Introduction
Coronary artery disease (CAD) or atherosclerotic
heart disease is one of the most common types of cardiovascular
disease with a high incidence of morbidity and mortality. It is
well acknowledged that atherosclerosis is the main cause of CAD. Of
the therapeutic measures currently available, percutaneous coronary
intervention [PCI or percutaneous transluminal coronary angioplasty
(PTCA)] has developed into a mature, well-established technique
used to treat CAD since it was first introduced in 1977 (1). PCI effectively improves coronary
blood flow, reduces angina pectoris and greatly improves the
quality of life of patients (2).
Another advantage of PCI is that it is a relatively easy technique
and is minimally invasive. However, its long-term therapeutic
effects are often compromised by the high incidence of vascular
restenosis (RS) following angioplasty. In clinical practice, the
incidence of RS is approximately 20–50% at 6 months post-PCI and
even with stent implantation, the incidence of RS is also
approximately 25–30% (3).
Therefore, vascular RS is a serious complication of PCI, and it is
necessary to fully elucidate the molecular mechanisms responsible
for the development of RS following angioplasty so that a novel
therapeutic methods can be found to prevent the occurrence of
vascular RS following angioplasty.
It has been suggested that the proliferation and
migration of vascular smooth muscle cells (VSMCs) may be key
factors involved in the development of RS following angioplasty
(4). Mature VSMCs are highly
differentiated, and they are principally responsible for
contraction. However, VSMCs also proliferate and produce the matrix
components of the blood vessel wall under specific
pathophysiological conditions, such as vasculogenesis (4). Moreover, VSMCs still retain
remarkable plasticity, and may undergo relatively rapid and
reversible changes in their phenotype in response to local
environmental stress (5).
Vascular RS at an early stage, often occurs several hours or
several days following PCI, and RS is mainly induced by vasospasm
or decreased vessel elasticity. Subsequently, some cytokines,
including endothelin, angiotensin II, basic fibroblast growth
factor, platelet-derived growth factor and transforming growth
factor are released from damaged endothelial cells and macrophages
which have invaded the endothelial sublayer. These cytokines
stimulate the migration and proliferation of VSMCs, and induce the
accumulation of extracellular matrix components, which leads to the
remodeling of the blood vessel wall and ultimately to vascular RS
(6). Although some intracellular
signaling pathways have been found to regulate the proliferation
and migration of VSMCs (2,7,8),
the role of direct intercellular communication pathways and gap
junction (GJ) channels in the proliferation and migration of VSMCs,
as well as their contribution to RS still requires further
investigation.
GJs are direct intercellular communication pathways,
which consist of assemblies of channel proteins known as connexins
(9). Ions, second messengers and
small metabolites of up to 1 kDa in molecular mass may be rapidly
exchanged through GJs (10).
However, the precise assembly of their component connexins
influences the properties of GJ channels, including permeability
and conductance (11). In
arteries, the endothelium expresses three connexin isotypes,
connexin (Cx)40, Cx37 and Cx43. By contrast, VSMCs predominantly
express Cx43 and, in some instances, Cx40 or Cx45 (12–14). It has been reported that the
expression of Cx43 is upregulated during the alteration of the VSMC
phenotype (15). Moreover, Cx43
remodeling involving changes in size, structure, quantity and
distribution may also occur in vascular lesions (16). Cx43 remodeling influences not only
the conductivity and permeability of the GJ itself, but also the
electrical, chemical and metabolic channels between adjacent cells
(17–19). In our previous study, we found
that Cx43 was involved in the development of vascular RS following
angioplasty-induced balloon injury (20). However, the underlying mechanisms
remain unclear.
In the present study, we futher examined whether the
knockdown of Cx43 attenuates vascular RS following
angioplasty-induced balloon injury through the inhibition of VSMC
proliferation and migration. Cx43-RNAi-LV, a lentiviral vector
expressing shRNA targeting Cx43, was used to silence the mRNA and
protein expression of Cx43 in the VSMCs. The results of
3-(4,5-dimethylthiazol-2yl-)-2,5-diphenyl tetrazolium bromide (MTT)
and Transwell assays revealed that the knockdown of Cx43 by
Cx43-RNAi-LV significantly inhibited the proliferation and
migration of VSMCs in vitro. In addition, the knockdown of
Cx43 effectively attenuated the development of vascular RS and
intimal hyperplasia following balloon injury in vivo. Our
data further indicate that Cx43 plays a role in the development of
vascular RS and intimal hyperplasia by regulating the proliferation
and migration of VSMCs.
Materials and methods
Experimental animals
Male Sprague-Dawley rats (purchased from the
Department of Animal Science, Nanchang University, Nanchang, China)
weighing 300–400 g were maintained on a regular chow diet prior to
the study. All animal experimental procedures were in accordance
with the current National Institutes of Health Guidelines, and they
were approved by the Ethics Committee for Animal Experiments at
Nanchang University.
Cell culture
The culture of the VMSCs was performed as previously
described (21). Briefly, the
rats were anesthetized with an intraperitoneal injection of Hydral
(10%, 3.5 ml/kg; Harbin Pharmaceutical Group Co., Ltd., Harbin,
China) and the carotid artery was quickly removed under aseptic
conditions. After stripping the extravascular connective tissue,
the vessels were longitudinally cut off and the internal membrane
was positioned upward in a glass culture dish. Endothelial
denudation was achieved by digestion with 0.25% trypsin. The tunica
media vasorum was cut into 1-mm2-sized tissue blocks.
The internal membrane was positioned upward in a plastic culture
dish. These tissue blocks were incubated at 37°C in a 5%
CO2 saturated humidity incubator for 1–2 h and submerged
in 2 ml medium after firm adhesion. After 5–7 days, the VSMCs had
migrated from the tissue blocks in Dulbecco's modified Eagle's
medium (DMEM) supplemented with 10% fetal calf serum (Trans Serum™;
Transgen Biotech, Beijing, China) and 1% penicillin/streptomycin
(P/S; Solarbio, Beijing, China). Third to fifth generation VSMCs
were used in the experiments.
Construction of GFP-Cx43-shRNA-lentiviral
vectors
To examine the role of Cx43 in vascular RS, a
lentiviral vector expressing shRNA targeting Cx43 (Cx43-RNAi-LV)
was constructed (GeneChem, Shanghai, China). Briefly, the shRNA
sequence for Cx43 (5′-AGAGCACGGCAAGGTGAAA-3′) was designed using
the manufacturer's RNA interference (RNAi) designer program, and
the negative control construct (control shRNA) was created using a
scrambled sequence (5′-TTCTCCGAACGTGTCACGT-3′), as described in a
previous study (22). DNA oligos
were chemically synthesized (GeneChem), annealed and inserted into
the expression vector by double digestion with AgeI and
EcoRI (New England Biolabs, Ipswich, MA, USA), and ligated
with T4 DNA ligase (Takara, Dalian, China) in accordance with the
manufacturer's instructions. The ligation was transformed into
competent E. coli cells and confirmed by restriction enzyme
analysis and DNA sequencing. The sequences were then cloned into
pGCSIL-GFP to generate lentiviral vectors. The expression vectors
and package vectors were transfected into 293T cells (ATCC,
Manassas, VA, USA) using Lipofectamine 2000 (Invitrogen, Carlsbad,
CA, USA). After 48 h of culture, the supernatants containing
lentiviruses, such as Cx43-RNAi-LV and NC-GFP-LV (negative control)
were harvestedy. Purification was then performed using
ultracentrifugation and the lentiviral titer was calculated (as
107 TU/µl).
Reverse transcription-polymerase chain
reaction (RT-PCR)
Total RNA was extracted from the rat carotid tissue
using TRIzol reagent (Tiangen, Beijing, China), and cDNA was
synthesized from the extracted total RNA using the SuperScript III
kit (Promega, Madison, WI, USA) following the manufacturer's
instructions. The specific primers for PCR were as follows: Cx43
sense, 5′-AAAGGCGTTAAGGAT CGCGTG-3′ and antisense,
5′-GTCATCAGGCCGAGG CCT-3′ (23);
β-actin sense, 5′-CCCATCTATGAGGGTT ACGC-3′ and antisense,
5′-TTTAATGTCACGCACGAT TTC-3′. All specific primers were chemically
synthesized (Generay Biotech Co. Ltd., Shanghai, China). The PCR
reactions were performed using the GeneAmp PCR System 9700 (Applied
Biosystems, Foster City, CA, USA). The amplification conditions for
Cx43 were as follows: 94°C for 2 min followed by 32 cycles at 94°C
for 45 sec, 58°C for 45 sec, and 72°C for 90 sec, and the final
extension at 72°C for 5 min. The PCR conditions for β-actin were as
follows: 94°C for 2 min followed by 28 cycles at 94°C for 30 sec,
56°C for 30 sec, and 72°C for 90 sec, and the final extension at
72°C for 5 min. The amplified RT-PCR products were separated on
1.2% (w/v) agarose containing ethidium bromide (both from Sangon
Biotech, Shanghai, China) for 30 min. The results of
electrophoresis were photographed using the Molecular
Imager® ChemiDoc™ XRS+ system, and the signal densities
of the gels were analyzed by Quantity One software (both from
Bio-Rad, Hercules, CA, USA).
Western blot analysis
The VSMCs were harvested and treated with
radioimmunoprecipitation assay (RIPA) lysis buffer (Sangon
Biotech). The whole cell lysate was resus-pended in sample buffer
containing 4% sodium dodecyl sulfate (SDS; Sangon Biotech).
Proteins were separated by SDS-polyacrylamide gel electrophoresis
(PAGE) and transferred onto nitrocellulose membranes (Millipore,
Bedford, MA, USA). The membranes were blocked in 5% skim milk for 1
h at room temperature, and incubated with rabbit polyclonal
anti-Cx43 antibody (71-0700; 1:250 in 5% skim milk; Zymed
Laboratories, San Francisco, CA, USA) or rabbit monoclonal
anti-GAPDH antibody (ABS16; 1:1,000 in 5% skim milk; Chemicon,
Temecula, CA, USA) overnight at 4°C. The membranes were then washed
3 times with TBST, and then incubated with the relative
HRP-conjugated IgG secondary antibody (BV-S8008; Zhongshan Golden
Bridge Biotechnology Co., Beijing, China) for 2 h at room
temperature, and washed in TBST 3 times. A chemiluminescence assay
was carried out with Amersham ECL Prime Western Blotting Detection
reagents, and the immunoblotting signal was detected using the
Molecular Imager® ChemiDoc™ XRS+ system (Bio-Rad). The
intensity of each Cx43 band was normalized to the GAPDH band, and
the relative expression of Cx43 following viral infection was
normalized to the control.
MTT assay
MTT assay was applied to assess the proliferation of
the VSMCs according to the manufacturer's instructions. Briefly,
5×104 cells were seeded in a 96-well tissue culture
plates and cultured in growth medium. The VSMCs were infected with
NC-GFP-LV or Cx43-RNAi-LV at an MOI 0, 50, 100, 150 and 200. At 24,
48, 72 and 96 h after viral infection, 20 µl of the MTT
solution were added to each well (5 mg/ml, 0.5% MTT) and the cells
were cultured for a further 4 h to detect the proliferation of the
VSMCs. Subsequently, 100 µl of dimethyl sulfoxide were added
to each well, and the culture plate was shaken at a low speed for
10 min until the crystals dissolved completely. The color intensity
was measured spectrophotometrically using a microplate reader
(Multiskan FC; Thermo Fisher Scientific, Waltham, MA, USA) at 492
nm. All assays were performed at least 3 times.
Transwell assay
Transwell assays were performed with Transwell
chambers (Corning Inc., Corning, NY, USA) as previously described
(24). Briefly,
5.0×104 VSMCs were seeded into the upper chamber with
200 µl of serum-free medium following infection with
NC-GFP-LV or Cx43-RNAi-LV at an MOI of 100. The bottom of the upper
chamber was incubated in 500 µl of complete medium
containing 10% fetal calf serum and 1% P/S. Following 12 h of
incubation, the cells on the top surface of the insert were gently
removed with a cotton swab. The migrated cells on the lower surface
were fixed with 4% paraformaldehyde and stained with crystal violet
(both from Sigma-Aldrich, St. Louis, MO, USA) for 30 min. The
migrated cells were photographed and counted in 4 random
fields.
Establishment of model of vascular RS
induced by balloon injury
The rats were anesthetized with an intraperitoneal
injection of Hydral (10%, 3.5 ml/kg; Harbin Pharmaceutical Group
Co., Ltd.). To establish a model of vascular RS, the angioplasty
balloon (1.5×20 mm; Cordis Corp., Miami, FL, USA) was inserted into
the rat common carotid artery through an incision in the left
external carotid artery as previously described (25). The balloon was then inflated
sufficiently in the carotid artery and was drawn 3 times
consistently from proximal area to the carotid bifurcation to
produce endothelial denudation. The external carotid was ligated
and blood flow in the common carotid was restored. To examine the
effects of Cx43-RNAi-LV on balloon injury-induced vascular RS, the
rats were randomly divided into 3 groups (n=6/group): control group
(no balloon injury); injury group (balloon injury only) and
Cx43-RNAi-LV-treated injury group (balloon injury + Cx43-RNAi-LV).
The lentivirus (5×108 TU/ml, 100 µl) expressing
shRNA was injected into the balloon-injured rat carotid arteries
using a polyethylene catheter, to knockdown Cx43 in the VSMCs.
Benzylpenicillin sodium (40×104 IU/day for 3 days;
Harbin Pharmaceutical Group Co., Ltd.,) was then administered by
intramuscular injection to prevent infection. All the rats were
euthanized by an overdose of Hydral at 28 days following balloon
injury. Three serial cryosections (5 µm thick) were prepared
from the middle portion of the rat common carotid arteries. The
slices were stained with hematoxylin and eosin (H&E; Sangon
Biotech, Shanghai, China), and histomorphologic observation was
performed under a light microscope (Olympus, Tokyo, Japan) to
examine the structure of the blood vessel wall following balloon
injury. The intimal and medial area of 3 serial cryosections from
each sample was measured using Image-Pro Plus 5.0 software (Media
Cybernetics, Inc., Houston, TX, USA) in order to evaluate vascular
remodeling.
Statistical analysis
The quantitative data are presented as the means ±
standard deviation (SD). Data were analyzed using the Student's
t-test to compare two conditions, and a value of P<0.05 was
considered to indicate a statistically significant difference.
Results
Lentivirus-mediated knockdown of Cx43 in
cultured VSMCs
In our previous study, it was found that Cx43 is
involved in the development of vascular RS and intimal hyperplasia
after balloon injury (20). As
the intimal hyperplasia and vascular RS which occur following
balloon injury are mainly induced by the proliferation and
migration of VSMCs, we used a lentiviral vector expressing shRNA
targeting Cx43 (Cx43-RNAi-LV) to silence Cx43 in the VSMCs. The
results of RT-PCR revealed that the mRNA level of Cx43 was
significantly decreased in the VSMCs infected with Cx43-RNAi-LV,
but not in the control (uninfected) or the NC-GFP-LV-infected
cells. Compared with the controls, the mRNA level of Cx43 in the
Cx43-RNAi-LV-infected VSMCs decreased to approximately 54.3%
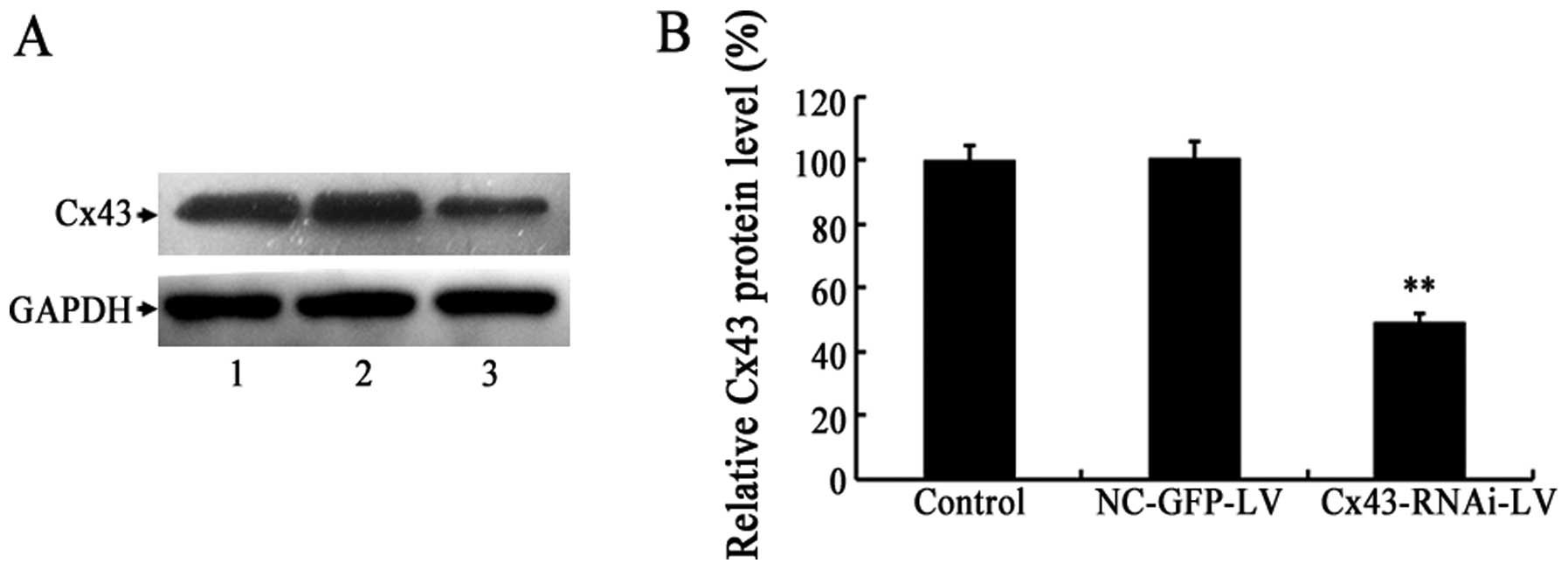
(Fig. 1A). Moreover, the protein
expression of Cx43 in the VSMCs was examined by western blot
analysis. Compared with the controls, the protein level of Cx43 in
the VSMCs infected with Cx43-RNAi-LV decreased to approximately
49.4% (Fig. 2). These results
indicated that the lentivirus-mediated knockdown of Cx43 in the
cultured VSMCs was achieved by infection Cx43-RNAi-LV.
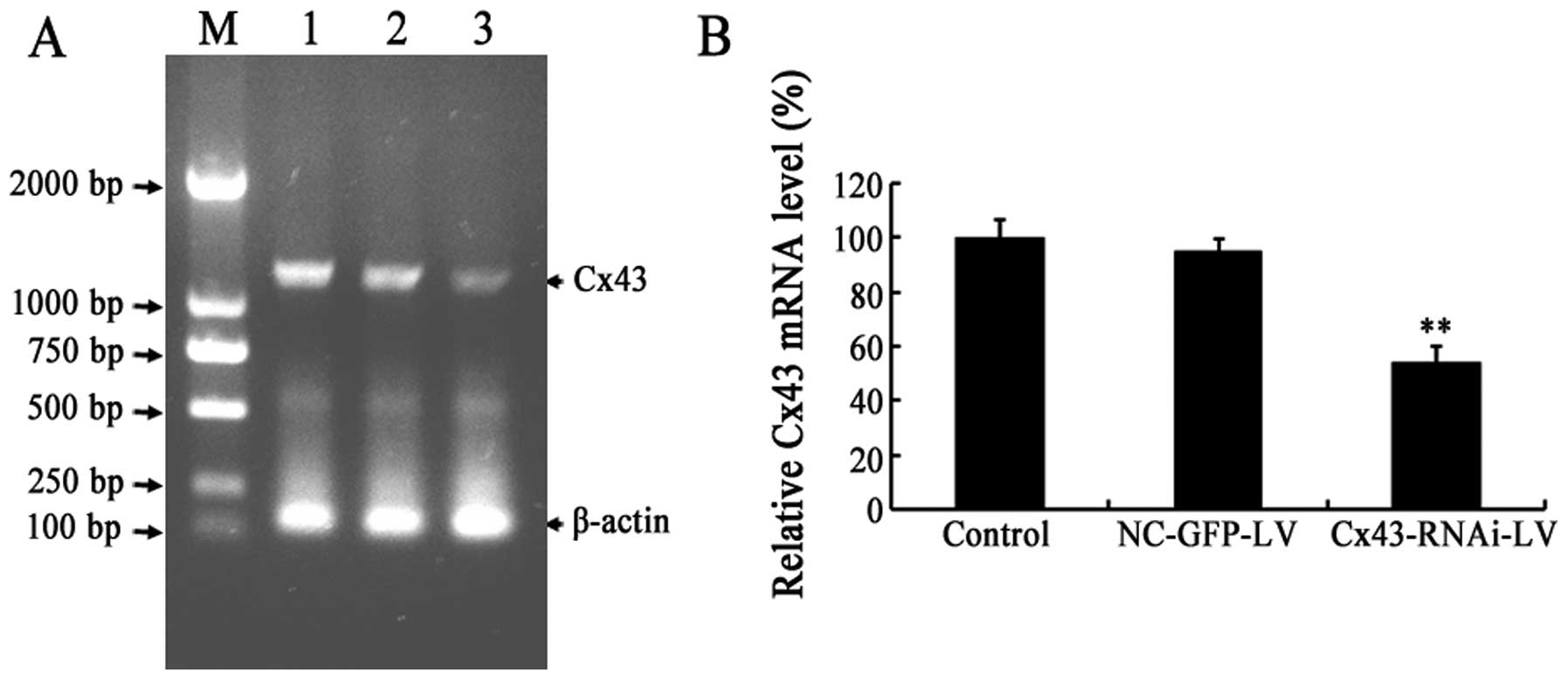 | Figure 1Silencing the mRNA expression of
connexin 43 (Cx43) in cultured vascular smooth muscle cells (VSMCs)
using Cx43-RNAi-LV. (A) Electrophoresis of RT-PCR products from
Cx43 and β-actin on an agarose gel. Lane M, DNA ladder (2,000,
1,000, 750, 500, 250 and 100 bp from top to bottom, respectively);
lane 1, control VSMCs; lane 2, NC-GFP-LV-infected VSMCs, lane 3,
Cx43-RNAi-LV-infected VSMCs. β-actin was used as an endogenous
control. (B) Quantification of the relative Cx43 mRNA level in each
group. The relative Cx43 mRNA level is indicated as the
normalization of the ratio of Cx43/β-actin in each sample to the
control. Data represent the means ± SD of at least 3 independent
experiments. **P<0.01 vs. controls (uninfected
cells). |
Effect of knockdown of Cx43 on the
proliferation of cultured VSMCs
Intimal hyperplasia and vascular RS which occur
following balloon injury are mainly induced by the proliferation
and migration of VSMCs, and the principal gap junction protein in
VSMCs, Cx43, plays an important role in the development of intimal
hyperplasia and vascular RS (4).
Thus, to examine the role of Cx43 in the proliferation of VSMCs,
Cx43-RNAi-LV was used to specifically silence Cx43 in the VSMCs,
and the lentiviral vector, NC-GFP-LV, was used as the negative
control. The proliferation of the VSMCs under each condition was
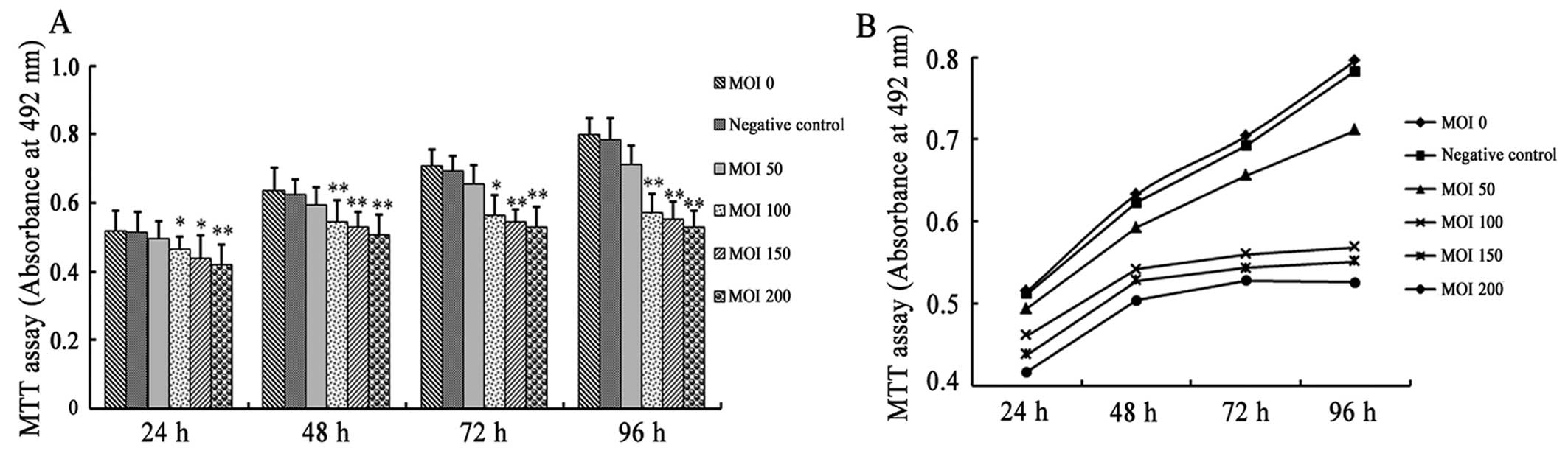
evaluated by MTT assay. Infection with Cx43-RNAi-LV at an MOI of
100, 150 and 200 significantly inhibited the proliferation of the
VSMCs at 24, 48, 72 and 96 h following infection (Fig. 3). By contrast, infection with
NC-GFP-LV and Cx43-RNAi-LV at an MOI of 50 had no significant
effect on the proliferation of the VSMCs. The results of MTT assay
indicated that the gap junction protein, Cx43, is important for the
proliferation of VSMCs.
Knockdown of Cx43 effectively inhibits
the migratory activity of the VSMCs
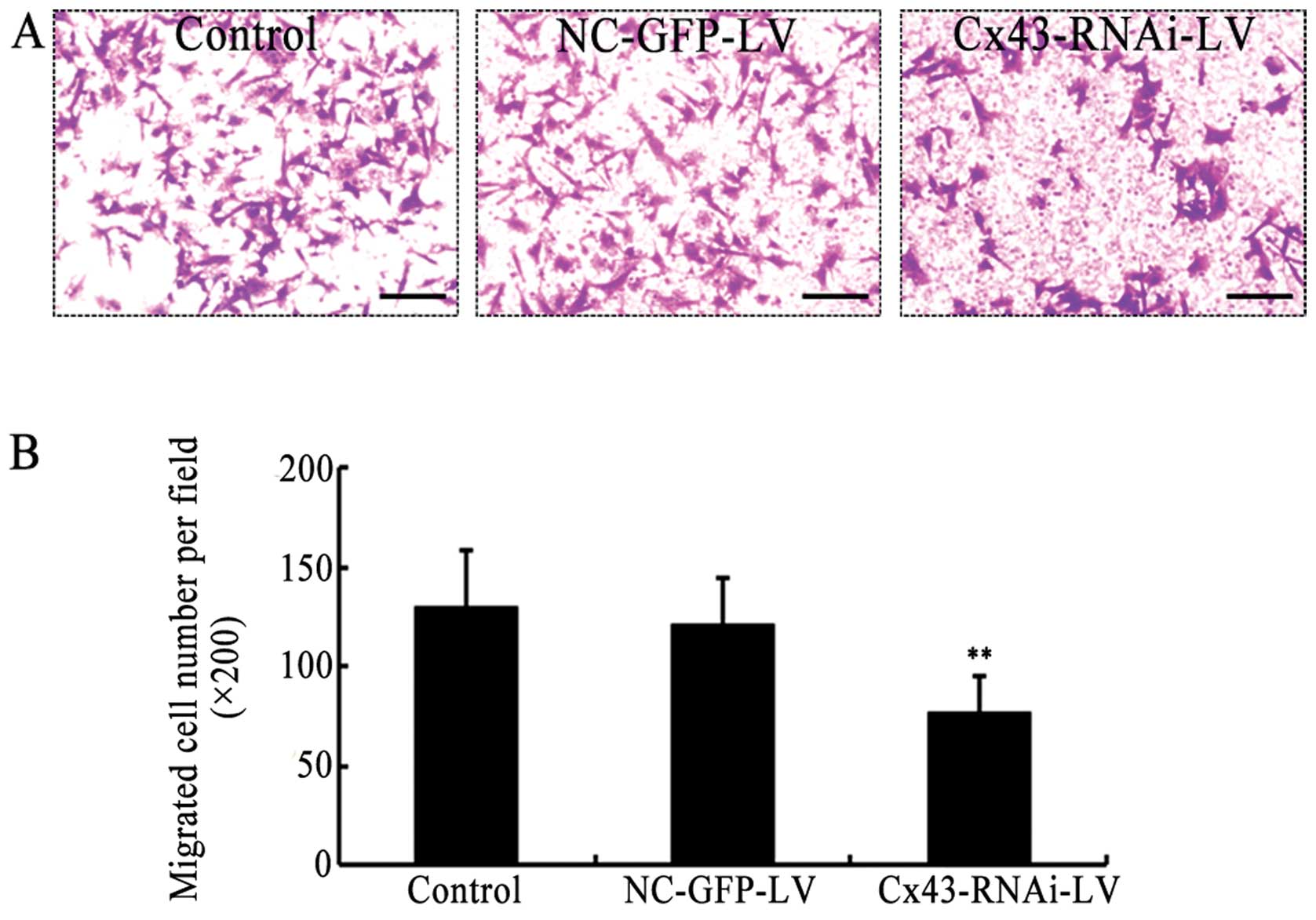
In addition, the migration of the VSMCs is involved
in the development of intimal hyperplasia and vascular RS (4). Thus, to examine the role of Cx43 in
the migration of VSMCs, Cx43 in the VSMCs was specifically silenced
by Cx43-RNAi-LV. The migratory activity of the VSMCs in which Cx43
was knocked down or not was evaluated by Transwell assay. Compared
with the control (uninfected) or the negative control (NC-GFP-LV),
the knockdown of Cx43 by Cx43-RNAi-LV significantly attenuated the
migration of the VSMCs (Fig. 4).
Taken together, the results of MTT assay and Transwell assay,
provide direct evidence that Cx43 is a vital regulatory factor in
the proliferation and migration of VSMCs. Our results also suggest
that Cx43 is involved in the development of intimal hyperplasia and
vascular RS which occur following balloon injury, by regulating the
proliferation and migration of VSMCs.
Knockdown of Cx43 attenuates the
development of intimal hyperplasia and vascular RS induced by
balloon injury in vivo
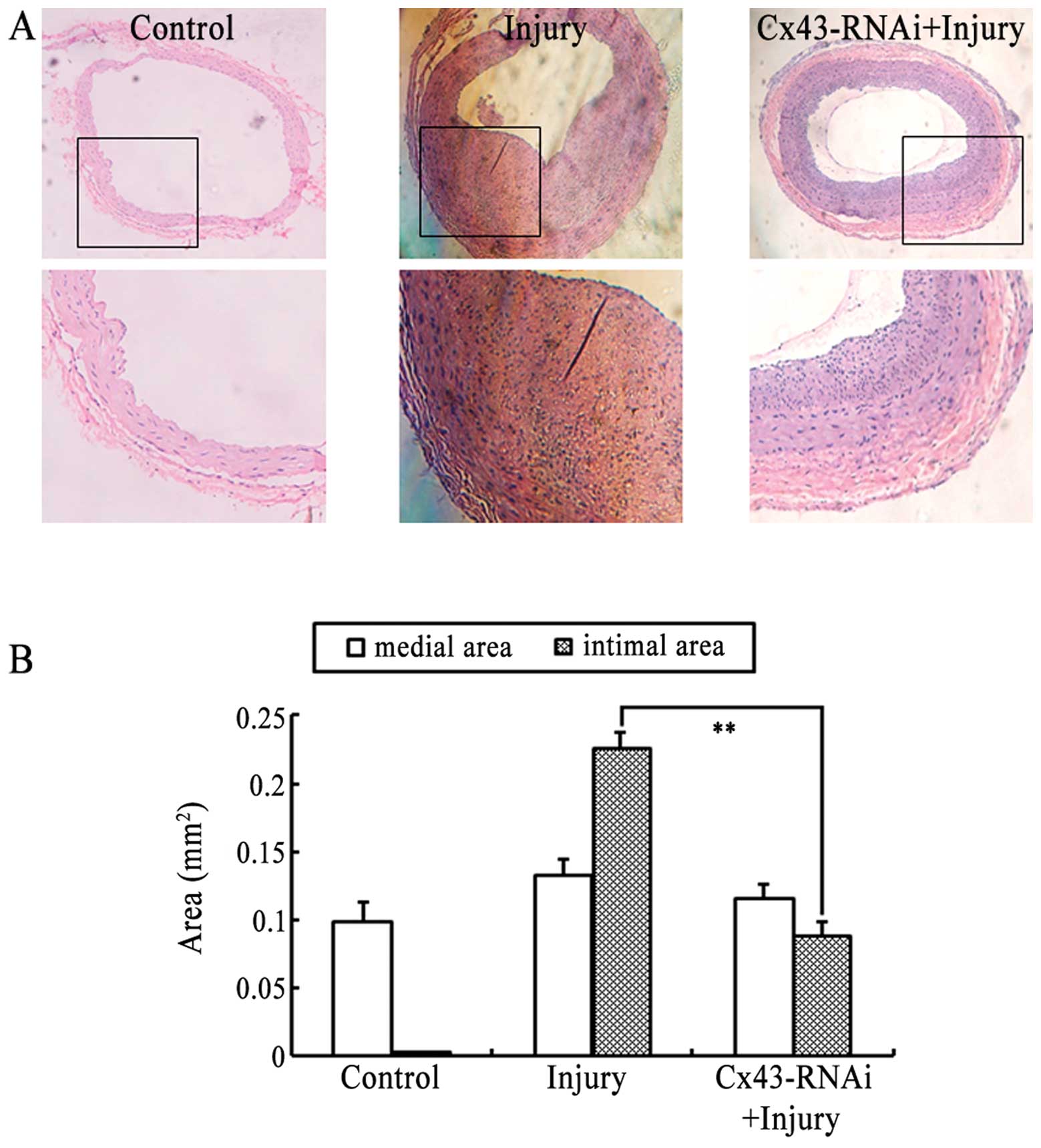
Since the proliferation and migration of VSMCs is
important in the development of intimal hyperplasia and vascular RS
following balloon injury, and the knockdown of Cx43 effectively
attenuated the proliferation and migration of the VSMCs (Figs. 3 and 4), we further examined whether the
knockdown of Cx43 using Cx43-RNAi-LV prevents the development of
intimal hyperplasia and vascular RS following balloon injury in
vivo. The histomorphological observations of the arterial
sections (Fig. 5A) and
statistical analysis of the intimal/medial areas (Fig. 5B) revealed that the
lentivirus-mediated knockdown of Cx43 by Cx43-RNAi-LV significantly
attenutated the development of balloon injury-induced intimal
hyperplasia and vascular RS. These results are consistent with the
results of MTT assay and Transwell assay in vitro, and
suggest that Cx43 is involved in the development of intimal
hyperplasia and vascular RS following balloon injury, through the
regulation of the proliferation and migration of VSMCs.
Discussion
Vascular RS and intimal hyperplasia are the main
adverse effects of PCI, which usually limit the clinical effects of
PCI. Evidence indicates the vital role of endothelial denudation,
as well as the proliferation and migration of VSMCs in vascular RS
and intimal hyperplasia (1,2).
The related molecular mechanisms, nevertheless, remain unclear. On
the other hand, studies have suggested the involvement of GJs in
the development of some vascular diseases, such as hypertension,
atherosclerosis and RS (24,26). Particularly, the principal GJ
protein, Cx43, in VSMCs may be involved in the development of
vascular RS and intimal hyperplasia (20,27). In transgenic mice, it was found
that a reduced Cx43 expression effectively inhibited acute
neointimal formation in hypercholesterolemic mice (28). In our previous study (20), we also found that the expression
of the GJ protein, Cx43, was upregulated in vessels following
balloon injury. The knockdown of Cx43 effectively prevented balloon
injury-induced vascular RS and intimal hyperplasia (20). Moreover, evidence suggests that
GJs are involved in the proliferation and migration-related
signaling pathways of VSMCs (22,29). The blockade of Cx43 hemichannels
has been shown to reduce neointima formation by inhibiting the
proliferation and phenotypic modulation of VSMCs (30). Cx43 is also involved in the
angiotensin II- or oxidized-phospholipid-induced migration and
proliferation of VSMCs (8,31,32).
However, direct evidence is still lacking as to the role of Cx43 in
the proliferation and migration of VSMCs induced by endothelial
denudation or balloon injury. Therefore, it is interesting to
investigate the direct effects of Cx43 silencing on the
proliferation and migration of VSMCs and the development of
vascular RS following PCI.
In the present study, we constructed a lentiviral
vector expressing shRNA targeting Cx43, Cx43-RNAi-LV, to silence
Cx43. Cx43-RNAi-LV effectively knocked down Cx43 in the cultured
VSMCs (Figs. 1 and 2). These results are consistent with the
in vivo results of our previous study (20). To examine the effects of
Cx43-RNAi-LV on the proliferation of VSMCs, the cells were infected
with Cx43-RNAi-LV at various MOIs (0, 50, 100, 150 and 200).
Compared with infection at an MOIof 0 or infection with the
negative control, infection with Cx43-RNAi-LV at MOIs of 100, 150
and 200 significantly inhibited the proliferation of VSMCs
(Fig. 3). In addition, the
migration of the cultured VSMCs under each experimental condition
was evaluated by Transwell assay. Infection with Cx43-RNAi-LV at an
MOI of 100 significantly attenuated the migratory activity of the
VSMCs (Fig. 4). By contrast,
infection with NC-GFP-LV had no significant effect on the migration
of the VSMCs. These in vitro results are consistent with
those of a previous study (30),
and provide direct evidence that Cx43 plays an important role in
regulating the proliferation and migration of VSMCs. On the other
hand, the lentivirus-mediated knockdown of Cx43 effectively
attenuated the development of vascular RS and intimal hyperplasia
following balloon injury (Fig.
5). Taken together, these results indicate that Cx43
contributes to the development of vascular RS and intimal
hyperplasia through the regulation of the proliferation and
migration of VSMCs. Although the detailed mechanisms involved
remain unclear, it has been suggested the MAPK signaling pathway
regulates the role of Cx43 in the proliferation and migration of
VSMCs (8,33). The MAPK-AP-1 signaling pathway has
been found to regulate the role of Cx43 in the angiotensin
II-induced migration and proliferation of saphenous vein smooth
muscle cells (8). Moreover, the
MAPK-mediated phosphorylation of Cx43 promotes the proliferation of
VSMCs through the facilitation of the binding of Cx43 to cell cycle
protein cyclin E (33).
In conclusion, our data demonstrate that the
knockdown of the principal GJ protein in VSMCs, Cx43, may be an
effective measure to prevent the development of intimal hyperplasia
and vascular RS through the inhibition of the proliferation and
migration of VSMCs. In the present study, we constructed a
lentiviral vector expressing shRNA targeting Cx43. Infection with
Cx43-RNAi-LV effectively silenced the mRNA and protein expression
of Cx43 in the VSMCs. In addition, the knockdown of Cx43 with
Cx43-RNAi-LV significantly inhibited the proliferation and
migration of VSMCs in vitro, and effectively attenuated the
development of vascular RS and intimal hyperplasia following
balloon injury in vivo. These results demonstrate that the
GJ protein, Cx43, plays an important role in the development of
vascular RS and intimal hyperplasia through the regulation of the
proliferation and migration of VSMCs. Thus, our data suggest that
Cx43 may be a novel and promising pharmacological target for
preventing the development of intimal hyperplasia and RS following
PCI.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (grant nos. 30760286 and 81241125), the
Jiangxi Province Natural Science Foundation (grant no.
20142BAB205022), and partially by the National Natural Science
Foundation of China (grant nos. 31360241 and 81472371), and the
Postgraduate Student Foundation for New Teachers from the Ministry
of Education of China (no. 20123601120001).
Abbreviations:
|
RS
|
restenosis
|
|
PCI
|
percutaneous coronary intervention
|
|
VSMCs
|
vascular smooth muscle cells
|
|
GJ
|
gap junctions
|
|
Cx43
|
connexin 43
|
|
RNAi
|
RNA interference
|
|
CAD
|
coronary artery disease
|
References
|
1
|
Trikalinos TA, Alsheikh-Ali AA, Tatsioni
A, Nallamothu BK and Kent DM: Percutaneous coronary interventions
for non-acute coronary artery disease: a quantitative 20-year
synopsis and a network meta-analysis. Lancet. 373:911–918. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Meads C, Cummins C, Jolly K, Stevens A,
Burls A and Hyde C: Coronary artery stents in the treatment of
ischaemic heart disease: a rapid and systematic review. Health
Technol Assess. 4:1–153. 2000.PubMed/NCBI
|
|
3
|
Odell A, Grip L and Hallberg LR:
Restenosis after percutaneous coronary intervention (PCI):
experiences from the patients' perspective. Eur J Cardiovasc Nurs.
5:150–157. 2006. View Article : Google Scholar
|
|
4
|
Zhang C, Chaturvedi D, Jaggar L, Magnuson
D, Lee JM and Patel TB: Regulation of vascular smooth muscle cell
proliferation and migration by human sprouty 2. Arterioscler Thromb
Vasc Biol. 25:533–538. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Owens GK: Regulation of differentiation of
vascular smooth muscle cells. Physiol Rev. 75:487–517.
1995.PubMed/NCBI
|
|
6
|
Crowley ST, Ray CJ, Nawaz D, Majack RA and
Horwitz LD: Multiple growth factors are released from mechanically
injured vascular smooth muscle cells. Am J Physiol.
269:H1641–H1647. 1995.PubMed/NCBI
|
|
7
|
Li F, Zhang C, Schaefer S, Estes A and
Malik KU: ANG II-induced neointimal growth is mediated via cPLA2-
and PLD2-activated Akt in balloon-injured rat carotid artery. Am J
Physiol Heart Circ Physiol. 289:H2592–H2601. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jia G, Cheng G, Gangahar DM and Agrawal
DK: Involvement of connexin 43 in angiotensin II-induced migration
and proliferation of saphenous vein smooth muscle cells via the
MAPK-AP-1 signaling pathway. J Mol Cell Cardiol. 44:882–890. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hervé JC, Phelan P, Bruzzone R and White
TW: Connexins, innexins and pannexins: bridging the communication
gap. Biochim Biophys Acta. 1719:3–5. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bruzzone R, White TW and Paul DL:
Connections with connexins: the molecular basis of direct
intercellular signaling. Eur J Biochem. 238:1–27. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Elfgang C, Eckert R, Lichtenberg-Fraté H,
Butterweck A, Traub O, Klein RA, Hülser DF and Willecke K: Specific
permeability and selective formation of gap junction channels in
connexin-transfected HeLa cells. J Cell Biol. 129:805–817. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
van Kempen MJ and Jongsma HJ: Distribution
of connexin37, connexin40 and connexin43 in the aorta and coronary
artery of several mammals. Histochem Cell Biol. 112:479–486. 1999.
View Article : Google Scholar
|
|
13
|
Hong T and Hill CE: Restricted expression
of the gap junctional protein connexin 43 in the arterial system of
the rat. J Anat. 192:583–593. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li X and Simard JM: Increase in Cx45 gap
junction channels in cerebral smooth muscle cells from SHR.
Hypertension. 40:940–946. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Matsushita T, Rama A, Charolidi N, Dupont
E and Severs NJ: Relationship of connexin43 expression to
phenotypic modulation in cultured human aortic smooth muscle cells.
Eur J Cell Biol. 86:617–628. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ram R, Wescott AP, Varandas K, Dirksen RT
and Blaxall BC: Mena associates with Rac1 and modulates connexin 43
remodeling in cardiomyocytes. Am J Physiol Heart Circ Physiol.
306:H154–H159. 2014. View Article : Google Scholar :
|
|
17
|
Kieken F, Mutsaers N, Dolmatova E, Virgil
K, Wit AL, Kellezi A, Hirst-Jensen BJ, Duffy HS and Sorgen PL:
Structural and molecular mechanisms of gap junction remodeling in
epicardial border zone myocytes following myocardial infarction.
Circ Res. 104:1103–1112. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Qu J, Volpicelli FM, Garcia LI, Sandeep N,
Zhang J, Márquez-Rosado L, Lampe PD and Fishman GI: Gap junction
remodeling and spironolactone-dependent reverse remodeling in the
hypertrophied heart. Circ Res. 104:365–371. 2009. View Article : Google Scholar :
|
|
19
|
Rucker-Martin C, Milliez P, Tan S, Decrouy
X, Recouvreur M, Vranckx R, Delcayre C, Renaud JF, Dunia I,
Segretain D and Hatem SN: Chronic hemodynamic overload of the atria
is an important factor for gap junction remodeling in human and rat
hearts. Cardiovasc Res. 72:69–79. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Han XJ, Chen M, Hong T, Zhu LY, He D, Feng
JG and Jiang LP: Lentivivirus-mediated RNAi knockdown of the gap
junction protein, Cx43, attenuates the development of vascular
restenosis following balloon injury. Int J Mol Med. 35:885–892.
2015.PubMed/NCBI
|
|
21
|
Zhang J, Guo C, Wang R, Huang L, Liang W,
Liu R and Sun B: An Egr-1-specific DNAzyme regulates Egr-1 and
proliferating cell nuclear antigen expression in rat vascular
smooth muscle cells. Exp Ther Med. 5:1371–1374. 2013.PubMed/NCBI
|
|
22
|
Ai Z, Yin L, Zhou X, Zhu Y, Zhu D, Yu Y
and Feng Y: Inhibition of survivin reduces cell proliferation and
induces apoptosis in human endometrial cancer. Cancer. 107:746–756.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Barac YD, Zeevi-Levin N, Yaniv G, Reiter
I, Milman F, Shilkrut M, Coleman R, Abassi Z and Binah O: The
1,4,5-inositol trisphosphate pathway is a key component in
Fas-mediated hypertrophy in neonatal rat ventricular myocytes.
Cardiovasc Res. 68:75–86. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Severs NJ, Rothery S, Dupont E, Coppen SR,
Yeh HI, Ko YS, Matsushita T, Kaba R and Halliday D:
Immunocytochemical analysis of connexin expression in the healthy
and diseased cardiovascular system. Microsc Res Tech. 52:301–322.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Meng QH, Yang G, Yang W, Jiang B, Wu L and
Wang R: Protective effect of hydrogen sulfide on balloon
injury-induced neointima hyperplasia in rat carotid arteries. Am J
Pathol. 170:1406–1414. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Brisset AC, Isakson BE and Kwak BR:
Connexins in vascular physiology and pathology. Antioxid Redox
Signal. 11:267–282. 2009. View Article : Google Scholar
|
|
27
|
Déglise S, Martin D, Probst H, Saucy F,
Hayoz D, Waeber G, Nicod P, Ris HB, Corpataux JM and Haefliger JA:
Increased connexin43 expression in human saphenous veins in culture
is associated with intimal hyperplasia. J Vasc Surg. 41:1043–1052.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Chadjichristos CE, Matter CM, Roth I,
Sutter E, Pelli G, Lüscher TF, Chanson M and Kwak BR: Reduced
connexin43 expression limits neointima formation after balloon
distension injury in hypercholesterolemic mice. Circulation.
113:2835–2843. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chadjichristos CE, Morel S, Derouette JP,
Sutter E, Roth I, Brisset AC, Bochaton-Piallat ML and Kwak BR:
Targeting connexin 43 prevents platelet-derived growth
factor-BB-induced phenotypic change in porcine coronary artery
smooth muscle cells. Circ Res. 102:653–660. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Song M, Yu X, Cui X, Zhu G, Zhao G, Chen J
and Huang L: Blockade of connexin 43 hemichannels reduces neointima
formation after vascular injury by inhibiting proliferation and
phenotypic modulation of smooth muscle cells. Exp Biol Med
(Maywood). 234:1192–1200. 2009. View Article : Google Scholar
|
|
31
|
Shi Y, Hou X, Zhang X, Wang Y, Chen Y and
Zou J: Inhibition of oxidized-phospholipid-induced vascular smooth
muscle cell proliferation by resveratrol is associated with
reducing Cx43 phosphorylation. J Agric Food Chem. 61:10534–10541.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Johnstone SR, Ross J, Rizzo MJ, Straub AC,
Lampe PD, Leitinger N and Isakson BE: Oxidized phospholipid species
promote in vivo differential cx43 phosphorylation and vascular
smooth muscle cell proliferation. Am J Pathol. 175:916–924. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Johnstone SR, Kroncke BM, Straub AC, Best
AK, Dunn CA, Mitchell LA, Peskova Y, Nakamoto RK, Koval M, Lo CW,
et al: MAPK phosphorylation of connexin 43 promotes binding of
cyclin E and smooth muscle cell proliferation. Circ Res.
111:201–211. 2012. View Article : Google Scholar : PubMed/NCBI
|