the
TIANamp Blood DNA kit (Tiangen, Beijing, China) according to the
manufacturer's instructions. The DNA concentration was quantified
using a spectrophotometer (NanoDrop 1000; Thermo Fisher Scientific,
Waltham, MA, USA).
Whole-exome sequencing
The library was prepared and the exome was captured
using the Illumina HiSeq 2000 platform based on the manufacturer's
instructions (9). In brief, a
minimum of 3 µg genomic DNA was sheared, end-repaired and
ligated with special devices. The genomic DNA of each subject was
sheared into fragments ranging from 350–400 base pairs. Following
adaptor ligation, the library was amplified according to standard
Illumina protocols, and the polymerase chain reaction (PCR) product
was validated using the Agilent Bioanalyzer (Santa Clara, CA, USA).
Capture enrichment was performed by twice hybridization and washing
using specially designed capture probes and streptavidin beads;
subsequently, the exome-targeted DNA library was enriched. In
short, PCR was used to amplify the enriched library, as in the
previous step. Subsequent to the final product being amplified and
validated, the library was enriched for sequencing on the Illumina
HiSeq 2000 sequencer.
Sequencing data analysis
Following sequencing on the Illumina HiSeq 2000
platform, the primary data were processed to retrieve high-quality
reads using the SolexaQA package and the cutadapt program
(http://code.google.com/p/cutadapt/).
The clean sequence reads were aligned to the reference human genome
(hg19) using the SOAPaligner program. Subsequently, the dataset
files, including the PCR duplicates that were removed and the
identified SNPs, were analyzed using the Picard software and
SOAPsnp program, respectively. Variants of the Indels were
identified using the GATK program. All the identified variants were
annotated by the exome-assistant program (10). In addition, the variants with a
frequency >1% were removed and the SIFT (http://sift.jcvi.org/www/SIFT_enst_submit.html),
PolyPhen (http://genetics.bwh.harvard.edu/pph2/) and
MutationTaster (http://www.mutationtaster.org/) programs were used to
predict the effects of the variants on the protein function. As the
pedigree in the study exhibited 'autosomal dominant' transmission,
the candidate variants that were located in the dominant inherited
alleles were first analyzed; however, no positive result was
obtained; subsequently, the recessive inherited variants in the
data were fully analyzed from the three whole-exome sequencing runs
and the candidate mutations were identified.
Candidate mutation confirmation by Sanger
sequencing
The candidate mutations were listed following the
above standard filtering strategy in the whole-exome sequencing
data of the three subjects. The specific primers were designed
using Primer3 to amplify each coding region of the potential
mutation. Following amplification by PCR, the products were
sequenced by Sanger sequencing to further validate the precision of
the candidate mutations. The Sanger sequencing data were analyzed
by Mutation Surveyor (SoftGenetics, State College, PA, USA).
Results
Clinical phenotype
In order to clinically characterize the patients, a
detailed family history was obtained and full ophthalmology
examinations were performed on all the patients. Night blindness
and peripheral visual field constriction were reported by every
patient. Two patients (II:2 and III:1) were characterized by
moderate progressive hearing loss with the onset of their second
decade, and one of these patients (III:1) exhibited impaired
vestibular function (Table I).
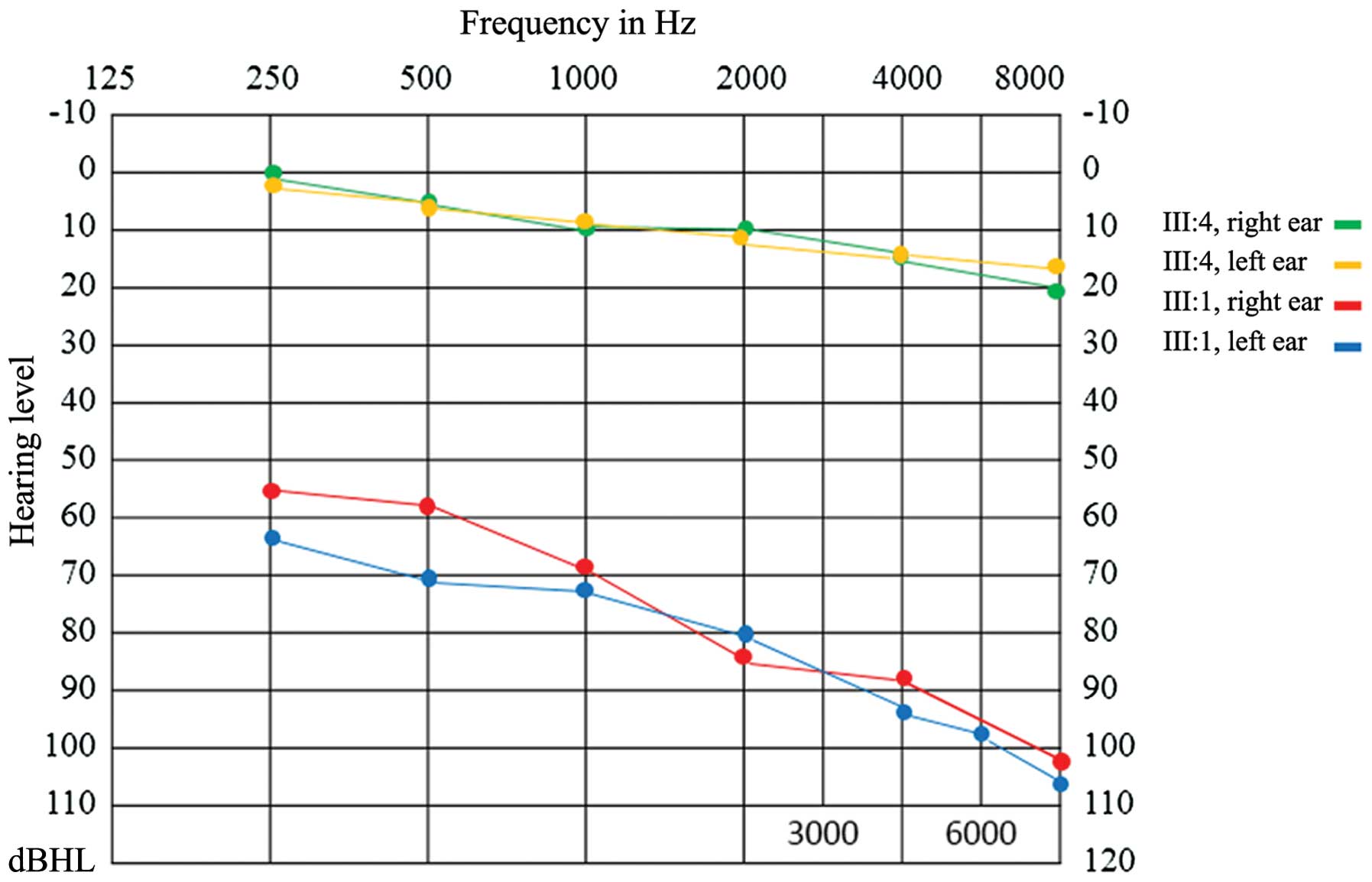
Audiogram showed bilateral downward-sloping moderate hearing loss
in patient III:1, while patient III:4 exhibited normal audiogram
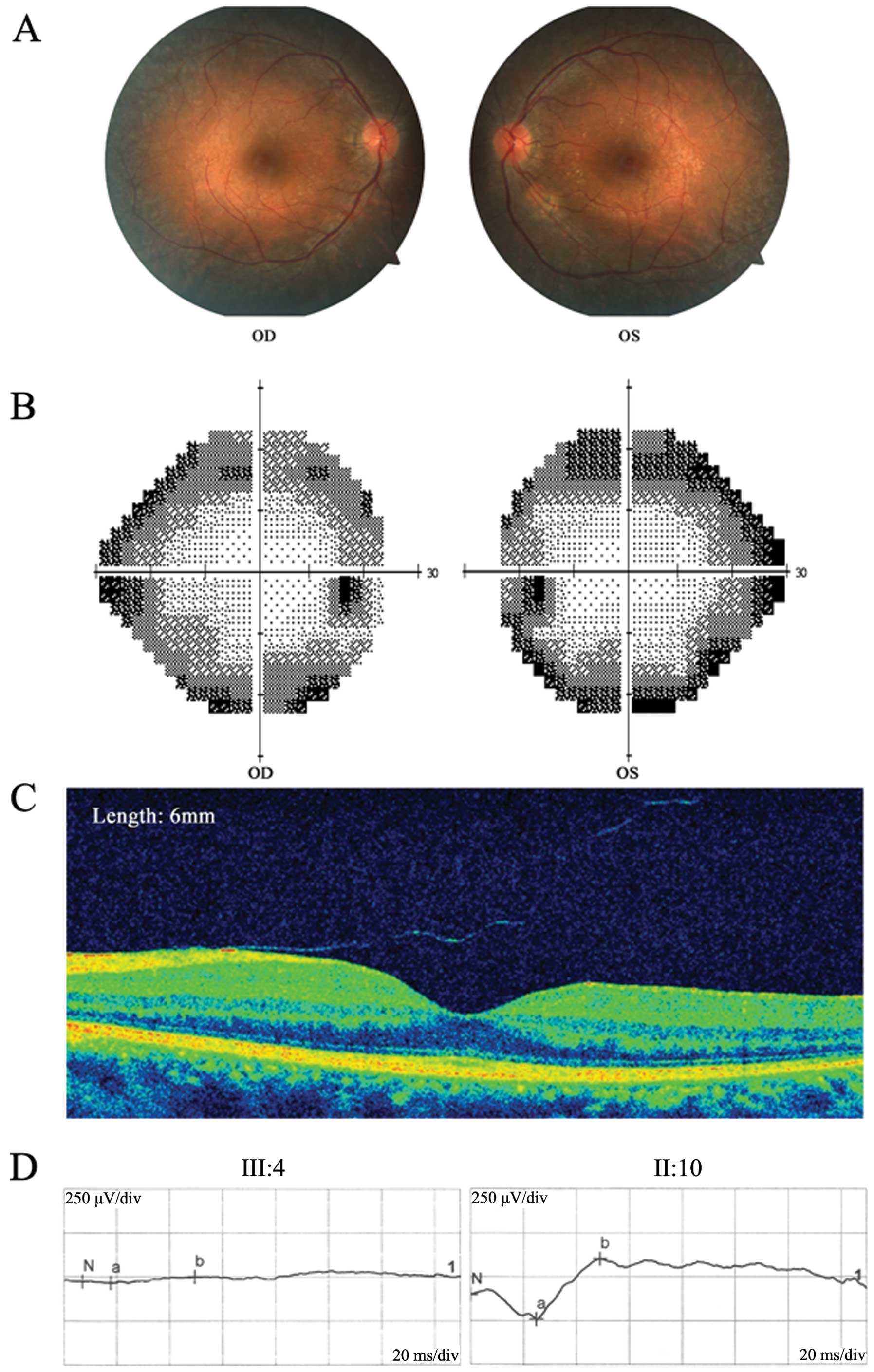
function (Fig. 2). Fundus
photography revealed typical RP signs, including mottling and
granularity of the RPE, attenuated retinal vessels and optic nerve
head pallor, in every patient (II:2, III:1 and III:4). The ERG
results clearly showed profound abnormalities with no detectable
rod response (Fig. 3). The static
visual field (Humphrey Field Analyzer 24-2) exhibited a greater
decrease in the retinal sensitivity in the far periphery. Color
vision defects and astigmatism were found in all the affected
patients. All the symptoms, particularly those typical of RP and
progressive deafness, in this pedigree supported a diagnosis of
USH. However, based on the clinical symptoms that were
aforementioned, it is difficult to diagnose which subtype of USH
this family belongs to. In addition, the inheritance pattern of USH
has only been reported as AR transmission; therefore, it was
suggested this pedigree is caused by a novel dominant gene.
 | Table IClinical phenotype of the patients in
the pedigree. |
Table I
Clinical phenotype of the patients in
the pedigree.
| Subjects | Gender | Age, years | Age at onset of
blindness | RP | Age at onset of
deafness | Vestibular
dysfunction | Astigmatism | BCVA, R/L |
|---|
| II:2 | F | 51 | <5 years | + | 20 years | − | + | 0.3/0.4 |
| III:1 | M | 33 | 15 years | + | 28 years | + | + | 0.5/0.7 |
| III:4 | F | 29 | 20 years | + | − | − | + | 0.7/0.9 |
Identification of candidate mutations by
whole-exome sequencing
Three subjects, including two affected individuals
(II:2 and III:1) and one unaffected individual (II:1), in the
pedigree were sequenced by whole-exome sequencing using the
Illumina HiSeq 2000 platform. From the whole-exome sequencing, the
average read depth of the targeted regions and the distribution of
the sequencing depth indicated a high sequencing quality (Table II and Fig. 4). Combining the pedigree clinical
phenotype and pathogenic genomic transmission mode, the candidate
mutations were finally identified. Through the aforementioned
filtering strategy, three candidate mutations were identified for
this pedigree.
| Table IIResult of exome sequencing data
analysis. |
Table II
Result of exome sequencing data
analysis.
| Data analysis | Patient II:1 | Patient II:2 | Patient III:1 |
|---|
| Total reads | 67,379,716 | 55,680,718 | 72,086,038 |
| Total yield, bp | 6,805,351,316 | 5,623,752,518 | 7,280,689,838 |
| Read length, bp | 101.0 | 101.0 | 101.0 |
| Target regions,
bp | 62,085,286 | 62,085,286 | 62,085,286 |
| Average throughput
depth of target regions | 109.6X | 90.6X | 117.3X |
| Mappable reads (reads
mapped to human genome) | 50,349,216 | 41,366,342 | 54,726,489 |
| Mappable yield,
bp | 4,933,335,174 | 4,051,988,166 | 5,371,273,701 |
| % Mappable reads | 74.7 | 74.3 | 75.9 |
| % Coverage of target
regions (>1X) | 94.7 | 94.9 | 94.6 |
| Number of on-target
genotypes (>1X) | 58,780,409 | 58,910,095 | 58,744,961 |
| % Coverage of target
regions (>10X) | 87.8 | 82.6 | 87.5 |
| Number of on-target
genotypes (>10X) | 54,531,850 | 51,291,897 | 54,349,898 |
| Median read depth of
target regions | 46.0X | 32.0X | 50.0X |
| Mean read depth of
target regions | 46.8X | 34.8X | 52.0X |
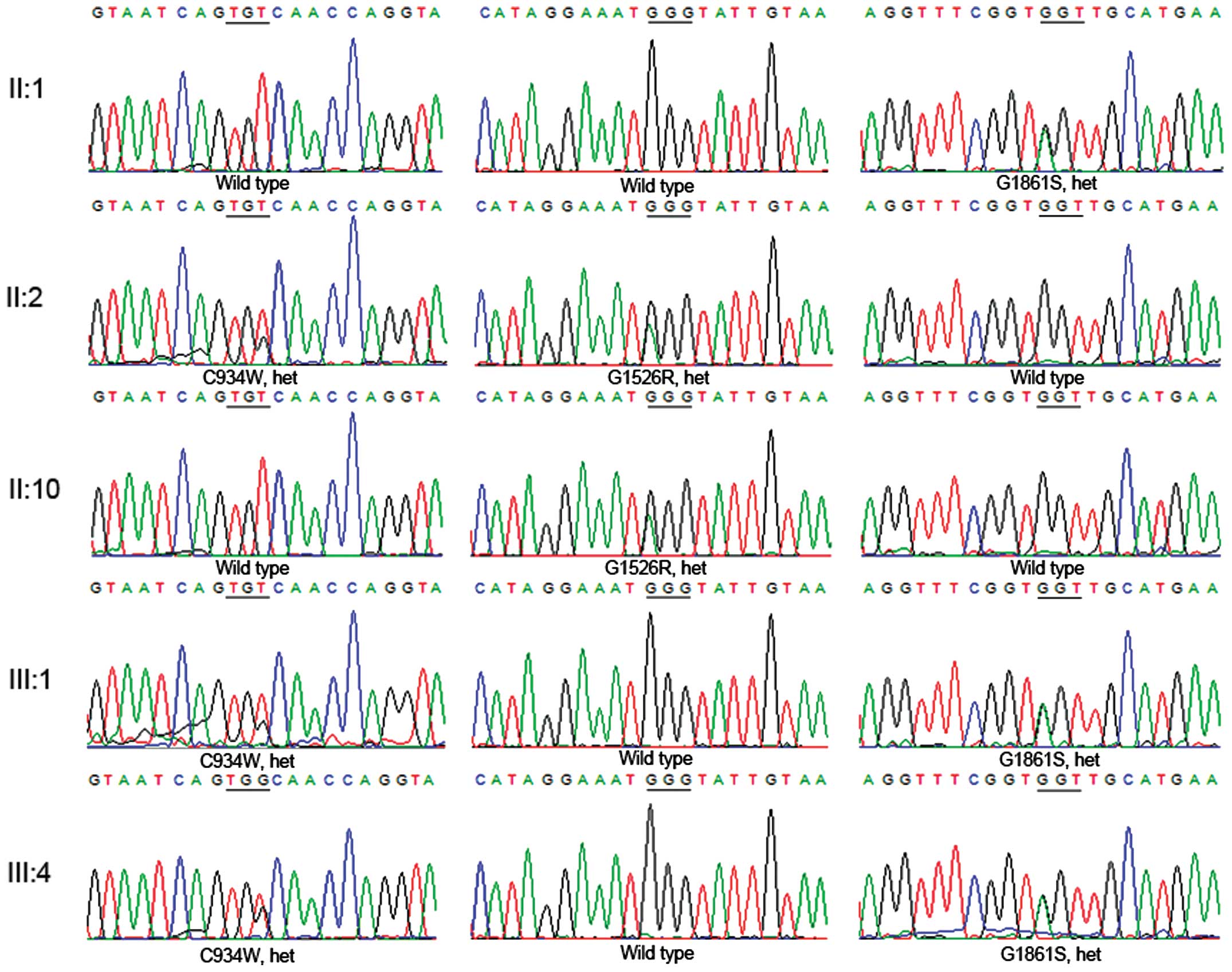
Mutation validation
The consistency of the Sanger sequencing results
confirmed all the mutations in the three subjects (II:1, II:2 and
III:1). Notably, the family showed a genetic continuity that
appeared to be a dominant inherited pedigree; one heterozygous
USH2A mutation (G1861S) was identified in the unaffected
father (II:1), two compound heterozygous USH2A mutations
(C934W and G1526R) in the affected mother (II:2) and two compound
heterozygous USH2A mutations (C934W and G1861S) in the
affected son (III:1) (Figs. 1 and
5; Table III). The validated results of
the pedigree indicate that the affected son (II:1) inherited one
heterozygous mutation (G1861S) from the father (II:1) and another
(C934W) from the mother (II:2), which contributed to the illness.
To further confirm these results, the familial validation was
expanded in the affected daughter (III:4). Via a traditional direct
sequencing of III:4, two compound heterozygous mutations (C934W and
G1861S) in the USH2A gene were successfully identified. All
the mutations of C934W and G1526R were confirmed in the mother
(II:2), who was an affected patient; however, only C934W was
transmitted to the two affected children (III:1 and III:4). The
healthy individual carried a heterozygous mutation (G1526R). All
these mutations were absent in 200 healthy controls. Taken
together, the candidate mutations were validated using Sanger
sequencing and a co-segregation trial.
| Table IIIIdentified mutations in the
pedigree. |
Table III
Identified mutations in the
pedigree.
| Subject | Affected | Mutation | Type | Amino acid | Reported | SIFT | PolyPhen-2 | MutationTaster |
|---|
| II:1 | − | c.5581G>A | Hetero | G1861S | Reported | Damaging | Probably
damaging | Disease causing |
| II:2 | + | c.2802T>G | Hetero | C934W | Reported | Damaging | Probably
damaging | Disease causing |
| c.4576G>A | Hetero | G1526R | Novel | Tolerated | Probably
damaging | Disease causing |
| II:10 | − | c.4576G>A | Hetero | G1526R | Novel | Tolerated | Probably
damaging | Disease causing |
| III:1 | + | c.2802T>G | Hetero | C934W | Reported | Damaging | Probably
damaging | Disease
causing |
| c.5581G>A | Hetero | G1861S | Reported | Damaging | Probably
damaging | Disease
causing |
| III:4 | + | c.2802T>G | Hetero | C934W | Reported | Damaging | Probably
damaging | Disease
causing |
| c.5581G>A | Hetero | G1861S | Reported | Damaging | Probably
damaging | Disease
causing |
Assessment of the pathogenicity of the
mutations
In the study, one novel and two known USH2A
gene mutations were identified in the pedigree and resulted in
single amino acid substitutions at protein positions 934, 1526 and
1861 (p.C934W, p.G1526R and p.G1861S). To predict whether these
amino acid changes were pathological, the combined evaluation of
different computer algorithms, including SIFT, PolyPhen and
MutationTaster (Table III),
were used. The novel missense mutations were predicted to be
deleterious. As all the missense mutations were also located in the
highly phylogenetically conserved regions (Fig. 1), it was strongly suggested that
the mutations in the USH2A gene were disease-causing and
contributed to the disorder in this pedigree.
Discussion
USH is a monogenic disorder with AR inheritance. The
worldwide prevalence of USH is relatively high, accounting for
>50% of patients who are both deaf and blind (11,12). Visual acuity loss due to typical
RP, congenital hearing impairment and variable vestibular
dysfunction are the primary clinical symptoms. Based on these
characteristics, USH could be divided clinically into three
subtypes. USH is also a genetically heterogeneous disorder, and
currently, 10 causative genes have been identified. In addition, 3
loci (USH1E, USH1H and USH1K) have been mapped. Type 1 USH is
caused by mutations in MYO7A, USH1C, USH1G,
CDH23, PCDH15 and CIB2; type 2 USH is caused
by mutations in USH2A, DFNB31 and GPR98; and
type 3 USH is caused by mutations in CLRN1. However, the
correlations between the phenotype and genotype are highly complex
(13,14). Among these correlations,
USH2A and CLRN1 were reported in AR RP; and
CDH23, CIB2, DFNB31, MYO7A,
PCDH15 and USH1C were identified in AR deafness alone
or as a syndrome. In the present pedigree, five individuals,
including three affected and two unaffected, were studied. Although
RP, the primary symptom of USH, was described in three of the
patients, the phenotype of each affected individual was distinct.
The mother (II:2), who was a patient, suffered from RP and
progressive deafness but no vestibular dysfunction, and the
affected son (III:1) suffered from RP, progressive deafness and
vestibular dysfunction. Notably, the affected daughter (III:4)
suffered from RP without deafness and vestibular dysfunction,
despite carrying the same genotype as that of the affected son
(III:1). From the characteristics of USH that are aforementioned,
it was suggested that different mutations in one gene could cause
different phenotypes, and that the same mutations could cause
intrafamilial phenotypic variability.
Taken together with the fact that more than a dozen
genes contribute to USH, Sanger sequencing is not a good method for
screening all the coding exons of all the causative genes due to
its huge workload and low efficiency. As the appearance of
next-generation sequencing, whole-exome sequencing has proven to be
an efficient diagnostic method for disorders with high degrees of
genetic heterogeneity. Whole-exome sequencing can sequence coding
regions of genomic DNA and can optimally screen huge number of
genes, particularly disease-causing loci. Three subjects were
selected, including two affected patients and one healthy relative,
to undergo exome sequencing. Through data analysis and Sanger
sequencing confirmation, one heterozygous mutation in the healthy
father, two compound heterozygous mutations in the affected mother
and two compound heterozygous mutations in the affected son were
quickly identified. By Sanger sequencing, two compound heterozygous
mutations were reconfirmed in the affected daughter (III:4) that
were the same as those in the affected son (III:1). From the above
results, it was revealed that the father is a carrier who
transmitted the defective allele to his children and that the
mother transmitted C934W to her children. This pedigree illustrates
a pseudo-dominant family. Considering a previous study of
USH2A mutations causing pseudo-dominant USH (6), it is possible that the carrier of
the USH2A mutation is relatively common in the Chinese
population. Thus, it may be a priority to screen USH2A when
a pseudo-dominant USH family is diagnosed. Taken together, the
present study successfully identified the USH2A mutations in
the family using whole-exome sequencing and demonstrated the
robustness of whole-exome sequencing to precisely diagnose USH.
In conclusion, one novel and two known USH2A
mutations were identified using whole-exome sequencing in a
pseudo-dominant USH family. These results highlight the possibility
that the USH2A gene has an important role in Chinese
pseudo-dominant USH and that whole-exome sequencing is a valuable
approach for the genomic diagnosis of disorders with high degrees
of genomic heterogeneity.
Acknowledgments
The authors gratefully acknowledge all the
participants in the study.
References
|
1
|
Licastro D, Mutarelli M, Peluso I,
Neveling K, Wieskamp N, Rispoli R, Vozzi D, Athanasakis E,
D'Eustacchio A, Pizzo M, et al: Molecular diagnosis of Usher
syndrome: Application of two different next generation
sequencing-based procedures. PLoS One. 7:e437992012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Millán JM, Aller E, Jaijo T, Blanco-Kelly
F, Gimenez-Pardo A and Ayuso C: An update on the genetics of usher
syndrome. J Ophthalmol. 2011:4172172011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bonnet C, Grati M, Marlin S, Levilliers J,
Hardelin JP, Parodi M, Niasme-Grare M, Zelenika D, Délépine M,
Feldmann D, et al: Complete exon sequencing of all known Usher
syndrome genes greatly improves molecular diagnosis. Orphanet J
Rare Dis. 6:212011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Vozzi D, Aaspõllu A, Athanasakis E, Berto
A, Fabretto A, Licastro D, Külm M, Testa F, Trevisi P, Vahter M, et
al: Molecular epidemiology of Usher syndrome in Italy. Mol Vis.
17:1662–1668. 2011.PubMed/NCBI
|
|
5
|
Jaijo T, Aller E, García-García G, Aparisi
MJ, Bernal S, Avila-Fernández A, Barragán I, Baiget M, Ayuso C,
Antiñolo G, et al: Microarray-based mutation analysis of 183
Spanish families with Usher syndrome. Invest Ophthalmol Vis Sci.
51:1311–1317. 2010. View Article : Google Scholar
|
|
6
|
Huang XF, Xiang P, Chen J, Xing DJ, Huang
N, Min Q, Gu F, Tong Y, Pang CP, Qu J, et al: Targeted exome
sequencing identified novel USH2A mutations in Usher syndrome
families. PLoS One. 8:e638322013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Xing DJ, Zhang HX, Huang N, Wu KC, Huang
XF, Huang F, Tong Y, Pang CP, Qu J and Jin ZB: Comprehensive
molecular diagnosis of Bardet-Biedl syndrome by high-throughput
targeted exome sequencing. PLoS One. 9:e905992014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jin ZB, Huang XF, Lv JN, Xiang L, Li DQ,
Chen J, Huang C, Wu J, Lu F and Qu J: SLC7A14 linked to autosomal
recessive retinitis pigmentosa. Nat Commun. 5:35172014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Soler VJ, Tran-Viet KN, Galiacy SD,
Limviphuvadh V, Klemm TP, St Germain E, Fournié PR, Guillaud C,
Maurer-Stroh S, Hawthorne F, et al: Whole exome sequencing
identifies a mutation for a novel form of corneal intraepithelial
dyskeratosis. J Med Genet. 50:246–254. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu Q, Shen E, Min Q, Li X, Wang X, Li X,
Sun ZS and Wu J: Exome-assistant: A rapid and easy detection of
disease-related genes and genetic variations from exome sequencing.
BMC Genomics. 13:6922012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Vernon M: Sociological and psychological
factors associated with hearing loss. J Speech Hear Res.
12:541–563. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Boughman JA, Vernon M and Shaver KA: Usher
syndrome: Definition and estimate of prevalence from two high-risk
populations. J Chronic Dis. 36:595–603. 1983. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Méndez-Vidal C, González-Del Pozo M,
Vela-Boza A, Santoyo-López J, López-Domingo FJ, Vázquez-Marouschek
C, Dopazo J, Borrego S and Antiñolo G: Whole-exome sequencing
identifies novel compound heterozygous mutations in USH2A in
Spanish patients with autosomal recessive retinitis pigmentosa. Mol
Vis. 19:2187–2195. 2013.PubMed/NCBI
|
|
14
|
Besnard T, Vaché C, Baux D, Larrieu L,
Abadie C, Blanchet C, Odent S, Blanchet P, Calvas P, Hamel C, et
al: Non-USH2A mutations in USH2 patients. Hum Mutat. 33:504–510.
2012. View Article : Google Scholar
|