Introduction
The p53 signaling pathway is activated in response
to a variety of stress signals to facilitate the expression of
genes mediating cell cycle arrest, DNA repair and apoptosis.
Mutations in the p53 gene are the most common genetic abnormality
found in human cancers (1) and
may be regarded as a hallmark of cancer cells (2). However, mutant p53 proteins in
cancer cells not only lose their tumor suppressor function, but
often gain additional oncogenic functions that endow cells with
growth and survival advantages, as well as metastastatic ability.
Resistance to chemotherapy is also a phenotypic gain-of-function
effect of p53 mutations. Mutant p53 proteins have been found in
drug-induced resistant cancer cell lines and in tumors following
exposure to anticancer drugs (3).
The p53 status has a significant impact on the resistance of cancer
cells to chemotherapy. Thus, research has focused on identifying
p53 mutations and their association with drug resistance (4).
Multidrug resistance (MDR) is the outcome of a
series of mechanisms through which cancer cells develop resistance
to chemotherapeutic agents. The mechanisms involved in MDR include
the activation of DNA repair pathways, the alteration of drug
targets, as well as a decrease in the uptake of chemotherapeutic
drugs (5). Drug efflux is
mediated by ATP-binding cassette (ABC) transporters, which are
members of a protein superfamily that reduces intracellular drug
concentrations. The upregulation of ABC transporter genes in cancer
cells results in the active efflux of drugs, which is an important
cause of MDR (6). MDR has been
linked to gene amplification and/or increased MDR1 gene expression
(7). The human MDR1 gene promoter
contains a number of recognition sites for SP1, nuclear factor
(NF)-Y, p53, NF-κB, and Y-box binding protein 1 (YB-1)
transcription factors, which upregulate MDR1 promoter activity
(8–11,21). As regards the effect of the p53
status on the acquisition of a MDR phenotype, an inhibitory role
for wild-type p53 on the MDR1 gene promoter has been demonstrated,
whereas mutant p53 acts as an activator (12). Mutant p53 has been reported to
upregulate NF-κB in cancer cells (13–15). The expression of mutant p53 and
NF-κB has been found in many types of cancer with a poor prognosis
(16,17). NF-κB is a key transcription factor
which plays a role in cancer progression and chemoresistance by
ativating a multitude of mediators and processes, including cell
growth, survival, transporters, anti-apoptotic genes and oncogenes
(18–21). The association between the p53
status and NF-κB and their role in the development of drug
resistance merits further investigation.
Epithelial-mesenchymal transition (EMT) is a process
through which epithelial cells lose their cell polarity to give
rise to matrix-producing fibroblasts and myofibroblasts from their
neighbors and migrate to distant regions during tumor cell
progression and metastasis. EMT in malignant cells is closely
related to MDR (22).
Transcription factors that lead to EMT and invasion orchestrate the
overexpression of drug transporters by directly modulating promoter
activity (23). Cadherin 1, type
1, E-cadherin (CDH1) is a suppressor of invasion and metastasis
(24). The downregulation of CDH1
is considered a hallmark of EMT. Slug, a member of the Snail family
of transcriptional repressors, is capable of repressing CDH1
expression and triggering EMT during malignant transformation and
metastatic progression in various types of cancer (25–27). It has been demonstrated that the
wild-type and mutant forms of p53 exert opposite effects on the
invasion-promoting factor, Slug, to regulate cancer invasion and
metastasis (26). During the
development of drug resistance, the p53 status may be important for
the activation of the EMT process, which mediates transformation,
invasion and cancer stem cell-like properties (28,29).
CD44 is a stem cell-like marker and its expression
is important in the progression of many types of cancer. It has
been demonstrated that CD44 plays a role in cell migration,
differentiation and survival, which is important to cancer stem
cells (CSCs) (30). When CD44 is
highly expressed in breast cancer cells, it generates a
microenvironment that facilitates-tumor progression and invasion
(30). It would be interesting to
determine whether mutant p53 plays a role in the process through
which cells acquire stem cell-like properties.
Missense mutations are the most common (75%) of p53
mutations. Mutation hotspots may be responsible for
gain-of-function effects (31).
However, we have previously established a series of MCF-7 cell
lines with incremental levels of resistance to doxorubicin and
found that only the cell line that overexpressed the MDR1 gene
contained a deleted p53 gene (Y127_K133 del p53) (32). The p53 deletion (designated as del
p53) leads to increased stability, overexpression and nuclear
localization of the protein. This deletion site has been found in
the MCF-7/adr cell lines following different induction procedures
by doxorubicin (33,34). del p53 was also found in U1285
lung cancer and OVCAR-8 ovarian cancer cell lines with low levels
of P-glyco protein (P-gp) (33,35–37). Ribophorin II (RPN2) which is
regulated by del p53 confers P-gp-mediated docetaxel resistance in
MCF-7/adr cells (38). Moreover,
oxidative stress-responsive heat shock factor (HSF)-1 and heat
shock protein (Hsp)27 have been shown to be inhibited in mutant
p53-expressing MCF-7/adr cells, leading to increased NF-κB activity
(15). Accordingly, the role of
the p53 gene with a 21- (bp) deletion within the DNA binding domain
warrants further investigation. The aim of this study was to
examine the gain-of-resistance and metastatic properties mediated
by del p53 in MCF-7 cells. Using a MCF-7 clone which stably
overexpressed del p53, we examined cell proliferation, the
expression levels of the genes related to drug resistance, the EMT
process and stem cell-like properties.
Materials and methods
Chemicals, cell lines and cell
culture
The MCF-7/adr cell line and its counterpart,
MCF-7/wt, were kindly provided by Dr Chih-Hsin Yang (National
Taiwan University Hospital, Taipei, Taiwan). The expression vector
pcDNA3.1, pGL3-Basic vector, TransFast™ Transfection reagent and
GoTaq Green Master Mix were purchased from Promega Corp. (Madison,
WI, USA). Lipofectamine™ 2000, TRIzol reagent, penicillin and
streptomycin were purchased from Invitrogen (Carlsbad, CA, USA).
G418 was purchased from Gene Teks Bioscience (New Taipei City,
Taiwan). Cyclosporin A (CsA; Sandimmune) was purchased from Roche
(Mannheim, Germany).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
solution, verapamil, rhodamine 123 and doxorubicin were purchased
from Sigma-Aldrich (St. Louis, MO, USA). Anti-MDR1 (SC-55510),
anti-p53 (DO-1; SC-126), anti-NF-κB (p65; SC-372), and anti-mouse
horseradish peroxide (HRP; SC-2005) and anti-rabbit
peroxidase-conjugated (SC-2004) secondary antibodies were purchased
from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). The
cells were maintained in Dulbecco's modified Eagle's medium (DMEM)
with 10% fetal bovine serum (FBS) and 100 ng/ml of penicillin and
streptomycin at 37°C in 5% CO2. The MCF-7/adr cells were
grown in medium containing 6–8 µg/ml of doxorubicin which
was removed 1 week prior to the assays.
Construction of the plasmid pcDNA3.1
containing del p53 and the establishment of the MCF-7/del p53 cell
line stably expressing del p53
Total RNA was extracted from the MCF-7/adr cells and
subjected to reverse transcription-polymerase chain reaction
(RT-PCR) to yield the 21-bp-deleted p53 cDNA (del p53). Briefly,
the EcoRI and KpnI restriction sites were linked onto
the forward and reverse primers as follows: del p53 (EcoRI)
forward, 5′-GGGAATTCATGGAGGAGCCGCAGT-3′ and del p53 (KpnI)
reverse, 5′-GGGGTACCGTCTGAGTCAGGCCCTT-3′. The PCR products were
visualized by ethidium bromide staining, followed by double
digestion with EcoRI and KpnI restriction enzymes to
yield a 1,104-bp fragment which was confirmed by DNA sequencing.
The del p53 sequence was then inserted into the eukaryotic
expression vector, pcDNA3.1, to generate a recombinant plasmid. The
resulting plasmid, pcDNA3.1-del p53, was transfected into the
MCF-7/wt cells, followed by the selection of stable clones
containing the del p53 gene. A vector expressing C-terminal
FLAG-tagged protein was constructed to confirm gene expression.
Briefly, the transfection of pcDNA3.1-del p53 was carried out using
Lipofectamine 2000 when the cells reached 80% cell fusion, as
previously described (39). DMEM
(250 µl) without serum and 4 µg of the plasmid
pcDNA3.1-del p53 per well were pre-incubated for 5–10 min at room
temperature, followed by the addition of 10 µl Lipofectamine
2000. The cells were then transfected according to the
manufacturer's instructions. The successfully transfected cells
were selected by 400 µg/ml G418 gradually for 1 month to
establish cell lines containing del p53 and cultured for at least
90 days to yield the stable MCF-7/del p53 cell line.
Full-length p53 cDNA sequencing
The primers used for amplifying p53 cDNA fragments
and overlapping the full-length p53 coding sequence were as
follows: codons 1–148, 5′-ATGGAGGAGCCGCAGTCA-3′ and
5′-ATCAACCCACAGCTGCACAGGG-3′; codons 118–353,
5′-GGGACAGCCAAGTCTGTGACT-3′ and 5′-CCTGGGCATCCTTGAGTT-3′; and
codons 253–393, 5′-ACCATCATCACACTGGAAGACTCC-3′ and
5′-ATGTCAGTCTGAGTCAGG-3′. PCR products were sequenced by Mission
Biotech (Taipei, Taiwan) as previously described (33,34).
Determination of gene expression by
RT-PCR
Total RNA was isolated from the cells using TRIzol
reagent. First strand cDNA was synthesized from the extracted RNA
using an oligo(dT) primer. After the cDNA was synthesized, the
primers of target genes were employed and GoTaq Green Master Mix
was used to amplify the genes. The gene products were separated on
a 2% agarose gel (2% agarose/TAE buffer 100 ml). DNA was stained by
ethidium bromide for 3 min, and then detected using a UVP BioDoc-It
imaging system (UVP, Inc., Upland, CA, USA). The primer sequences
are listed in Table I.
 | Table IPrimer sequences used for RT-PCR. |
Table I
Primer sequences used for RT-PCR.
| Gene | | Sequence | Size (bp) |
|---|
| del p53 mutant | Forward | 5′-
GAAGACCCAGGTCCAGATGA -3′ | 222 |
| Reverse | 5′-
TGGCAAAACATCGTGCAAGTC -3′ | |
| p53 wild-type | Forward | 5′-
GAAGACCCAGGTCCAGATGA -3′ | 228 |
| Reverse | 5′-
CTTGTTGAGGGCAGGGGAGTA -3′ | |
| β-actin | Forward | 5′-
ACAGCTGAGGGAAATCGTGGG -3′ | 150 |
| Reverse | 5′-
ACTTGCGCTCAGGAGGAGCAATG -3′ | |
| Slug | Forward | 5′-
AGATGCATATTCGGACCCAC -3′ | 257 |
| Reverse | 5′-
CCTCATGTTTGTGCAGGAGA -3′ | |
| Twist1 | Forward | 5′-
GGAGTCCGCAGTCTTACGAG -3′ | 201 |
| Reverse | 5′-
TCTGGAGGACCTGGTAGAGG -3′ | |
| Snail | Forward | 5′- GAA
AGGCCTTCAACTGCAAA -3′ | 249 |
| Reverse | 5′-
TGACATCTGAGTGGGTCTGG -3′ | |
| VIM | Forward | 5′-
GGAAGCTGCTGGAAGGCGA -3′ | 159 |
| Reverse | 5′-
CCTGTCCATCTCTAGTTTCAACCGTCTTA -3′ | |
| CDH2 | Forward | 5′-
AATGACAATCCTCCAGAGTTTACTGCC -3′ | 210 |
| Reverse | 5′-
GGTGACTAACCCGTCGTTGCT -3′ | |
| CDH1 | Forward | 5′-
TCACAGCAGAACTAACACACGGG -3′ | 165 |
| Reverse |
5′-GTGGTCACTTGGTCTTTATTCTGGTTATCC-3′ | |
MTT assay
The cells were seeded in 96-well plates at a density
of 5×103 cells/well. The cytotoxicity of doxorubicin to
the MCF-7/wt, MCF-7/del p53 and MCF-7/adr cells was determined by
MTT assay after the cells were incubated with doxorubicin
(10−8–10−4 M) for 3–5 days.
Western blot analysis of P-gp, p53 and
NF-κB
The MCF-7/wt, MCF-7/pcDNA, MCF-7/del p53 or
MCF-7/adr cells were seeded into 6-cm plates at a density of
1×106 cells/well. Following incubation for 2 days, total
protein was isolated using RIPA cell lysis buffer, containing 150
mM NaCl, 1.0% (v/v) Triton X-100, 0.5% (v/v) sodium deoxycholate,
0.1% (w/v) SDS and 50 mM Tris (pH 8.0). Protein concentrations were
determined using a Bradford assay with the Bio-Rad protein assay
kit (Richmond, CA, USA). Protein samples were loaded onto a 10%
SDS-polyacryamide gel, and then transfered onto an Immobilon NC
membrane (Millipore Corp., Bedford, MA, USA) with transfer buffer
[25 mM Tris, 190 mM glycine, 20% (v/v) methanol]. The membranes
were then blocked in 5% milk TBST (Tris-buffered saline Tween-20)
at room temperature for 1 h. Proteins were labeled with anti-MDR1,
anti-p53 (DO-1) and anti-NF-κB (p65) antibodies. The p53 antibody
(DO-1) was used for the detection of wild-type and mutant p53 by
epitope mapping of amino acid residues 11–25 of p53. Immunoreactive
bands were detected by anti-mouse HRP or anti-rabbit
peroxidase-conjugated secondary antibody. Protein bands were
visualized using an enhanced chemiluminescence (ECL) detection kit
(GE Healthcare, Little Chalfont, UK) and detected using a UVP
BioDoc-IT imaging system.
Construction of the expression vector
containing the MDR1 promoter
The sequences of the primers used for plasmid
construction are as follows: primer 1,
5′-GCGCTAGCCTAGAGAGGTGCAACG-3′ (−198 to −182); primer 2,
5′-GCAGATCTGCGGCCTCTGCTTCTT-3′ (+28 to +43). The 241-bp MDR1
promoter fragment (residues −198 to +43) was amplified by PCR using
primers 1 and 2. The pGL3-Basic vector was digested with
NheI and BglII. The gel-purified PCR product was
digested with NheI and BglII and cloned into the
vector, as previously described (40). The accuracy of the pGL3-promoter
vector was confirmed by direct sequencing.
Transient expression assay
The cells (3×105 cells/well) were seeded
into 6-well plates and grown in 5 ml of DMEM with 10% FCS for 24 h
prior to transfection. Using TransFast Transfection reagent, the
cells were transfected with 2 µg/well plasmid. Luciferase
activity was measured using a Bright-Glo Luciferase Assay system
(Promega Corp.). Luminescence was measured using a Berthold
Microplate Luminometer (Berthold Technologies GmbH, Bad Wildbad,
Germany).
Determination of drug efflux by flow
cytometric analysis
The cells were pre-treated with or without 4
µM of the P-gp inhibitor, verapamil, for 2 h. The cells were
then incubated with 10 µM rhodamine 123 in the dark at 37°C
for 1 h and were then trypsinized from the subfluent monolayer, and
the pellet was washed twice with ice-cold PBS. Rhodamine 123
accumulation in the cells was analyzed immediately using a
FACSCaliber flow cytometer (BD Biosciences San Jose, CA, USA). The
fluorescence of rhodamine 123 was measured using a FL1 band-pass
filter.
Wound healing assay
The cells in medium containing 10% FBS were seeded
into 24-multiwell plates. After the cells grew to confluence,
wounds were made using sterile pipette tips. The cells were washed
with PBS and refreshed with medium with or without 10% FBS.
Following overnight incubation at 37°C, the cells were photographed
using a Nikon Coolpix 995 digital camera (Nikon Corp., Tokyo,
Japan). The migration distances were quantified by Inage J software
which is a Java-based image analysis package.
Flow cytometric analysis of the CSC
markers, CD44 and CD24
The MCF-7 cells were analyzed after staining with
CD24-PE (Cat. no. 555428) or CD44-FITC (Cat. no. 555478) antibody
that were purchased from BD Pharmingen (San Diego, CA, USA). At
least 1×105 cells were centrifuged at 500 × g for 3 min
at 4°C, resuspended in 10 µl of FITC-conjugated anti-CD44
and 10 µl of PE-conjugated anti-CD24, and then incubated at
4°C in the dark for 30 min. As a negative control, cells were
incubated with the isotype of CD44 or CD24. The labeled cells were
washed 3 times and then analyzed using a FACSCaliber flow cytometer
(BD Biosciences). Each analysis detected 10,000 cells.
Colony formation assay
The cells (5×103) in 1.5 ml 0.35%
agarose-containing growth medium were overlaid with 1.5 ml 0.5%
agarose-containing growth medium, and the cells were incubated for
10–14 days. The whole-well images were photographed using a Nikon
Coolpix 995 digital camera (Nikon Corp.) and 3 fields (×10
magnification) of each well were imaged.
Statistical analysis
Data are statistically presented as the means ± SEM
for the indicated number of separate experiments. Comparisons
between groups were made using Student's t-tests. Probability
values of P<0.05 were considered to indicate statistically
significant differences.
Results
Establishment of the MCF-7/del p53 cell
line stably expressing del p53
Total RNA was extracted from the MCF-7/adr cells and
subjected to RT-PCR to yield a 1,104-bp full-length mutant p53
cDNA, designated as del p53. del p53 was then inserted into the
eukaryotic expression vector, pcDNA3.1. The resulting plasmid,
pcDNA3.1-del p53, was transfected into the MCF-7/wt cells, and this
was followed by selection of stable clones containing the del p53
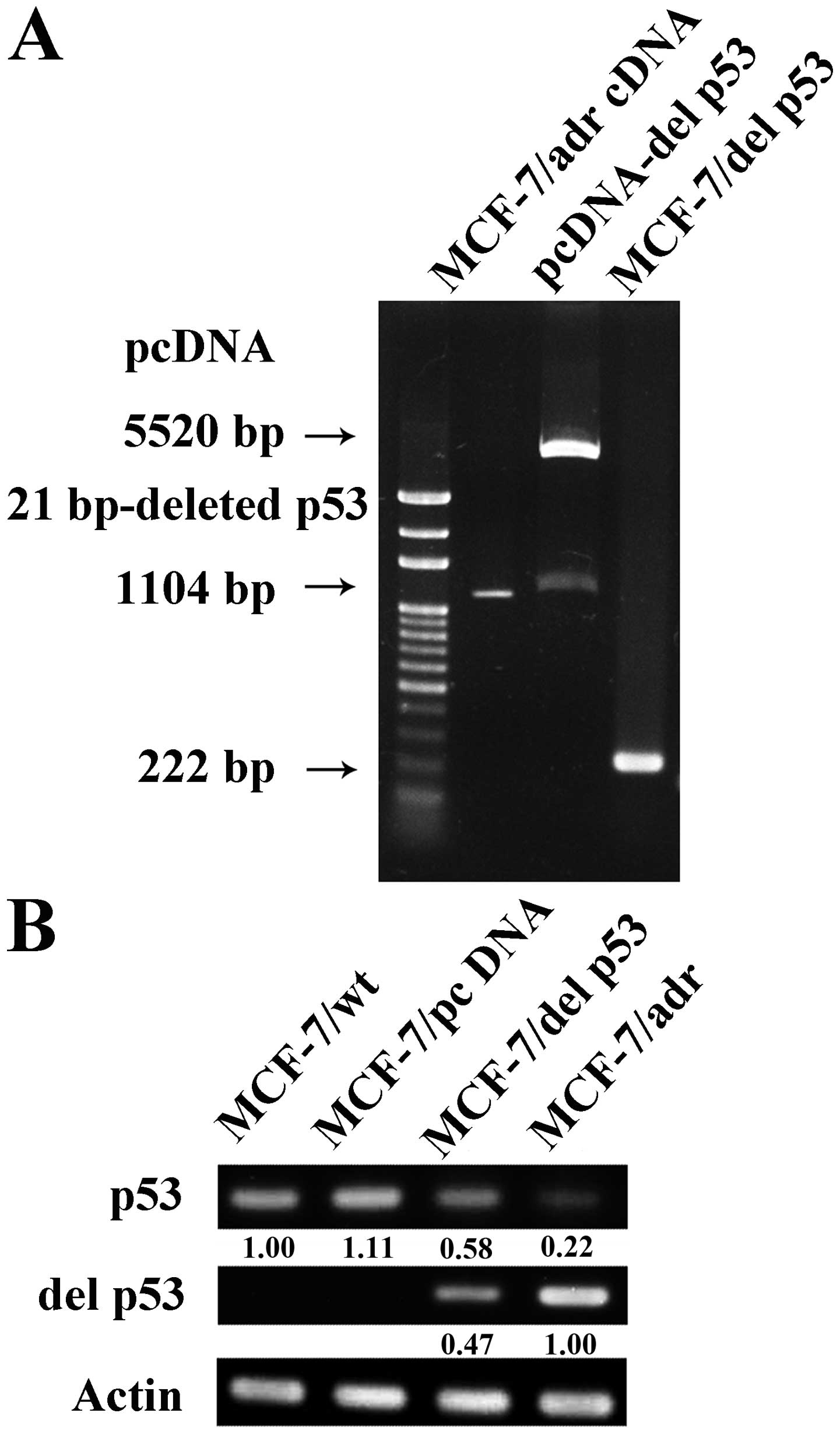
gene which was confirmed by a 222-bp RT-PCR product (Fig. 1A). The MCF-7/del p53 cell line is
one of the stable clones with similar characteristics. DNA
sequencing was performed to confirm a 21-bp deletion of p53 in the
MCF-7/adr and MCF-7/del p53 cells. We then determined the mRNA
expression levels of p53 in the various cell lines by RT-PCR.
MCF-7/wt did not express del p53 mRNA, whereas the MCF-7/del p53
cells stably expressed both wild-type p53 and del p53 mRNA
(Fig. 1B). The expression level
of the del p53 gene in the MCF-7/del p53 cells was approximately
47% lower than the level in the MCF-7/adr cells.
MCF-7/del p53 cells acquire resistance to
doxorubucin
In our previous study, the mutant p53 gene with a
21-bp deletion was detected in doxorubicin-resistant MCF-7 cell
lines following various induction processes (32). In this study, we wished to examine
the role of del p53 in the acquired resistance of MCF-7 cells to
doxorubicin. The degree of resistance to doxorubicin was assessed
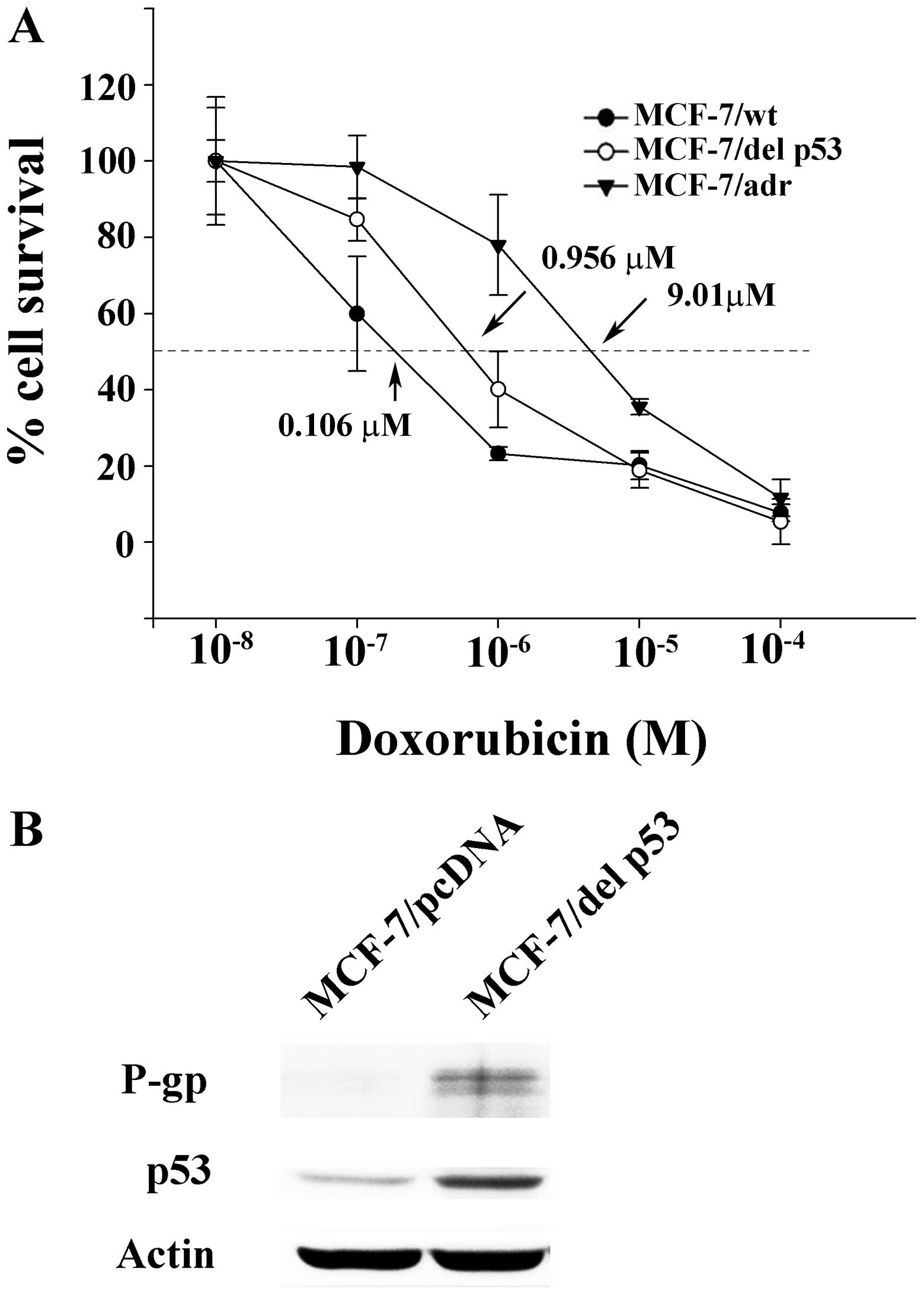
by MTT assay in the MCF-7/wt, MCF-7/del p53 and MCF-7/adr cells.
The concentrations of doxorubicin that inhibited cell survival by
50% (IC50) were extrapolated from cell survival plots.
The IC50 values in the MCF-7/wt, MCF-7/del p53 and
MCF-7/adr cells were 0.106, 0.956 and 8.179 µM, respectively
(Fig. 2A). The resistance index
is defined as the ratio of the IC50 value of MCF-7/del
p53 or MCF-7/adr to the IC50 value of MCF-7/wt.
Accordingly, the resistance indexes of the MCF-7/adr and MCF-7/del
p53 cells are 77.16 and 9.01, respectively, compared with the
MCF-7/wt cells. The acquisition of resistance to doxorubicin may be
related to the transfection of del p53 into the MCF-7 cells. The
gain-of-resistance activity may be associated with the capability
of del p53 to stimulate the expression of an alternate set of
endogenous genes that potentially promotes tumor progression and
induces drug resistance. The expression of the MDR1 gene was also
examined to examine the hypothesis that MDR1 is the endogenous
target of del p53 and that the activation of MDR1 is mediated by
del p53. Western blot analysis revealed that the p53 protein was
detected by the antibody against both wild-type p53 and del p53 in
the MCF-7/del p53 cells, whereas only low levels of p53 protein
were detected in the MCF-7/pcDNA control. It is probable that the
del p53 protein, with a longer half-life compared with wild-type
p53, was stably expressed in the MCF-7/del p53 cells (Fig. 2B). The MDR marker, P-gp, was also
mildly expressed in the MCF-7/del p53 cells. The low level of P-gp
was consistent with the degree of the increase in the resistance
index of MCF-7/del p53 cells.
del p53-mediated P-gp function is
associated with MDR1 promoter activation and NF-κB expression
To examine the positive role of del p53 on the MDR1
promoter in MCF-7 cells, we inserted the MDR1 promoter (residues
−198 to +43, 241 bp) into a luciferase-expressing pGL3-basic vector
upstream of the luciferase gene to generate the pGL3-MDR vector.
The cells were transiently transfected with pGL3-MDR. The activity
of the MDR1 promoter was measured as a function of luciferase.
Luciferase activity in the MCF-7/del p53 and MCF-7/adr cells was
significantly higher than that in the MCF-7/wt cells (Fig. 3A). To rule out the interaction of
other endogenous factors with possible regulatory sites on the MDR1
promoter region in pGL3-MDR, the MCF-7/wt cells were co-transfected
with the pGL3-MDR and pcDNA3.1-del p53 vector. A high luciferase
activity was also observed in the co-transfected cells. It is
probable that the del p53 protein contributed to the activation of
the MDR1 promoter and its downstream genes. We then assessed
intracellular rhodamine 123 accumulation, to determine whether the
P-gp efflux function was enhanced by del p53 protein. The results
revealed a moderate decrease in rhodamine 123 accumulation in the
MCF-7/del p53 cells compared with the MCF-7/wt cells (Fig. 3B). By adding the P-gp inhibitor,
verapamil, the intracellular rhodamine 123 level was restored in
the MCF-7/del p53 cells. As the degree of MDR1 promoter activation
is not projected to the P-gp efflux function in MCF-7/del p53, we
hypothesized that additional factors may be required for P-gp
expression and function. The ubiquitous transcription factor,
NF-κB, controls the expression of numerous genes in apoptotic
pathways and induces drug resistance in cancer cells (21). Thus, to determine whether del p53
protein expression is in accordance with the increase in NF-κB, the
NF-κB protein levels were measured by western blot analysis. The
results revealed that the NF-κB and P-gp levels were moderately
increased in the MCF-7/del p53 cells in comparison with the parent
MCF-7/wt cells (Fig. 3C). This
increase in expression was suppressed by treatment with the NF-κB
inhibitor, CsA. Cell survival assays demonstrated that the addition
of CsA sensitized the MCF-7/del p53 and MCF-7/adr cells to
doxorubicin toxicity (Fig. 3D).
Thus, P-gp expression may involve the upregulation of NF-κB in
MCF-7/del p53 cells.
EMT is induced in MCF-7/del p53
cells
We examined cellular changes, including morphology
and EMT markers, that may be mediated by del p53 in MCF-7/del p53
cells. The MCF-7/wt cells were arranged in a tightly-packed layer,
which is characteristic of epithelial cells and exhibited limited
cell spreading. Unlike the MCF-7/wt cells, the MCF-7/del p53 cells
had a flattened morphology and had lost cell-cell contacts, which
was similar to the MCF-7/adr cells (Fig. 4A). Changes in the morphological
characteristics of epithelial cells may lead to the cells losing
their epithelial characteristics and can increase their metastatic
and invasive potential (41). The
induction of EMT leads to the acquisition of mesenchymal traits
(42). In this study, the
expression of a series of EMT-related transcription factors was
determined by RT-PCR. The expression of mesenchymal markers was
upregulated, including that of Slug and vimentin in the MCF-7/del
p53 cells, and the expression of the epithelial marker, CDH1, was
downregulated (Fig. 4B). However,
the mesenchymal marker, CDH2, was not observed in the MCF-7/del p53
cells. The overexpression of EMT-inducing transcription factors has
been associated with chemoresistance and the depletion of these
factors has been shown to increase drug sensitivity (23). Thus, we wished to determine
whether the upregulation of the transcription factors, Slug and
vimentin, enhances the invasive ability of the MCF-7/del p53 cells.
The results of wound healing assay demonstrated that the migration
ability of the MCF-7/del p53 cells increased by 2.5-fold, compared
with the MCF-7/wt cells (Fig. 4C and
D). The longer migration distances may be due to the activation
of the mesenchymal markers in the MCF-7/del p53 cells.
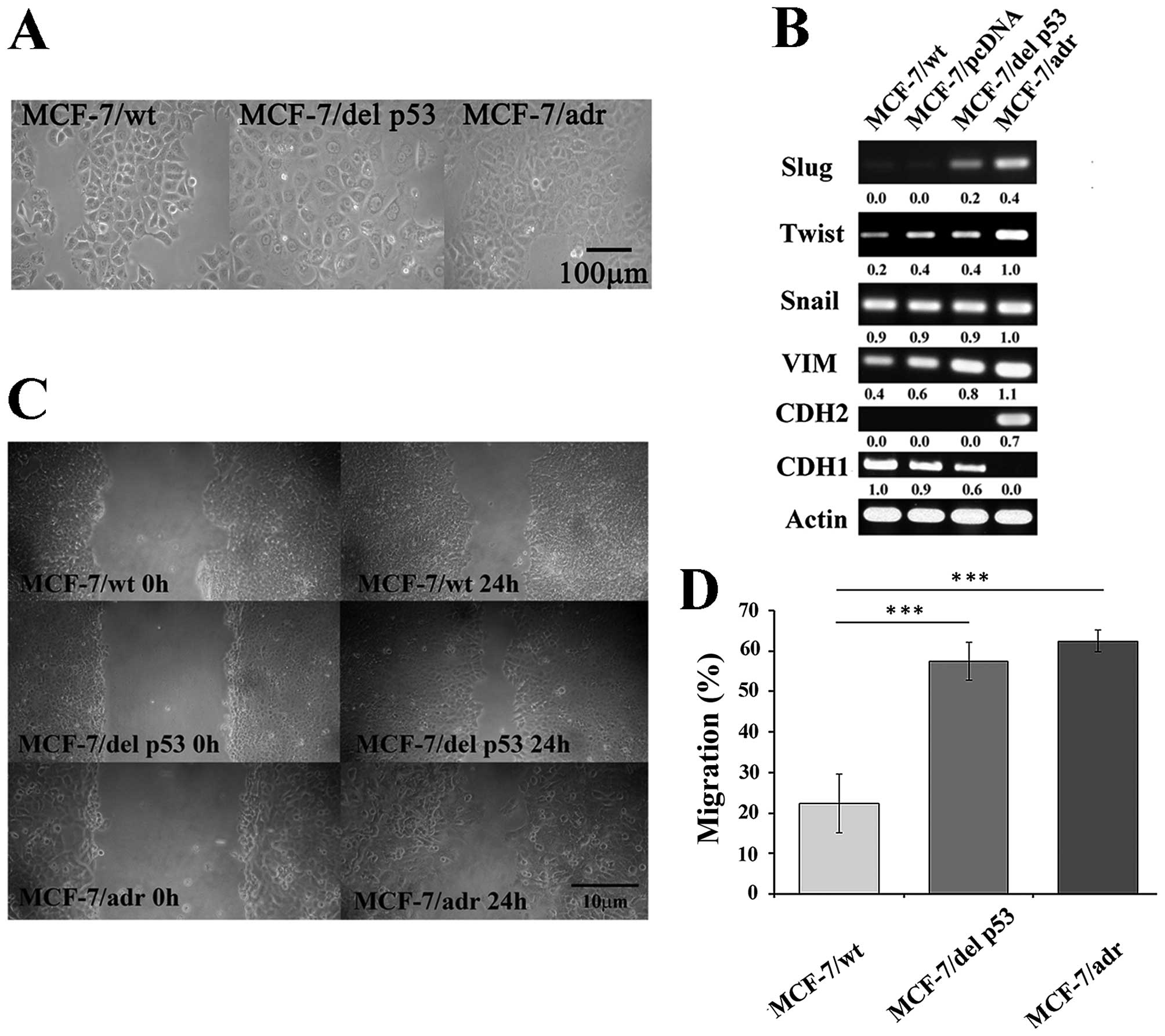 | Figure 4del p53 is involved in the EMT
process. (A) Phase-contrast microscopic images of MCF-7/wt,
MCF-7/del p53 and MCF-7/adr cells (x200 magnification), scale bar,
100 µm. (B) Expression of EMT-related transcription factors
Slug, Twist, Snail, VIM, CDH1 and CDH2. RT-PCR products were run on
a 1% agarose gel to show the expression levels of transcription
factors. (C) Photomicrographs show cell migration by wound healing
assay. (D) Graph represents relative cell migration distances
measured at 0 and 24 h in MCF-7/wt, MCF-7/del p53 and MCF-7/adr
cells, using Image J software, n=3. Error bars denote ± SEM.
***P<0.001. |
MCF-7/del p53 cells exhibit CSC-like
properties
Alterations in CD44/CD24 configuration are
associated with human breast CSCs and normal mammary epithelial
stem cells. When CD44 is highly expressed in breast cancer cells,
it generates a microenvironment to facilitate tumor progression and
invasion (30). Thus, we detected
the CD44/CD24 subpopulations in MCF-7/del p53 cells by flow
cytometry. As shown by our results, the MCF-7/wt cells exhibited a
CD44low subpopulation, whereas the MCF-7/adr cells
exhibited a CD44high/CD24high subpopulation.
The MCF-7/del p53 cells were thus in a transitional phase,
exhibiting an increase in the
CD44high/CD24high subpopulation (Fig. 5A). The number of colonies formed
in the MCF-7/del p53 cells was 5.7-fold higher than that in the
MCF-7/wt cells (Fig 5B and
C).
Discussion
It is well known that p53, which is involved in DNA
repair, plays an important role in the maintenance of genome
integrity in response to a variety of anticancer drugs (43). Mutations in p53 are linked with
the defects in growth arrest, apoptosis after DNA damage and
sensitivity to anticancer agents (44). Mutant p53 not only disrupts
sequence-specific transactivation, but also confers
gain-of-function effects, such as the overexpression of the drug
resistance gene, MDR1, in tumors (12,45). Wild-type p53 has an inhibitory
effect on the MDR1 gene promoter, whereas mutant p53 disrupts the
DNA binding domain and acts as an activator (12). Transcription factors, such as
NF-κB, SP1, NF-Y, C/EBP-β and activator protein (AP)-1, bind to the
MDR1 promoter to regulate MDR1 gene expression in a complex manner,
since transcription factors may act through competitive or
cooperative interactions with the MDR1 promoter (46). Mutant p53 directly binds to the
MDR1 promoter to transactivate MDR1 expression or indirectly
interact with other transcription factors, such the proto-oncogenic
factor ETS-1, to modulate MDR1 expression. The results from the
present study demonstrated a high transient expression of the MDR1
promoter in MCF-7/del p53 or MCF-7/wt cells co-transfected with the
MDR1 promoter and del p53, indicating that MDR1 promoter activity
is exclusively mediated by either internal or external del p53.
Other possible factors acting on the regulatory sites in our
pGL3-MDR construct were ruled out. Increased MDR1 expression is
mediated by mutated p53 in a variety of cancer cell lines. In
Caco-2 cells, the MDR1 promoter was activated by p53 mutants at
R175H and D281G, whereas wild-type p53 had either no effect or an
inhibitory effect on the promoter (12). In Saos-2 cells, the MDR1 promoter
was activated by p53 mutants at R175H and R248Q (47). In Hep3B cells, p53 mutants at
R248Q and R273C activated the MDR1 promoter (48). Mutant p53 with a 21-bp deletion in
exon 5 has been found in MCF-7/adr cells, as well as in U1285 and
OVCAR-8 cells (33,35,36). High levels of P-gp have been found
in MCF-7/adr cells, whereas low levels of P-gp have been detected
in U1285 and OVCAR-8 cells (36,49). MCF-7/del p53 cells also weakly
expressed P-gp in our study. This implies that, in addition to del
p53, other factors may be recruited to accomplish P-gp expression
and induce MDR. The transfection of NF-κB has been reported to
induce P-gp expression (50). In
a previous study, NF-κB-induced drug resistance was suppressed by
the NF-κB inhibitor, CsA, through decreased P-gp expression
(21). Our results revealed a
higher level of NF-κB expression in MCF-7/del p53 cells. This
expression was suppressed by CsA, and accompanied by a diminished
P-gp expression. In this way, the MCF-7/del p53 cells were
significantly sensitized to doxorubicin toxicity. We therefore
hypothesized that the activation of the MDR1 promoter by del p53
may be partly related to the upregulation of NF-κB. It is possible
that del p53 is an important upstream factor for NF-κB activation
which aids the aggressive growth and drug resistance of cancer
cells. However, the fact that CsA exerted less of an inhibitory
effect on cell survival in MCF-7/adr cells, compared with that in
MCF-7/del p53 cells, illustrates that complex factors are involved
in the acquisition of drug resistance.
EMT is considered an essential process in the
metastatic cascade. The loss of epithelial markers, including CDH1,
ZO-1, occludin, as well as a corresponding increase in mesenchymal
markers, such as vimentin, Slug, SMA, fibronectin and CDH2, are
critical events signaling the loss of the epithelial phenotype and
the commencement of mesenchymalization (24). Usually, the EMT process is
mediated by the inhibition of CDH1 expression, leading to the
induction of CDH2 expression, and this has been associated with
tumor invasiveness and CSC-like properties. EMT transcription
factors, such as Twist, Snail and Slug, play a regulatory role
repressing CDH1 gene expression. The expression of Slug is
associated with MDR (22,51). Emerging evidence has suggested
that mutant p53 induces drug resistance and mediates invasiveness
through the positive regulation of Slug, an invasion-promoting
factor in EMT (26). The aberrant
expression of Slug also contributes to the invasive behavior of
glioma and melanoma cells (25,52). Yet, little is known about the
molecular mechanisms of mutant p53 linking the two phenomena. The
present study found that the transcription factors, Slug and
vimentin, were upregulated and CDH1 was downregulated in the
MCF-7/del p53 cells. In addition, gain of oncogenic function by p53
mutants has been shown to regulate EMT-related gene expression in
colon and endometrial cancers (29,53). The expression of Slug has also
been shown to contribute to cisplatin resistance in ovarian cancer
and to activate the transforming growth factor-β signaling pathway
in MCF-7 cells (54,55). However, in this study, the
ultimate mesenchymal marker, CDH2, which is a key factor for the
commencement of the EMT process was not detected in the MCF-7/del
p53 cells. Moreover, unlike the MCF-7/adr cells with no CDH1
expression, the MCF-7/del p53 cells still presented with low levels
of CDH1. This indicates that the 21-bp-deleted p53 may participate
in the initiation of EMT, although the subsequent recruitment of
additional factors is necessary for the upregulation of CDH2
expression. The identity of the additional factors remains unknown.
As previousy demonstrated, the mouse double minute 2 homolog
(MDM2)-mediated degradation of Slug may be inhibited by mutant p53
to result in cancer cell invasion (26). However, further investigations are
warranted.
The induction of an EMT in immortalized human
mammary epithelial cells result in not only the acquisition of
mesenchymal traits, but also increases the expression of stem-cell
markers. Slug is involved in MDR which is mediated by stem cell
factor (SCF)/c-Kit in malignant mesothelioma cells. Slug gene
expression is also part of a hypoxia-induced genetic program which
sets up a basal/stem cell-like, aggressive phenotype in breast
cancer cells (27,56). CD44 is a cell surface marker that
is expressed in the progression of many tumors, as well as in CSCs.
The overexpression of EMT-related genes, such as Slug and Gli-2,
can transform MCF-7 cells from a
CD44low/CD24high phenotype to the stem
cell-like properties of a CD44high/CD24low
phenotype (30,57). Our data demonstrated a
transitional manner of CD44/CD24 configuration in MCF-7/del p53
cells compared with the
CD44high/CD24high-enriched MCF-7/adr cells
and the CD44low-enriched MCF-7/wt cells. This indicated
that the cell subpopulations of MCF-7/del p53 shifted to CSC-like
cells.
The cell migration ability of the MCF-7/del p53
cells was significantly greater than that of the MCF-7/wt cells,
indicating that 21-bp-deleted p53 may enable the cells to acquire
CSC-like properties. Breast cancers are classified into luminal,
basal, mesenchymal, ErbB2-positive and myoepithelial, according to
molecular profiling studies. Cell lines that contain
CD44high/CD24low populations are in the
basal/mesenchymal or the myoepithelial group (58–60). In this regard, the co-existence of
aberrant Slug and CD44high markers suggests that
MCF-7/del p53 cells may emerge through an EMT process in which the
cells lose their epithelial characteristics and gain mesenchymal
properties, and then progress toward an MDR phenotype. Further
studies are required in order to provide the necessary
evidence.
In conclusion, 21-bp-deleted p53 found in a variety
of cancer cells is a gain-of-function mutation which partly
participates in the acquisition of chemoresistance in MCF-7 cells.
The underlying mechanisms involve the transactivation of the MDR1
promoter through NF-κB upregulation, the increased expression of
the mesenchymal markers, Slug and vimentin, as well as the stem
cell-like marker, CD44, in MCF-7 cells stably expressing
21-bp-deleted p53. Thus, the 21-bp deletion of p53 has potential
for use as a therapeutic target with which to inhibit
gain-of-resistance in cancers.
Acknowledgments
This study was supported by a grant from the Taiwan
Cancer Foundation (no. TCF101-TM02-B4).
Abbreviations:
|
ABC transporters
|
ATP binding cassette transporters
|
|
CSC
|
cancer stem cell
|
|
EMT
|
epithelial-mesenchymal transition
|
|
MDR
|
multidrug resistance
|
|
P-gp/ABCB1
|
P-glycoprotein
|
References
|
1
|
Muller PA and Vousden KH: p53 mutations in
cancer. Nat Cell Biol. 15:2–8. 2013. View
Article : Google Scholar
|
|
2
|
Petitjean A, Achatz MI, Borresen-Dale AL,
Hainaut P and Olivier M: TP53 mutations in human cancers:
functional selection and impact on cancer prognosis and outcomes.
Oncogene. 26:2157–2165. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Keshelava N, Zuo JJ, Waidyaratne NS,
Triche TJ and Reynolds CP: p53 mutations and loss of p53 function
confer multidrug resistance in neuroblastoma. Med Pediatr Oncol.
35:563–568. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sturm I, Bosanquet AG, Hermann S, Güner D,
Dörken B and Daniel PT: Mutation of p53 and consecutive selective
drug resistance in B-CLL occurs as a consequence of prior
DNA-damaging chemotherapy. Cell Death Differ. 10:477–484. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pritchard JR, Lauffenburger DA and Hemann
MT: Understanding resistance to combination chemotherapy. Drug
Resist Updat. 15:249–257. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Glavinas H, Krajcsi P, Cserepes J and
Sarkadi B: The role of ABC transporters in drug resistance,
metabolism and toxicity. Curr Drug Deliv. 1:27–42. 2004. View Article : Google Scholar
|
|
7
|
Warmann S, Hunger M, Teichmann B, Flemming
P, Gratz KF and Fuchs J: The role of the MDR1 gene in the
development of multidrug resistance in human hepatoblastoma:
clinical course and in vivo model. Cancer. 95:1795–1801. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bargou RC, Jürchott K, Wagener C, Bergmann
S, Metzner S, Bommert K, Mapara MY, Winzer KJ, Dietel M, Dörken B
and Royer HD: Nuclear localization and increased levels of
transcription factor YB-1 in primary human breast cancers are
associated with intrinsic MDR1 gene expression. Nat Med. 3:447–450.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chin KV, Ueda K, Pastan I and Gottesman
MM: Modulation of activity of the promoter of the human MDR1 gene
by Ras and p53. Science. 255:459–462. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Okamura H, Yoshida K, Sasaki E, Morimoto H
and Haneji T: Transcription factor NF-Y regulates mdr1 expression
through binding to inverted CCAAT sequence in drug-resistant human
squamous carcinoma cells. Int J Oncol. 25:1031–1037.
2004.PubMed/NCBI
|
|
11
|
Rohlff C and Glazer RI: Regulation of the
MDR1 promoter by cyclic AMP-dependent protein kinase and
transcription factor Sp1. Int J Oncol. 12:383–386. 1998.PubMed/NCBI
|
|
12
|
Sampath J, Sun D, Kidd VJ, Grenet J,
Gandhi A, Shapiro LH, Wang Q, Zambetti GP and Schuetz JD: Mutant
p53 cooperates with ETS and selectively up-regulates human MDR1 not
MRP1. J Biol Chem. 276:39359–39367. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Arlt A and Schäfer H: NFkappaB-dependent
chemoresistance in solid tumors. Int J Clin Pharmacol Ther.
40:336–347. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Deb D, Scian M, Roth KE, Li W, Keiger J,
Chakraborti AS, Deb SP and Deb S: Hetero-oligomerization does not
compromise 'gain of function' of tumor-derived p53 mutants.
Oncogene. 21:176–189. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kanagasabai R, Krishnamurthy K, Druhan LJ
and Ilangovan G: Forced expression of heat shock protein 27 (Hsp27)
reverses P-glycoprotein (ABCB1)-mediated drug efflux and MDR1 gene
expression in Adriamycin-resistant human breast cancer cells. J
Biol Chem. 286:33289–33300. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cooks T, Pateras IS, Tarcic O, Solomon H,
Schetter AJ, Wilder S, Lozano G, Pikarsky E, Forshew T, Rosenfeld
N, et al: Mutant p53 prolongs NF-κB activation and promotes chronic
inflammation and inflammation-associated colorectal cancer. Cancer
Cell. 23:634–646. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ferris RL and Grandis JR: NF-kappaB gene
signatures and p53 mutations in head and neck squamous cell
carcinoma. Clin Cancer Res. 13:5663–5664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li Y, Ahmed F, Ali S, Philip PA, Kucuk O
and Sarkar FH: Inactivation of nuclear factor kappaB by soy
isoflavone genistein contributes to increased apoptosis induced by
chemotherapeutic agents in human cancer cells. Cancer Res.
65:6934–6942. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Braeuer SJ, Büneker C, Mohr A and Zwacka
RM: Constitutively activated nuclear factor-kappaB, but not induced
NF-kappaB, leads to TRAIL resistance by up-regulation of X-linked
inhibitor of apoptosis protein in human cancer cells. Mol Cancer
Res. 4:715–728. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Godwin P, Baird AM, Heavey S, Barr MP,
O'Byrne KJ and Gately K: Targeting nuclear factor-kappa B to
overcome resistance to chemotherapy. Front Oncol. 3:1202013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Bentires-Alj M, Barbu V, Fillet M, Chariot
A, Relic B, Jacobs N, Gielen J, Merville MP and Bours V: NF-kappaB
transcription factor induces drug resistance through MDR1
expression in cancer cells. Oncogene. 22:90–97. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Iseri OD, Kars MD, Arpaci F, Atalay C, Pak
I and Gunduz U: Drug resistant MCF-7 cells exhibit
epithelial-mesenchymal transition gene expression pattern. Biomed
Pharmacother. 65:40–45. 2011. View Article : Google Scholar
|
|
23
|
Saxena M, Stephens MA, Pathak H and
Rangarajan A: Transcription factors that mediate
epithelial-mesenchymal transition lead to multidrug resistance by
upregulating ABC transporters. Cell Death Dis. 2:e1792011.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Voulgari A and Pintzas A:
Epithelial-mesenchymal transition in cancer metastasis: mechanisms,
markers and strategies to overcome drug resistance in the clinic.
Biochim Biophys Acta. 1796:75–90. 2009.PubMed/NCBI
|
|
25
|
Yang HW, Menon LG, Black PM, Carroll RS
and Johnson MD: SNAI2/Slug promotes growth and invasion in human
gliomas. BMC Cancer. 10:3012010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wang SP, Wang WL, Chang YL, Wu CT, Chao
YC, Kao SH, Yuan A, Lin CW, Yang SC, Chan WK, et al: p53 controls
cancer cell invasion by inducing the MDM2-mediated degradation of
Slug. Nat Cell Biol. 11:694–704. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Storci G, Sansone P, Trere D, Tavolari S,
Taffurelli M, Ceccarelli C, Guarnieri T, Paterini P, Pariali M,
Montanaro L, et al: The basal-like breast carcinoma phenotype is
regulated by SLUG gene expression. J Pathol. 214:25–37. 2008.
View Article : Google Scholar
|
|
28
|
Lowe SW, Bodis S, McClatchey A, Remington
L, Ruley HE, Fisher DE, Housman DE and Jacks T: p53 status and the
efficacy of cancer therapy in vivo. Science. 266:807–810. 1994.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dong P, Karaayvaz M, Jia N, Kaneuchi M,
Hamada J, Watari H, Sudo S, Ju J and Sakuragi N: Mutant p53
gain-of-function induces epithelial-mesenchymal transition through
modulation of the miR-130b-ZEB1 axis. Oncogene. 32:3286–3295. 2013.
View Article : Google Scholar :
|
|
30
|
Sheridan C, Kishimoto H, Fuchs RK,
Mehrotra S, Bhat-Nakshatri P, Turner CH, Goulet R Jr, Badve S and
Nakshatri H: CD44+/CD24− breast cancer cells
exhibit enhanced invasive properties: an early step necessary for
metastasis. Breast Cancer Res. 8:R592006. View Article : Google Scholar
|
|
31
|
van Oijen MG and Slootweg PJ:
Gain-of-function mutations in the tumor suppressor gene p53. Clin
Cancer Res. 6:2138–2145. 2000.PubMed/NCBI
|
|
32
|
Tsou SH, Chen TM, Hsiao HT and Chen YH: A
critical dose of doxorubicin is required to alter the gene
expression profiles in MCF-7 cells acquiring multidrug resistance.
PLoS One. 10:e01167472015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ogretmen B and Safa AR: Expression of the
mutated p53 tumor suppressor protein and its molecular and
biochemical characterization in multidrug resistant MCF-7/Adr human
breast cancer cells. Oncogene. 14:499–506. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yu ST, Chen TM, Tseng SY and Chen YH:
Tryptanthrin inhibits MDR1 and reverses doxorubicin resistance in
breast cancer cells. Biochem Biophys Res Commun. 358:79–84. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Berglind H, Pawitan Y, Kato S, Ishioka C
and Soussi T: Analysis of p53 mutation status in human cancer cell
lines: a paradigm for cell line cross-contamination. Cancer Biol
Ther. 7:699–708. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Nygren P, Larsson R, Gruber A, Peterson C
and Bergh J: Doxorubicin selected multidrug-resistant small cell
lung cancer cell lines characterised by elevated cytoplasmic
Ca2+ and resistance modulation by verapamil in absence
of P-glycoprotein overexpression. Br J Cancer. 64:1011–1018. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Norberg T, Klaar S, Lindqvist L, Lindahl
T, Ahlgren J and Bergh J: Enzymatic mutation detection method
evaluated for detection of p53 mutations in cDNA from breast
cancers. Clin Chem. 47:821–828. 2001.PubMed/NCBI
|
|
38
|
Takahashi RU, Takeshita F, Honma K, Ono M,
Kato K and Ochiya T: Ribophorin II regulates breast tumor
initiation and metastasis through the functional suppression of
GSK3β. Sci Rep. 3:24742013. View Article : Google Scholar
|
|
39
|
Li W, Liu C, Tang Y, Li H, Zhou F and Lv
S: Overexpression of Snail accelerates adriamycin induction of
multidrug resistance in breast cancer cells. Asian Pac J Cancer
Prev. 12:2575–2580. 2011.
|
|
40
|
Ogretmen B and Safa AR: Negative
regulation of MDR1 promoter activity in MCF-7, but not in multidrug
resistant MCF-7/Adr, cells by cross-coupled NF-kappa B/p65 and
c-Fos transcription factors and their interaction with the CAAT
region. Biochemistry. 38:2189–2199. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Tsai JH and Yang J: Epithelial-mesenchymal
plasticity in carcinoma metastasis. Genes Dev. 27:2192–2206. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Nurwidya F, Takahashi F, Murakami A and
Takahashi K: Epithelial mesenchymal transition in drug resistance
and metastasis of lung cancer. Cancer Res Treat. 44:151–156. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Menon V and Povirk L: Involvement of p53
in the repair of DNA double strand breaks: multifaceted Roles of
p53 in homologous recombination repair (HRR) and non-homologous end
joining (NHEJ). Subcell Biochem. 85:321–336. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Wang Z and Sun Y: Targeting p53 for novel
anticancer therapy. Transl Oncol. 3:1–12. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Oka M, Kounoura K, Narasaki F, Sakamoto A,
Fukuda M, Matsuo I, Ikeda K, Tsurutani J, Ikuno N, Omagari K, et
al: P-glycoprotein is positively correlated with p53 protein
accumulation in human colorectal cancers. Jpn J Cancer Res.
88:738–742. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Labialle S, Gayet L, Marthinet E, Rigal D
and Baggetto LG: Transcriptional regulators of the human multidrug
resistance 1 gene: recent views. Biochem Pharmacol. 64:943–948.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang LH, Okaichi K, Ihara M and Okumura Y:
Sensitivity of anticancer drugs in Saos-2 cells transfected with
mutant p53 varied with mutation point. Anticancer Res. 18A:321–325.
1998.
|
|
48
|
Chan KT and Lung ML: Mutant p53 expression
enhances drug resistance in a hepatocellular carcinoma cell line.
Cancer Chemother Pharmacol. 53:519–526. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Sosa AJ, Chavez P and Dyke KV: Inibitory
effect of tetrandrine and paxlitaxel or doxorubicin on
multi-drug-resistant (MDR) cancer cells associated with MDR-ATPase.
Int J Pharmacother. 4:102014.
|
|
50
|
Kim HG, Hien TT, Han EH, Hwang YP, Choi
JH, Kang KW, Kwon KI, Kim BH, Kim SK, Song GY, et al: Metformin
inhibits P-glycoprotein expression via the NF-κB pathway and CRE
transcriptional activity through AMPK activation. Br J Pharmacol.
162:1096–1108. 2011. View Article : Google Scholar :
|
|
51
|
Kajita M, McClinic KN and Wade PA:
Aberrant expression of the transcription factors snail and slug
alters the response to genotoxic stress. Mol Cell Biol.
24:7559–7566. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Fenouille N, Tichet M, Dufies M, Pottier
A, Mogha A, Soo JK, Rocchi S, Mallavialle A, Galibert MD, Khammari
A, et al: The epithelial-mesenchymal transition (EMT) regulatory
factor SLUG (SNAI2) is a downstream target of SPARC and AKT in
promoting melanoma cell invasion. PLoS One. 7:e403782012.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Roger L, Jullien L, Gire V and Roux P:
Gain of oncogenic function of p53 mutants regulates E-cadherin
expression uncoupled from cell invasion in colon cancer cells. J
Cell Sci. 123:1295–1305. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Haslehurst AM, Koti M, Dharsee M, Nuin P,
Evans K, Geraci J, Childs T, Chen J, Li J, Weberpals J, et al: EMT
transcription factors snail and slug directly contribute to
cisplatin resistance in ovarian cancer. BMC Cancer. 12:912012.
View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Dhasarathy A, Phadke D, Mav D, Shah RR and
Wade PA: The transcription factors Snail and Slug activate the
transforming growth factor-beta signaling pathway in breast cancer.
PLoS One. 6:e265142011. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Catalano A, Rodilossi S, Rippo MR, Caprari
P and Procopio A: Induction of stem cell factor/c-Kit/slug signal
transduction in multidrug-resistant malignant mesothelioma cells. J
Biol Chem. 279:46706–46714. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Bhat-Nakshatri P, Appaiah H, Ballas C,
Pick-Franke P, Goulet R Jr, Badve S, Srour EF and Nakshatri H:
SLUG/SNAI2 and tumor necrosis factor generate breast cells with
CD44+/CD24− phenotype. BMC Cancer.
10:4112010. View Article : Google Scholar
|
|
58
|
Sørlie T, Perou CM, Tibshirani R, Aas T,
Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey
SS, et al: Gene expression patterns of breast carcinomas
distinguish tumor subclasses with clinical implications. Proc Natl
Acad Sci USA. 98:10869–10874. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Perou CM, Sørlie T, Eisen MB, van de Rijn
M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA,
et al: Molecular portraits of human breast tumours. Nature.
406:747–752. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Gordon LA, Mulligan KT, Maxwell-Jones H,
Adams M, Walker RA and Jones JL: Breast cell invasive potential
relates to the myoepithelial phenotype. Int J Cancer. 106:8–16.
2003. View Article : Google Scholar : PubMed/NCBI
|