Introduction
Severe acute pancreatitis (SAP) is an extremely
dangerous acute abdominal disorder which causes multiple
complications, and has a high mortality rate (1,2).
Despite significant improvements to the methods of diagnosis and
management of SAP, the mortality rate has not declined
significantly in the past few decades (3). Therefore, the treatment of SAP
remains problematic. It is thus critical to investigate the
molecular mechanisms responsible for the development of this
complex disease, particularly those that activate the innate immune
response (4). Some previous
studies have suggested that the severity and outcome of
pancreatitis may be determined by events that occur after acinar
cell injury (4,5).
The high-motility group box protein 1 (HMGB1), with
a low molecular weight (approximately 30 kDa), was originally
identified as a non-histone DNA-binding nuclear protein which is
involved in both intracellular and extracellular activities
(6–10). Extracellular HMGB1 with
cytokine-like properties (7,10)
is a proximal trigger that induces the release of other cytokines,
including tumor necrosis factor-α (TNF-α), interleukin (IL)-1β and
IL-6, which are classically associated with mediating the
inflammatory response (11,12). We, as well as others have
previously indicated that HMGB1 is elevated in pancreatic tissue
during acute pancreatitis, and this elevation is closely linked
with the severity of the disease (1,3,13,14). These results suggest that HMGB1
plays a pivotal role in the pathogenesis of SAP. However, the
mechanisms underlying this strong correlation remain unclear.
Toll-like receptor (TLR)4 is one of the least common
11 mammalian pattern-recognition receptors that comprise the innate
immune response. It is activated by the prototypical
pathogen-associated molecular pattern (PAMP) and damage-associated
molecular pattern (DAMP) proteins, such as heat shock protein 70
(HSP70) and HMGB1 (15,16). Extracellular PAMPs or DAMPs that
bind to TLR4 cause the myeloid differentiation primary response
gene 88 (MyD88) to activate nuclear factor-κ-B (NF-κB). Activated
NF-κB is transported to the nucleus from the cytoplasm, where it
induces the expression of inflammatory factors, including TNF-α,
IL-1β and IL-6 (17–19). Extracellular HMGB1 functions as a
damage-associated molecular pattern molecule, and it activates
pro-inflammatory signaling pathways by activating pattern
recognition receptors, including TLR2/4 and the receptor for
advanced glycation end-products (RAGE) (20,21). Previous studies have shown that
TLR4 plays an important role in the pathogenesis of HMGB1-mediated
acute lung injury (6,22). Moreover, TLR4 is widely
distributed in the tissue and vascular endothelial cells of the
pancreas, and has been reported to be associated with pancreatic
injury during acute pancreatitis (4,5,23).
The triggering of the TLR4 signaling pathway by HMGB1 activates
NF-κB, which subsequently induces the expression of inflammatory
factors (6,22). Excessive cytokine-mediated
inflammation plays a fundamental role in the pathogenesis of
SAP.
Based upon these findings, we hypothesized that
HMGB1 is involved in the pathogenesis of SAP, and that its
downstream TLR4-mediated NF-κB signaling pathway acts as an
important mediator in the development of pancreatic injury. In this
study, a murine model of acute pancreatitis, induced by the
intraperitoneal injection of L-arginine, was used. To examine the
role of TLR4 in the pathogenesis of HMGB1-induced pancreatic
injury, TLR4-deficient mice were used.
Materials and methods
Experimental animals
Male C57BL/6 mice (Shandong University Experimental
Animal Center, Jinan, China) and male C57BL/10ScNJ
TLR4−/− mice (Jackson Laboratories, Bar Harbor, ME, USA)
were used in this study. The mice were housed and bred in
micro-isolators under specific pathogen-free conditions, in a
climate-controlled enrivonment with an ambient temperature of 22°C
and a 12:12 h light/dark cycle. They were fed standard laboratory
chow, and drinking water was available ad libitum. All
experiments were performed using wild-type (WT) and deficient mice
that were 6–8 weeks old and weighed 20–30 g. All experimental
protocols were approved by the Ethics Review Board of Shandong
University. All animals received care in accordance with the
guidelines for animal care published by the United States National
Institutes of Health (NIH) for animal care (Guide for the Care and
Use of Laboratory Animals, Department of Health and Human Services,
NIH Publication no. 86–23, revised 1985).
Animal model and experimental groups
Induction of pancreatitis
The model of L-arginine-induced SAP was created as
previously described (24). Male
C57BL/6 mice were used and were randomly divided into the following
2 groups: i) the sham injection group (sham): animals in this group
received 2 sham intraperitoneal injections of sterile saline alone,
with a 1-h interval between injections; and ii) the SAP group:
animals in this group received 2 intraperitoneal injections of 8%
L-arginine, which were both at concentrations of 400 mg/100 g body
weight, with a 1-h interval between injections. Under
intraperitoneal anesthesia with 10% chloral hydrate, the mice were
sacrificed by exsanguination 48 h after the second injection
(n=6).
rhHMGB1 stimulation model
To create the model of rhHMGB1-induced pancreatic
injury, C57BL/6 (TLR4+/+) mice were randomly divided
into the following 3 groups (n=6/group) and were administered a
pancreas-targeted injection. The mice were divided into the
following 3 groups: i) the sham injection group (sham): animals in
this group received a pancreas-targeted ultrasound-guided injection
of 0.25 ml sterile saline alone; ii) the low-dose group (HM-LD):
animals in this group received a pancreas-targeted
ultrasound-guided injection of rhHMGB1 (SinoBio, Shanghai, China;
50 µg/kg body weight), diluted in 0.25 ml sterile saline;
and iii) the high-dose group (HM-HD): animals in this group
received a pancreas-targeted ultrasound-guided injection of rhHMGB1
(100 µg/kg body weight), diluted in 0.25 ml sterile saline.
Under intraperitoneal anesthesia with 10% chloral hydrate, the
animals were sacrificed by exsanguination 48 h after treatment.
TLR4-deficient model
The C57BL/6 (WT) and C57BL/10ScNJ (TLR4-deficient;
TLR4−/−) mice were independently and randomly divided
into the WT and TLR4-deficient groups (n=6/group): i) the WT group:
the C57BL/6 mice in this group received a pancreas-targeted
ultrasound-guided injection of rhHMGB1 (100 µg/kg body
weight), diluted in 0.25 ml sterile saline; ii) the TLR4-deficient
group (TLR4−/−): C57BL/10ScNJ mice in this group
received the same treatment as the mice in the WT group. Under
intraperitoneal anesthesia with 10% chloral hydrate, the animals
were sacrificed by exsanguination 48 h post-treatment.
Sample collection
The animals were anesthetized with an
intraperitoneal injection of pentobarbital sodium (100 mg/kg). The
serum and pancreas were stored at −80°C until further use. Tissues
for histopathological analysis were fixed in 10% buffered
formaldehyde solution overnight. The tissue was then embedded the
following day.
Histopathological analysis
For assessing the changes occurring in the
pancreatic tissue at the morphological level, 5-µm-thick
sections of pancreatic tissue were stained with hematoxylin and
eosin (H&E). The severity of SAP in the pancreatic tissue was
measured using the improved methods of Schmidt et al
(25) and Pozsar et al
(26) with i) edema: 0, null; 1,
interlobar space broadened gently; 2, interlobar space broadened
severely; 3, interacinous space broadened; 4, intercellular space
broadened; ii) necrosis: 0, null; 1, 1–10% necrotic area; 2, 11–20%
necrotic area; 3, 21–30% necrotic area; and 4, >30% necrotic
area; iii) hemorrhage: 0, negative; and 1, positive; and iv)
inflammatory cell infiltration: the number of leucocytes in the
lobule and around blood vessels was counted in a high-power field
with 0, 0–1; 1, 2–10; 2, 11–20; 3, 21–30; and 4, >30 or
micro-abscesses. Moreover, a total pancreatic injury score was
calculated as the sum of the 4 components. Five fields of each
section were counted, and the average score of these 5 fields was
the pathological injury score of this section.
Western blot analysis
The levels of pancreatic HMGB1, TLR4 and NF-κB p65
were measured by western blot analysis. The pancreas was harvested,
washed 3 times in sterile saline and then homogenized in RIPA
buffer (Beyotime Institute of Biotechnology, Suzhou, China)
containing a protease inhibitor cocktail (Thermo Scientific,
Rockford, IL, USA). Nucleus extraction from the pancreatic tissue
was prepared using the Pierce Nucleus and Cytoplasmic Extraction
Reagent kit (Thermo Scientific) containing a protease inhibitor
cocktail and phosphatase inhibitor cocktail (Roche, Basel, Germany)
according to the manufacturers' instructions. Following
centrifugatoin at 16,000 × g at 4°C for 30 min, the supernatant was
collected.
Protein was quantified using a BCA Protein Assay kit
(Beyotime Institute of Biotechnology). Samples of 40 µg were
run on 8% SDS-PAGE (TLR4) and 10% SDS-PAGE (HMGB1 and NF-κB p65)
gels. Proteins were then electrotransferred onto polyvinylidene
difluoride (PVDF) membranes (Beyotime Institute of Biotechnology).
The membranes were incubated in TBST containing 5% non-fat dried
milk for 1 h at 25°C. The blots were then incubated overnight at
4°C with primary antibodies to HMGB1 (ab79823), TLR4 (ab22048), p65
(ab16502; all from Abcam, Boston, MA, USA), β-actin (sc-47778;
ZSGB-BIO, Beijing, China) or histone H3 (ab76307; Abcam).
Subsequently, they were incubated with the appropriate horseradish
peroxidase (HRP)-conjugated anti-rabbit (ZB-2301) or anti-mouse
(ZB-2305) secondary antibodies (ZSGB-BIO) for 1 h at 25°C and
visualized with an enhanced chemiluminescence assay (Thermo
Scientific). The bands were quantified using MultiGauge version 3.2
software. Experiments were repeated independently 3 times, and the
relative expression of the target protein was normalized to the
level of β-actin or histone H3 in the same sample.
Enzyme-linked immunosorbent assay
(ELISA)
The levels of serum amylase and lipase were detected
by the laboratory of Shandong Provincial Hospital affiliated to
Shandong University. The serum levels of HMGB1 were detected using
commercial ELISA kits (Shino-Test Corp., Tokyo, Japan) according to
the manufacturer's instructions. Blood samples were collected from
the eye veins of the mice after they were anesthetized, and the
samples were centrifuged at 3,000 × g for 10 min at 4°C to collect
the serum. The serum was kept at −80°C until analysis.
The TNF-α and IL-1β levels in the pancreatic tissue
were detected using commercial ELISA kits (ExCell Bio, Shanghai,
China) according to the instructions provided by the manufacturer.
Protein extraction and concentration determination were performed
using the same procedures as described for western blot
analysis.
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from the pancreatic tissue
using TRIzol reagent (Takara, Tokyo, Japan). Using 1 µg
total RNA, first-strand cDNA was synthesized by the AMV enzyme in
20 µl reaction mixture (Takara). Using 2 µl reverse
transcriptase products, quantitative PCR was performed in a final
volume of 20 µl using gene-specific primers. The following
primers designed by Takara were used: mouse TLR4, 5′-CATGGATCA
GAAACTCAGCAAAGTC-3′ (sense), and 5′-CATGCCAT GCCTTGTCTTCA-3′
(antisense); mouse HMGB1, 5′-TTTA GATAGCCCTGTCCTGGTGGTA-3′ (sense),
and 5′-GTGCA CCAACAAGAACCTGCTTTA-3′ (antisense); and mouse actin,
5′-CATCCGTAAAGACCTCTATGCCAAC-3′ (sense), and
5′-ATGGAGCCACCGATCCACA-3′ (antisense). Amplification was carried
out as follows: 95°C, 30 sec, 1 cycle; 95°C, 3 sec and 60°C 30 sec
for 40 cycles; subsequently, the melting curve was determined. Gene
transcripts were quantified with SYBR Premix Ex Taq kit (Takara).
Data were calculated using the 2−ΔΔCT method and
presented as the fold change of transcripts for the HMGB1 and TLR4
genes in the pancreatic tissue of the other groups compared with
the control group (defined as 1.0-fold). Mouse actin was used as a
constitutive control. The relative expression of the target gene
was normalized to the level of actin in the same cDNA.
Statistical analysis
All values are expressed as the means ± SD. Analysis
of variance (ANOVA) followed by Tukey's multiple comparison tests
were used. A P-value <0.05 was considered to indicate a
statistically significant difference.
Results
Mouse model of SAP induced by
L-arginine
The murine model of acute pancreatitis induced by
L-arginine is a widely accepted model (4,24,27–29). In the present study, 48 h
following treatment with 8% L-arginine, SAP was observed, according
to the morphological characteristics and serum amylase and lipase
levels. In the SAP group, the pancreatic tissue exhibited marked
edema and was infiltrated by inflammatory cells; a mass of necrotic
acinar cells and the disappearance of normal structure in the
pancreatic lobes were also observed in the mice in this group
(Fig. 1A, panel b). As shown in
Fig. 1C, in the mice in the SAP
group, but not those which received the sham injection, a marked
increase in the serum amylase and lipase levels was noted.
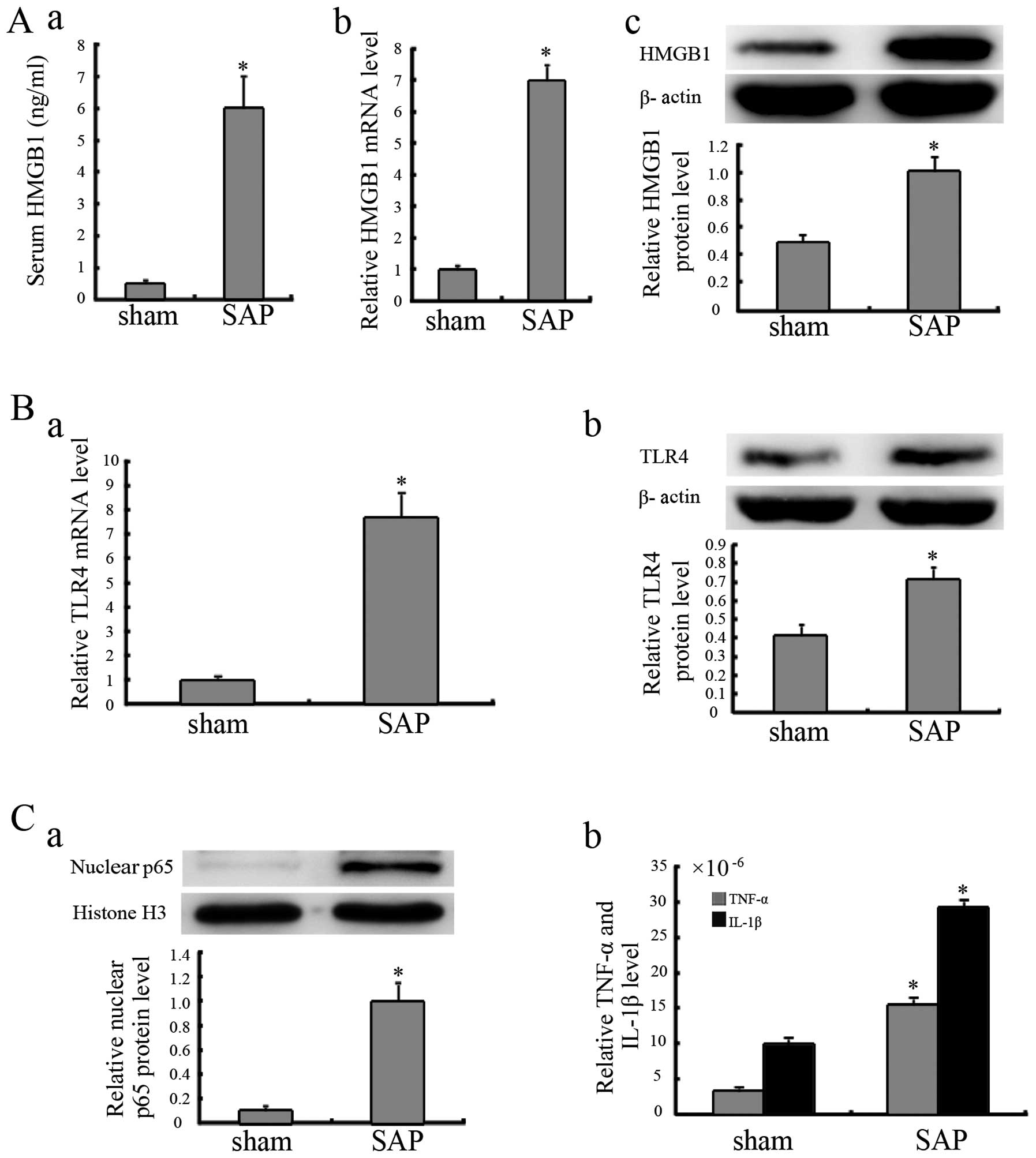
Elevated HMGB1 and TLR4 expression in
mice with SAP
Compared with the animals treated with the sham
injection, the mice with SAP exhibited increased HMGB1 mRNA and
protein levels in pancreatic tissue (Fig. 2A). The mice with SAP also
exhibited increased serum HMGB1 levels (Fig. 2A, panel a), which indicated that
HMGB1 was likely secreted into the extracellular space. Compared
with the mice which received the sham injection, increased TLR4
mRNA and protein levels were observed in the mice with SAP
(Fig. 2B).
NF-κB is activated during SAP
As shown in Fig.
2C, compared with the mice treated with the sham injection, the
protein levels of nuclear p65 in the pancreatic tissue from the
mice in the SAP group were significantly increased. The results
from ELISA revealed that the TNF-α and IL-1β levels in the
pancreatic tissue of the mice in the SAP group were higher than
those in the tissue of the mice in the sham injection group
(Fig. 2C).
rhHMGB1 induces pancreatic injury in
mice
WT mice (C57BL/6) were administered a
pancreas-targeted ultrasound-guided injection of rhHMGB1 to
determine whether it contributes to pancreatic injury. Histological
analysis of the pancreas was performed 48 h following treatment. As
shown in Fig. 3A, the pancreatic
tissue from the mice in the sham injection group had a normal
structure. By contrast, the pancreatic tissue from the mice in the
rhHMGB1 groups exhibited characteristics of pancreatic injury,
including considerable edema, neutrophil infiltration and acinar
cell necrosis. More intense accumulation of neutrophils and a mass
of necrotic acinar cells were also observed in the high-dose group,
indicating a change in the histology in response to various doses
of rhHMGB1. The serum amylase and lipase levels were also increased
in a dose-dependent manner in the rhHMGB1 groups compared to the
sham injection group.
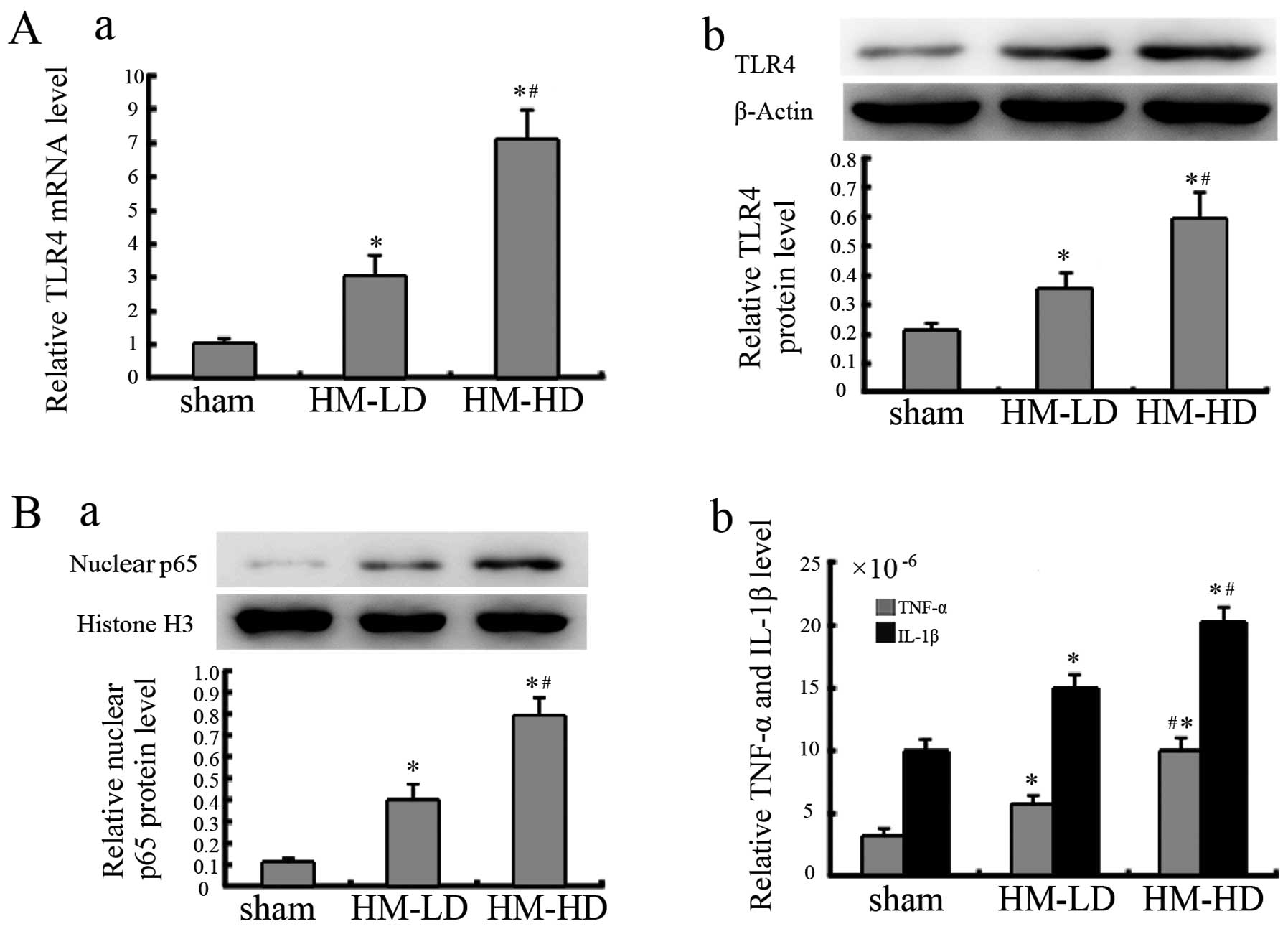
rhHMGB1 increases TLR4 expression and
activates NF-κB
Following the administration of rhHMGB1, the TLR4
mRNA expression level in the pancreatic tissue was significantly
increased in the rhHMGB1 groups compared to sham injection group.
Similar results were also observed in relation to the protein
expression levels (Fig. 4A).
Following the administration of rhHMGB1, as shown in
Fig. 4B, NF-κB was activated in
the rhHMGB1 groups, as observed from the increase in nuclear p65
expression and its downstream TNF-α and IL-1β expression.
Additionally, in relation to rhHMGB1 stimulation, TLR4 expression
and NF-κB activation was noted to be dependant on the concentration
of rhHMGB1.
TLR4 mediates rhHMGB1-induced pancreatic
injury
The assessment was performed at 48 h following the
high-dose administration of rhHMGB1. The pancreatic tissue from the
mice in the WT group exhibited characteristics of pancreatic
injury, including considerable edema, infiltration of inflammatory
cells, a mass of necrotic acinar cells and the disappearance of the
normal pancreatic lobe structure (Fig. 5A). However, in the TLR4-deficient
group, there was a significant reduction in pancreatic injury
(Fig. 5A, panel b). The serum
amylase and lipase levels of mice in the deficient group were also
significantly reduced compared to the mice in the WT group
(Fig. 5B, panel b).
As shown in Fig.
5C, NF-κB in the TLR4-deficient group was moderately activated
compared with the intense activation in the WT groups following the
administration of rhHMGB1. These results are clear from nuclear p65
expression and its downstream concentrations of TNF-α and IL-1β in
both groups of mice.
Discussion
SAP is an acute necrotic inflammation process that
suddenly occurs in the peripheral and internal areas of the
pancreas. At present, the pathogenesis of SAP remains incompletely
understood, leading to a lack of proper strategies for treating
SAP. However, the previously proposed inflammatory mediator theory
provides new information for SAP research (2). The increase in the levels of
inflammatory cytokines is closely associated with the severity of
SAP, and these inflammatory cytokines cause systemic inflammatory
response syndrome SIRS), multiple organ dysfunction syndrome (MODS)
and death.
We, as well as others have previously demonstrated
that HMGB1 is involved in the systematic inflammatory response of
SAP, that it appeares in the late phase and has a long duration
(1,3,13);
however, the underlying mechanisms have not yet been fully
elucidated. Yang et al (6)
and Deng et al (22) found
that TLR4 mediated HMGB1-induced acute lung injury. However, to the
best of our knowledge, no studies to date have demonstrated the
potential role of TLR4 in HMGB1-induced pancreatic injury.
The aim of this study was to further investigate
whether HMGB1 is involved as a stimulating factor, and whether its
downstream TLR4-mediated NF-κB signaling pathway mediates the
development of experimental pancreatic injury, with the aim of
developing further treatments for SAP. In this study, C57BL/6 mice
exposed to L-arginine had detectable SAP, as observed at the
pathological level and through serum amylase and lipase levels,
which is consistent with previous reports on L-arginine-induced SAP
(4,24,27–29). Moreover, significantly elevated
levels of HMGB1, TLR4 and NF-κB activation in the pancreatic tissue
were observed in the mice with SAP. These results from our study
are in agreement with previous observations (3,12–14,23). Therefore, we suggest that the
HMGB1 and the TLR4-mediated NF-κB signaling pathway play a
cooperative role in the development of pancreatic injury during
acute pancreatitis.
HMGB1, which was originally identified as a
DNA-binding protein, exhibits pro-inflammatory cytokine-like
properties when it is excreted into the extracellular space. In
previous studies, HMGB1 was found to be a regulator and inducer
that is involved in a number of diseases, such as sepsis,
ischemia-reperfusion injury, rheumatoid arthritis, thromboangiitis
obliterans, abdominal aortic aneurysm and acute pancreatitis
(12,14,30–35). In addition, anti-HMGB1-based
therapy using HMGB1 inhibitors, such as neutralizing anti-HMGB1
antibody, A box and the anti-inflammatory agents, ethyl pyravate
and sodium butyrate, has been shown to have beneficial effects on
HMGB1-related diseases (12,14,30–35). In the present study, we noted that
in mice with SAP, serum HMGB1 levels significantly increased,
indicating that HMGB1 was secreted into the extracellular space. To
determine whether HMGB1 alone induced pancreatic injury in mice, we
used a pancreas-targeted ultrasound-guided injection of various
doses of rhHMGB1 in mice and then observed pancreatic histology 48
h after administration. We observed considerable edema,
infiltration of inflammatory cells, a mass of necrotic acinar
cells, disappearance of the normal pancreas lobe structure and
significantly elevated serum amylase and lipase levels in the mice
in the rhHMGB1 groups, accounting for pancreatic injury. These
results are similar to previous observations made in relation to
mice and rats, which showed that acute lung inflammatory injury was
prompted by the intratracheal instillation of HMGB1 (12,22,36). In the present study, we also found
that the severity of pancreatic injury was dose dependent. These
results suggest that extracellular HMGB1 alone induces pancreatic
injury in a dose-dependent manner. Recently, Kang et al
(37) found that intracellular
HMGB1 inhibited inflammatory nucleosome release and exerted a
protective function in cases of acute pancreatitis. This was due to
the fact that nuclear HMGB1 directly interacted with the nucleosome
to maintain chromosomal structure, function and stability and
regulate DNA damage responses (38–43). Kang et al (37) also found that extracellular HMGB1
acted as a pro-inflammatory cytokine in cases of acute
pancreatitis, which was consistent with our finding.
TLR4 plays an important role in the innate immune
response. It is both an immune recognition receptor on the cell
surface and a transmembrane signal transduction molecule. When TLR4
is activated by PAMPs or DAMPs, it activates the MyD88- and
TRIF-dependent pathways, activating intracellular signaling
molecules, such as the interleukin-1 receptor-associated kinases
(IRAKs), tumor necrosis factor receptor-associated factors (TRAFs)
and TAK1. Therefore, it ultimately forms the primary and secondary
signal waves that activate NF-κB, which induces the transcription
and translation of inflammatory cytokines and leads to the massive
release of inflammatory mediators (17–19,44). In this study, using a murine model
of pancreatitis, the expression of TLR4 and NF-κB activation were
elevated in pancreatic tissue, and these results suggest that the
TLR4-mediated NF-κB signaling pathway plays a role in this form of
pancreatic injury. HMGB1 activates inflammatory pathways by
stimulating TLR4 in many types of tissue injury (6,22,45,46). However, it remains to be
determined whether the TLR4-mediated NF-κB signaling pathway
mediates HMGB1-induced pancreatic injury. To evaluate this form of
pancreatic injury, we administered pancreas-targeted rhHMGB1 to WT
and TLR4-deficient mice and then harvested pancreatic tissues 48 h
after treatment. In the WT group, we noted that rhHMGB1 upregulated
TLR4 expression, activated NF-κB and induced pancreatic injury.
These results suggest that rhHMGB1 alone upregulates TLR4
expression and activates the TLR4-mediated NF-κB signaling pathway
to induce pancreatic injury. However, in TLR4-deficient mice, the
extent of pancreatic injury was significantly reduced, and the
activation of NF-κB was lower than that in the WT mice. In spite of
the modest effect on pancreatic injury in TLR4-deficient mice after
the rhHMGB1 injection, we found that HMGB1-induced pancreatic
injury is predominantly mediated by the TLR4-mediated NF-κB
signaling pathway.
In conclusion, our results suggest that HMGB1 is
involved in the development of SAP. These findings contribute to a
better understanding of the mechanisms underlying the pathogenesis
of SAP. Manipulation of the interaction between HMGB1 and TLR4 will
likely ultimately lead to the development of novel therapies for
treating and preventing the progression of SAP.
Acknowledgments
This study was supported by the National Science
Fund for Distinguished Young Scholars (no. 81000186).
References
|
1
|
Yuan H, Jin X, Sun J, Li F, Feng Q, Zhang
C, Cao Y and Wang Y: Protective effect of HMGB1 a box on organ
injury of acute pancreatitis in mice. Pancreas. 38:143–148. 2009.
View Article : Google Scholar
|
|
2
|
Felderbauer P, Müller C, Bulut K, Belyaev
O, Schmitz F, Uhl W and Schmidt WE: Pathophysiology and treatment
of acute pancreatitis: new therapeutic targets - a ray of hope?
Basic Clin Pharmacol Toxicol. 97:342–350. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhang ZW, Zhang QY, Zhou MT, Liu NX, Chen
TK, Zhu YF and Wu L: Antioxidant inhibits HMGB1 expression and
reduces pancreas injury in rats with severe acute pancreatitis. Dig
Dis Sci. 55:2529–2536. 2010. View Article : Google Scholar
|
|
4
|
Sharif R, Dawra R, Wasiluk K, Phillips P,
Dudeja V, Kurt-Jones E, Finberg R and Saluja A: Impact of toll-like
receptor 4 on the severity of acute pancreatitis and
pancreatitis-associated lung injury in mice. Gut. 58:813–819. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ding SQ, Li Y, Zhou ZG, Wang C, Zhan L and
Zhou B: Toll-like receptor 4-mediated apoptosis of pancreatic cells
in cerulein-induced acute pancreatitis in mice. Hepatobiliary
Pancreat Dis Int. 9:645–650. 2010.PubMed/NCBI
|
|
6
|
Yang Z, Deng Y, Su D, Tian J, Gao Y, He Z
and Wang X: TLR4 as receptor for HMGB1-mediated acute lung injury
after liver ischemia/reperfusion injury. Lab Invest. 93:792–800.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li J, Wang H, Mason JM, Levine J, Yu M,
Ulloa L, Czura CJ, Tracey KJ and Yang H: Recombinant HMGB1 with
cytokine-stimulating activity. J Immunol Methods. 289:211–223.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Andersson U, Erlandsson-Harris H, Yang H
and Tracey KJ: HMGB1 as a DNA-binding cytokine. J Leukoc Biol.
72:1084–1091. 2002.PubMed/NCBI
|
|
9
|
Dumitriu IE, Baruah P, Manfredi AA,
Bianchi ME and Rovere-Querini P: HMGB1: guiding immunity from
within. Trends Immunol. 26:381–387. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang H, Wang H, Czura CJ and Tracey KJ:
HMGB1 as a cytokine and therapeutic target. J Endotoxin Res.
8:469–472. 2002. View Article : Google Scholar
|
|
11
|
Lotze MT, Zeh HJ, Rubartelli A, Sparvero
LJ, Amoscato AA, Washburn NR, Devera ME, Liang X, Tör M and Billiar
T: The grateful dead: damage-associated molecular pattern molecules
and reduction/oxidation regulate immunity. Immunol Rev. 220:60–81.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim JY, Park JS, Strassheim D, Douglas I,
Diaz del Valle F, Asehnoune K, Mitra S, Kwak SH, Yamada S, Maruyama
I, et al: HMGB1 contributes to the development of acute lung injury
after hemorrhage. Am J Physiol Lung Cell Mol Physiol.
288:L958–L965. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sawa H, Ueda T, Takeyama Y, Yasuda T,
Shinzeki M, Nakajima T and Kuroda Y: Blockade of high mobility
group box-1 protein attenuates experimental severe acute
pancreatitis. World J Gastroenterol. 12:7666–7670. 2006.PubMed/NCBI
|
|
14
|
Luan ZG, Zhang XJ, Yin XH, Ma XC, Zhang H,
Zhang C and Guo RX: Downregulation of HMGB1 protects against the
development of acute lung injury after severe acute pancreatitis.
Immunobiology. 218:1261–1270. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bianchi ME: DAMPs, PAMPs and alarmins: all
we need to know about danger. J Leukoc Biol. 81:1–5. 2007.
View Article : Google Scholar
|
|
16
|
Kawai T and Akira S: The role of
pattern-recognition receptors in innate immunity: update on
Toll-like receptors. Nat Immunol. 11:373–384. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Noreen M, Shah MA, Mall SM, Choudhary S,
Hussain T, Ahmed I, Jalil SF and Raza MI: TLR4 polymorphisms and
disease susceptibility. Inflamm Res. 61:177–188. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wullaert A: Role of NF-kappaB activation
in intestinal immune homeostasis. Int J Med Microbiol. 300:49–56.
2010. View Article : Google Scholar
|
|
19
|
Luo H, Guo P and Zhou Q: Role of
TLR4/NF-κB in damage to intestinal mucosa barrier function and
bacterial translocation in rats exposed to hypoxia. PLoS One.
7:e462912012. View Article : Google Scholar
|
|
20
|
Hori O, Brett J, Slattery T, Cao R, Zhang
J, Chen JX, Nagashima M, Lundh ER, Vijay S, Nitecki D, et al: The
receptor for advanced glycation end products (RAGE) is a cellular
binding site for amphoterin. Mediation of neurite outgrowth and
co-expression of rage and amphoterin in the developing nervous
system. J Biol Chem. 270:25752–25761. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Park JS, Svetkauskaite D, He Q, Kim JY,
Strassheim D, Ishizaka A and Abraham E: Involvement of Toll-like
receptors 2 and 4 in cellular activation by high mobility group box
1 protein. J Biol Chem. 279:7370–7377. 2004. View Article : Google Scholar
|
|
22
|
Deng Y, Yang Z, Gao Y, Xu H, Zheng B,
Jiang M, Xu J, He Z and Wang X: Toll-like receptor 4 mediates acute
lung injury induced by high mobility group box-1. PLoS One.
8:e643752013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Y, Zhou ZG, Xia QJ, Zhang J, Li HG, Cao
GQ, Wang R, Lu YL and Hu TZ: Toll-like receptor 4 detected in
exocrine pancreas and the change of expression in cerulein-induced
pancreatitis. Pancreas. 30:375–381. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Dawra R, Sharif R, Phillips P, Dudeja V,
Dhaulakhandi D and Saluja AK: Development of a new mouse model of
acute pancreatitis induced by administration of L-arginine. Am J
Physiol Gastrointest Liver Physiol. 292:G1009–G1018. 2007.
View Article : Google Scholar
|
|
25
|
Schmidt J, Lewandrowsi K, Warshaw AL,
Compton CC and Rattner DW: Morphometric characteristics and
homogeneity of a new model of acute pancreatitis in the rat. Int J
Pancreatol. 12:41–51. 1992.PubMed/NCBI
|
|
26
|
Pozsar J, Berger Z, Simon K, Kovacsai A,
Marosi E and Pap A: Biphasic effect of prostaglandin E1 on the
severity of acute pancreatitis induced by a closed duodenal loop in
rats. Pancreas. 12:159–164. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wang Y, Guo W, Li Y, Pan X, Lv W, Cui L,
Li C, Wang Y, Yan S, Zhang J and Liu B: Hypothermia induced by
adenosine 5′-mono-phosphate attenuates injury in an
L-arginine-induced acute pancreatitis rat model. J Gastroenterol
Hepatol. 29:742–748. 2014. View Article : Google Scholar
|
|
28
|
Jamdar S, Babu BI, Nirmalan M, Jeziorska
M, McMahon RF and Siriwardena AK: Administration of human
recombinant activated protein C is not associated with pancreatic
parenchymal haemorrhage in L-arginine-induced experimental acute
pancreatitis. JOP. 14:610–617. 2013.PubMed/NCBI
|
|
29
|
Yenicerioglu A, Cetinkaya Z, Girgin M,
Ustundag B, Ozercan IH, Ayten R and Kanat BH: Effects of
trimetazidine in acute pancreatitis induced by L-arginine. Can J
Surg. 56:175–179. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Chung KY, Park JJ and Kim YS: The role of
high-mobility group box-1 in renal ischemia and reperfusion injury
and the effect of ethyl pyruvate. Transplant Proc. 40:2136–2138.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yang H, Ochani M, Li J, Qiang X, Tanovic
M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, et al:
Reversing established sepsis with antagonists of endogenous
high-mobility group box 1. Proc Natl Acad Sci USA. 101:296–301.
2004. View Article : Google Scholar :
|
|
32
|
Kokkola R, Li J, Sundberg E, Aveberger AC,
Palmblad K, Yang H, Tracey KJ, Andersson U and Harris HE:
Successful treatment of collagen-induced arthritis in mice and rats
by targeting extracellular high mobility group box chromosomal
protein 1 activity. Arthritis Rheum. 48:2052–2058. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Andrassy M, Volz HC, Igwe JC, Funke B,
Eichberger SN, Kaya Z, Buss S, Autschbach F, Pleger ST, Lukic IK,
et al: High-mobility group box-1 in ischemia-reperfusion injury of
the heart. Circulation. 117:3216–3226. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kong X, Yuan H, Wu X, Zhang J, Zhou H,
Wang M, Liu Y and Jin X: High-mobility-group box protein 1A box
reduces development of sodium laurate-induced thromboangiitis
obliterans in rats. J Vasc Surg. 57:194–204. 2013. View Article : Google Scholar
|
|
35
|
Kohno T, Anzai T, Kaneko H, Sugano Y,
Shimizu H, Shimoda M, Miyasho T, Okamoto M, Yokota H, Yamada S, et
al: High-mobility group box 1 protein blockade suppresses
development of abdominal aortic aneurysm. J Cardiol. 59:299–306.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ueno H, Matsuda T, Hashimoto S, Amaya F,
Kitamura Y, Tanaka M, Kobayashi A, Maruyama I, Yamada S, Hasegawa
N, et al: Contributions of high mobility group box protein in
experimental and clinical acute lung injury. Am J Respir Crit Care
Med. 170:1310–1316. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Kang R, Zhang Q, Hou W, Yan Z, Chen R,
Bonaroti J, Bansal P, Billiar TR, Tsung A, Wang Q, et al:
Intracellular Hmgb1 inhibits inflammatory nucleosome release and
limits acute pancreatitis in mice. Gastroenterology. 146:1097–1107.
2014. View Article : Google Scholar :
|
|
38
|
Lange SS, Mitchell DL and Vasquez KM: High
mobility group protein B1 enhances DNA repair and chromatin
modification after DNA damage. Proc Natl Acad Sci USA.
105:10320–10325. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Kawase T, Sato K, Ueda T and Yoshida M:
Distinct domains in HMGB1 are involved in specific intramolecular
and nucleosomal interactions. Biochemistry. 47:13991–13996. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Cato L, Stott K, Watson M and Thomas JO:
The interaction of HMGB1 and linker histones occurs through their
acidic and basic tails. J Mol Biol. 384:1262–1272. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Giavara S, Kosmidou E, Hande MP, Bianchi
ME, Morgan A, d'Adda di Fagagna F and Jackson SP: Yeast Nhp6A/B and
mammalian Hmgb1 facilitate the maintenance of genome stability.
Curr Biol. 15:68–72. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Celona B, Weiner A, Di Felice F, Mancuso
FM, Cesarini E, Rossi RL, Gregory L, Baban D, Rossetti G, Grianti
P, et al: Substantial histone reduction modulates genomewide
nucleosomal occupancy and global transcriptional output. PLoS Biol.
9:e10010862011. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Bonaldi T, Längst G, Strohner R, Becker PB
and Bianchi ME: The DNA chaperone HMGB1 facilitates
ACF/CHRAC-dependent nucleosome sliding. EMBO J. 21:6865–6873. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Covert MW, Leung TH, Gaston JE and
Baltimore D: Achieving stability of lipopolysaccharide-induced
NF-kappaB activation. Science. 309:1854–1857. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yang H, Hreggvidsdottir HS, Palmblad K,
Wang H, Ochani M, Li J, Lu B, Chavan S, Rosas-Ballina M, Al-Abed Y,
et al: A critical cysteine is required for HMGB1 binding to
Toll-like receptor 4 and activation of macrophage cytokine release.
Proc Natl Acad Sci USA. 107:11942–11947. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Dobrovolskaia MA, Medvedev AE, Thomas KE,
Cuesta N, Toshchakov V, Ren T, Cody MJ, Michalek SM, Rice NR and
Vogel SN: Induction of in vitro reprogramming by Toll-like receptor
(TLR)2 and TLR4 agonists in murine macrophages: effects of TLR
'homotolerance' versus 'heterotolerance' on NF-kappa B signaling
pathway components. J Immunol. 170:508–519. 2003. View Article : Google Scholar
|