Introduction
Systemic lupus erythematosus (SLE) is a systemic
autoimmune disease that affects different organs and systems and
has a complex genetic inheritance (1). SLE has a complex etiology and is
affected by both genetic and environmental factors (2). The major histocompatibility complex
(MHC) located on chromosome 6p21 is one of the key factors that
contribute to the development of SLE (3). The human leukocyte antigen (HLA) has
been shown to be associated with susceptibility to SLE. Genome-wide
association studies have demonstrated that variants within the MHC
region confer the greatest genetic risk of developing SLE in
European and Chinese populations. However, the causal variants
remain elusive due to the tight linkage disequilibrium across
disease-associated MHC haplo-types, the highly polymorphic nature
of several MHC genes, and the heterogeneity of SLE phenotypes. The
loci include HLA-DPB1, HLA-G and MSH5, which
are independent of each other and HLA-DRB1 alleles. These
data highlight the usefulness of mapping disease susceptibility
loci using a transancestral approach, particularly in a region as
complex as the MHC, and offer a springboard for further
fine-mapping, resequencing and transcriptomic analysis (4). Certain studies have suggested an
association between MHC class I and II (HLA-A*29,
HLA-B*51, HLA-DRB1*15 and
HLA-DQB1*06) and susceptibility to SLE in the
Saudi population (5). In
African-American women, a single-nucleotide polymorphism (SNP)
which is closely associated with SLE, rs9271366, was found near the
HLA-DRB1 gene (6).
Although genetic variations within the MHC are
associated with the development of SLE, its role in the development
of the clinical manifestations and autoantibody production has not
been well defined. A meta-analysis of 4 independent European SLE
case collections was previously performed in an effort to identify
associations between SLE sub-phenotypes and MHC SNP genotypes, HLA
alleles, and variant HLA amino acids. The results provided strong
evidence for a multilevel risk model for
HLA-DRB1*03:01 in SLE, wherein the association
with anti-Ro and anti-La antibody-positive SLE is much stronger
than with SLE without these autoantibodies (7). Despite the research which has been
performed to date, a complete picture of these correlations has not
yet been painted, and further studies are still needed to shed
light on the associations between SLE and its susceptibility genes.
In addition, novel methods should be used to investigate this
subject.
Using a predictive bioinformatics algorithm, in a
previous study, Mantila Roosa et al created a linear model
of gene expression and identified 44 transcription factor-binding
motifs and 29 miRNA-binding sites that were predicted to regulate
gene expression across a time course. In addition to known sites,
novel transcription factor-binding motifs and several novel
miRNA-binding sites were identified throughout the time course.
These time-dependent regulatory mechanisms may be important for
controlling the loading-induced bone formation process (8). This integrated bioinformatics
analysis method was also used in this study. Although the link
between MHC and SLE has been proven, further investigations are
likely to reveal the involvement of MHC in simple and complex
genetic diseases, such as SLE. Indeed, we are interested in
studying MHC, CpG methylation and transcribed ultra-conserved
region (T-UCR) as a first step toward a better understanding of the
regulation of gene expression in SLE. In the present study, we
provide an extensive view of SLE based on an integrated
bioinformatics analysis of MHC, CpG methylation and T-UCR
datasets.
Materials and methods
Patients and controls
Whole blood samples from 15 patients with SLE (8
females; 7 males; aged 18–50 years, with an average age of
35.64±11.27 years) and 15 normal healthy controls (8 females; 7
males; aged 20–45 years, with an average age of 33.47±9.61 years)
were collected from the 181st Hospital of Guilin, China, between
January and September 2011. The SLE diagnoses were confirmed based
on pathological and clinical evidence according to the American
Rheumatism Association classification criteria (9,10).
Written informed consent was obtained from all the subjects or
their guardians. The use of biopsy material for studies beyond
routine diagnosis was approved by the Ethics Committee of the 181st
Hospital of Guilin. This study abides by the Helsinki Declaration
on Ethical Principles for Medical Research Involving Human
Subjects.
MHC gene capture, hMeDIP-chip and T-UCR
microarray analysis
Genomic DNA was isolated from peripheral blood
samples. According to the MHC genomic sequence, a completely
complementary probe was designed and fixed on a support and then
applied to the genomic DNA after coupling with a probe connector.
The unhybridized probe was washed away, and the probe that had
hybridized with the DNA was eluted to directly build a library for
DNA sequencing (HiSeq 2000 high-throughput sequencing). The MHC
region capture technology was based on the NimbleGen SeqCap EZ
Choice Library, enabling the deep sequencing coverage of the human
MHC region. The data were analyzed using the Chi-squared test with
Yates' correction for continuity.
Genomic DNA was extracted using a DNeasy Blood and
Tissue kit (Qiagen, Fremont, CA, USA). The sonicated genomic DNA (1
µg) was used for immunoprecipitation with a mouse monoclonal
antibody. For DNA labeling, a NimbleGen Dual-Color DNA Labeling kit
was used according to the manufacturer's instructions detailed in
the NimbleGen hMeDIP-chip protocol (NimbleGen Systems, Inc.,
Madison, WI, USA). The microarrays were hybridized in Nimblegen
hybridization buffer/hybridization component A in a hybridization
chamber (Hybridization System-Nimblegen Systems, Inc., Madison, WI,
USA). For array hybridization, the NimbleGen Promoter plus CpG
Island array (Roche, Basel, Switzerland) was used.
The Arraystar Human T-UCR Microarray profiles the
expression of 1,518 long non-coding RNAs (lncRNAs) and 2,261 mRNAs
with transcription units (TU) that overlap UCRs in either the sense
or antisense orientation. Sample RNA labeling and array
hybridization were performed according to the Agilent One-Color
Microarray-Based Gene Expression Analysis protocol (Agilent
Technology, Santa Clara, CA, USA), with minor modifications. The
hybridized arrays were washed, fixed and scanned, using Agilent DNA
Microarray Scanner (part no. G2505C). Agilent Feature Extraction
software (version 11.0.1.1) was used to analyze the acquired array
images. Quantile normalization and subsequent data processing were
performed using the GeneSpring GX v12.1 software package (Agilent
Technologies).
Bioinformatics analysis
CpG methylation enrichment in the MHC
segment and analysis for differential enrichment
The MHC gene capture sequencing segment was
chr6:28477797-33448354. To search the enrichment location, we
analyzed the CpG peaks in the MHC segment.
T-UCR expression in the MHC
segment
To search for the location of transcripts, we
analyzed T-UCR expression in the MHC segment.
Effect of the CpG methylation level
and T-UCR expression level in immunological processes
We analyzed all methylated CpGs, T-UCR, and their
corresponding genes, and then analyzed the related genes with
regard to immunological processes. To further examine the functions
of these genes, we used the Online Gene Ontology Tool EASE
(http://david.abcc.ncifcrf.gov/ease/ease1.htm). The
differentially expressed genes were classified with regard to
biological processes. Gene Ontology (GO) and KEGG pathway mapping
of the genes was performed using the web-accessible DAVID
annotation system.
Correlation of MHC mutation with CpG
methylation
To identify correlations, we calculated the data of
differential CpG methylation and MHC mutation and analyzed the
correlation coefficients.
Results
Capturing the number of genes and SNP
loci in the MHC region
We obtained 150 genes and 27,066 SNPs by MHC gene
capture and high-throughput sequencing in the patients with SLE
compared with the normal controls (data not shown).
hMeDIP-chip
The 3,826 genes with CpG islands had significantly
different methylation levels in the patients with SLE compared with
the normal controls, as was also previously noted (11).
T-UCR microarray analysis
To identify potential differentially expressed
T-UCRs, we performed fold change filtering of the SLE patients
compared with the normal controls. We found a signature of 8
upregulated T-UCRs and 29 downregulated T-UCRs (data not
shown).
CpG peak in the MHC segment
To search for enrichment locations, we analyzed CpG
peaks in the MHC segment. The results indicated the enrichment of
12 CpG-methylated sites (Table
I), with 6 in the patients with SLE and 6 in the normal
controls. One CpG-methylated enrichment site was found to be
located in the HLA-B promoter region
(chr6:31323946-31325211, 1,265 bp) (Table II) in the patients with SLE;
another site was located in the HLA-DPB2 intragenic region
(chr6:32975684-32975926, 242 bp) (Table II) in the control group.
 | Table ITwelve CpG-methylated enrichment
sites in the MHC segment. |
Table I
Twelve CpG-methylated enrichment
sites in the MHC segment.
| CpG name
(hg19) | Length (bp) | Control | SLE | Gene name | Location |
|---|
|
chr6:30042918-30043500 | 582 | | 1 | RNF39 | Promoter |
|
chr6:31323946-31325211 | 1,265 | | 1 | HLA-B | Promoter |
|
chr6:31695894-31698245 | 2,351 | | 1 | DDAH2 | Promoter |
|
chr6:31695894-31698245 | 2,351 | | 1 | LY6G6C | Promoter |
|
chr6:31695894-31698245 | 2,351 | | 1 | MSH5 | Promoter |
|
chr6:32935896-32936792 | 896 | | 1 | BRD2 | Promoter |
|
chr6:29521110-29521833 | 723 | 1 | | UBD | Intergenic |
|
chr6:30538983-30539487 | 504 | 1 | | ABCF1 | Promoter |
|
chr6:30684836-30685503 | 667 | 1 | | MDC1 | Promoter |
|
chr6:30684836-30685503 | 667 | 1 | | TUBB | Promoter |
|
chr6:31548436-31549277 | 841 | 1 | | LST1 | Promoter |
|
chr6:32975684-32975926 | 242 | 1 | |
HLA-DPB2 | Intragenic |
| Table IIHLA-B promoter region and
HLA-DPB2 intragenic region in patients with SLE. |
Table II
HLA-B promoter region and
HLA-DPB2 intragenic region in patients with SLE.
| HLA-B
promoter region (chr6:31323946-31325211, 1265 bp) |
CGAAGTCCCAGGTCCCGGACGGGGCTCTCAGGGTCTCAGGCTCCGAGGGCCGCGTCTGCAATGGGGAGGCGCAG
CGTTGGGGATTCCCCACTCCCCTGAGTTTCACTTCTTCTCCCAACTTGTGTCGGGTCCTTCTTCCAGGATACTCGTG
ACGCGTCCCCACTTCCCACTCCCATTGGGTATTGGATATCTAGAGAAGCCAATCAGCGTCGCCGCGGTCCCAGTTC
TAAAGTCCCCACGCACCCACCCGGACTCAGAGTCTCCTCAGACGCCGAGATGCTGGTCATGGCGCCCCGAACCGT
CCTCCTGCTGCTCTCGGCGGCCCTGGCCCTGACCGAGACCTGGGCCGGTGAGTGCGGGTCGGGAGGGAAATGGC
CTCTGCCGGGAGGAGCGAGGGGACCGCAGGCGGGGGCGCAGGACCTGAGGAGCCGCGCCGGGAGGAGGGTCGG
GCGGGTCTCAGCCCCTCCTCACCCCCAGGCTCCCACTCCATGAGGTATTTCTACACCTCCGTGTCCCGGCCCGGCC
GCGGGGAGCCCCGCTTCATCTCAGTGGGCTACGTGGACGACACCCAGTTCGTGAGGTTCGACAGCGACGCCGCG
AGTCCGAGAGAGGAGCCGCGGGCGCCGTGGATAGAGCAGGAGGGGCCGGAGTATTGGGACCGGAACACACAGA
TCTACAAGGCCCAGGCACAGACTGACCGAGAGAGCCTGCGGAACCTGCGCGGCTACTACAACCAGAGCGAGGCC
GGTGAGTGACCCCGGCCCGGGGCGCAGGTCACGACTCCCCATCCCCCACGTACGGCCCGGGTCGCCCCGAGTCTC
CGGGTCCGAGATCCGCCTCCCTGAGGCCGCGGGACCCGCCCAGACCCTCGACCGGCGAGAGCCCCAGGCGCGTT
TACCCGGTTTCATTTTCAGTTGAGGCCAAAATCCCCGCGGGTTGGTCGGGGCGGGGCGGGGCTCGGGGGACTGGG
CTGACCGCGGGGCCGGGGCCAGGGTCTCACACCCTCCAGAGCATGTACGGCTGCGACGTGGGGCCGGACGGGCG
CCTCCTCCGCGGGCATGACCAGTACGCCTACGACGGCAAGGATTACATCGCCCTGAACGAGGACCTGCGCTCCTG
GACCGCCGCGGACACGGCGGCTCAGATCACCCAGCGCAAGTGGGAGGCGGCCCGTGAGGCGGAGCAGCGGAGA
GCCTACCTGGAGGGCGAGTGCGTGGAGTGGCTCCGCAGATACCTGGAGAACGGGAAGGACAAGCTGGAGCGCGC |
| HLA-DPB2
intragenic region (chr6:32975684-32975926, 242 bp) |
CGAGGCCGTGTGGCGTCTGCCTGAGTTTGGTGACTTTGCCCGCTTTGACCCGCAGGGCGGGCTGGCCGGCATCGC
CGCAATCAAAGCCCATCTGGACATCCTGGTGGAGCGCTCCAACCGCAGCAGAGCCATCAACGGTACCGGCCCTCC
CTCTGCCCACCCAGTCAGGCGGGAAGGTCCAGAGAAACTTCCTCCCAGTTCCTAGGCTCCCATCACTCTGGGGCG
CGCTCTCAGCGCCCGCGC |
T-UCR expression in the MHC segment
In the present study, we analyzed the expression of
all T-UCRs; however, UCR-overlapping and UCR-proximal genes were
not discovered in the MHC segment.
Effect of the CpG methylation level on
immunological processes
We annotated corresponding CpG-methylated genes in
the patients with SLE with GO schemes using the DAVID gene
annotation tool. The genes produced a total of 97 GO terms in the
patients with SLE (Table III).
However, no significant enrichment was found for immune-correlated
process GO terms, such as GO:0006955 - immune response (48 genes:
TNFAIP8 L2, ITGAL, GALNT2, YWHAZ, LST1, TOLLIP, IFITM2, IFITM3,
SUSD2, TLR2, NLRX1, VTN, TLR5, CX3CL1, PYDC1, FTH1, IGF1R, TUBB,
IL2RG, CFD, SPON2, APLN, SPN, DNAJA3, POLL, TRPM4, DBNL, IL18R1,
SMAD6, EOMES, CNPY3, STXBP2, POLR3A, HLA-B, VAV1, WAS, CD1D, LAT,
CYBA, PRELID1, GPI, SARM1, ULBP1, TGFBR3, ADAM17, TCF12, ICOSLG,
TNFAIP1, P=0.999999, FDR >0.05).
| Table IIIGO term annotations of corresponding
CpG-methylated genes in SLE patients. |
Table III
GO term annotations of corresponding
CpG-methylated genes in SLE patients.
| GO term | Gene count | P-value | FDR |
|---|
| GO:0030182 - neuron
differentiation | 113 | 1.39E-13 | 2.58E-10 |
| GO:0051252 -
regulation of RNA metabolic process | 319 | 2.37E-10 | 4.41E-07 |
| GO:0006355 -
regulation of transcription, DNA-dependent | 313 | 2.50E-10 | 4.66E-07 |
| GO:0045449 -
regulation of transcription | 429 | 8.22E-10 | 1.53E-06 |
| GO:0000904 - cell
morphogenesis involved in differentiation | 66 | 3.46E-09 | 6.45E-06 |
| GO:0048666 - neuron
development | 82 | 1.08E-08 | 2.01E-05 |
| GO:0007409 -
axonogenesis | 55 | 1.18E-08 | 2.19E-05 |
| GO:0006350 -
transcription | 349 | 2.66E-08 | 4.96E-05 |
| GO:0048667 - cell
morphogenesis involved in neuron differentiation | 57 | 3.36E-08 | 6.26E-05 |
| GO:0000902 - cell
morphogenesis | 83 | 5.01E-08 | 9.34E-05 |
| GO:0007389 -
pattern specification process | 66 | 1.46E-07 | 2.73E-04 |
| GO:0048812 - neuron
projection morphogenesis | 56 | 1.71E-07 | 3.18E-04 |
| GO:0031175 - neuron
projection development | 63 | 3.43E-07 | 6.39E-04 |
| GO:0048858 - cell
projection morphogenesis | 61 | 3.52E-07 | 6.56E-04 |
| GO:0032989 -
cellular component morphogenesis | 87 | 4.22E-07 | 7.86E-04 |
| GO:0032990 - cell
part morphogenesis | 62 | 7.71E-07 | 0.001437153 |
| GO:0006357 -
regulation of transcription from RNA polymerase II promoter | 138 | 1.20E-06 | 0.002241345 |
| GO:0030030 - cell
projection organization | 80 | 1.79E-06 | 0.003344582 |
| GO:0003002 -
regionalization | 50 | 2.55E-06 | 0.004748657 |
| GO:0021953 -
central nervous system neuron differentiation | 18 | 2.92E-06 | 0.005449045 |
| GO:0030900 -
forebrain development | 41 | 4.82E-06 | 0.008984544 |
| GO:0048663 - neuron
fate commitment | 18 | 6.52E-06 | 0.012150091 |
| GO:0048598 -
embryonic morphogenesis | 67 | 1.19E-05 | 0.022252245 |
| GO:0021954 -
central nervous system neuron development | 15 | 1.47E-05 | 0.02743134 |
| GO:0045944 -
positive regulation of transcription from RNA polymerase II
promoter | 77 | 1.68E-05 | 0.031223255 |
| GO:0007411 - axon
guidance | 31 | 1.93E-05 | 0.035915973 |
| GO:0045165 - cell
fate commitment | 37 | 2.06E-05 | 0.038453597 |
| GO:0021872 -
generation of neurons in the forebrain | 10 | 2.17E-05 | 0.040470383 |
| GO:0045893 -
positive regulation of transcription, DNA-dependent | 93 | 2.77E-05 | 0.051580919 |
| GO:0051254 -
positive regulation of RNA metabolic process | 93 | 3.85E-05 | 0.071644433 |
| GO:0006928 - cell
motion | 92 | 3.98E-05 | 0.074087062 |
| GO:0007423 -
sensory organ development | 52 | 4.33E-05 | 0.08062919 |
| GO:0007169 -
transmembrane receptor protein tyrosine kinase signaling
pathway | 51 | 4.81E-05 | 0.089665676 |
| GO:0021879 -
forebrain neuron differentiation | 9 | 4.99E-05 | 0.092920613 |
| GO:0045935 -
positive regulation of nucleobase, nucleoside, nucleotide and
nucleic acid metabolic process | 114 | 6.21E-05 | 0.115713935 |
| GO:0051173 -
positive regulation of nitrogen compound metabolic process | 117 | 6.28E-05 | 0.116921469 |
| GO:0060284 -
regulation of cell development | 47 | 8.31E-05 | 0.154746845 |
| GO:0045941 -
positive regulation of transcription | 104 | 9.33E-05 | 0.173822702 |
| GO:0050767 -
regulation of neurogenesis | 40 | 1.00E-04 | 0.186897834 |
| GO:0045664 -
regulation of neuron differentiation | 34 | 1.11E-04 | 0.206978981 |
| GO:0010628 -
positive regulation of gene expression | 106 | 1.20E-04 | 0.223869384 |
| GO:0010557 -
positive regulation of macromolecule biosynthetic process | 117 | 1.23E-04 | 0.228227922 |
| GO:0016192 -
vesicle-mediated transport | 105 | 1.36E-04 | 0.253359896 |
| GO:0051960 -
regulation of nervous system development | 44 | 1.46E-04 | 0.271915907 |
| GO:0031328 -
positive regulation of cellular biosynthetic process | 121 | 1.62E-04 | 0.302155781 |
| GO:0009891 -
positive regulation of biosynthetic process | 122 | 1.98E-04 | 0.368213146 |
| GO:0007167 - enzyme
linked receptor protein signaling pathway | 68 | 2.08E-04 | 0.386237807 |
| GO:0045892 -
negative regulation of transcription, DNA-dependent | 70 | 2.33E-04 | 0.432663991 |
| GO:0030817 -
regulation of cAMP biosynthetic process | 27 | 3.08E-04 | 0.572491129 |
| GO:0002009 -
morphogenesis of an epithelium | 27 | 3.08E-04 | 0.572491129 |
| GO:0030902 -
hindbrain development | 19 | 3.48E-04 | 0.647196595 |
| GO:0021537 -
telencephalon development | 20 | 3.49E-04 | 0.648170377 |
| GO:0021761 - limbic
system development | 13 | 3.51E-04 | 0.6529104 |
| GO:0051253 -
negative regulation of RNA metabolic process | 70 | 3.83E-04 | 0.7106764 |
| GO:0045934 -
negative regulation of nucleobase, nucleoside, nucleotide and
nucleic acid metabolic process | 93 | 3.88E-04 | 0.7213528 |
| GO:0010604 -
positive regulation of macromolecule metabolic process | 144 | 3.97E-04 | 0.737772867 |
| GO:0030814 -
regulation of cAMP metabolic process | 27 | 4.29E-04 | 0.795879925 |
| GO:0009792 -
embryonic development ending in birth or egg hatching | 65 | 5.31E-04 | 0.984899092 |
| GO:0008285 -
negative regulation of cell proliferation | 69 | 5.90E-04 | 1.094286156 |
| GO:0051172 -
negative regulation of nitrogen compound metabolic process | 93 | 6.13E-04 | 1.136642775 |
| GO:0009890 -
negative regulation of biosynthetic process | 101 | 6.30E-04 | 1.168027793 |
| GO:0010558 -
negative regulation of macromolecule biosynthetic process | 97 | 6.65E-04 | 1.231652838 |
| GO:0009952 -
anterior/posterior pattern formation | 33 | 6.76E-04 | 1.252734624 |
| GO:0031327 -
negative regulation of cellular biosynthetic process | 99 | 6.87E-04 | 1.273119736 |
| GO:0043583 - ear
development | 25 | 6.92E-04 | 1.282237143 |
| GO:0051339 -
regulation of lyase activity | 26 | 7.49E-04 | 1.385942497 |
| GO:0048732 - gland
development | 32 | 7.50E-04 | 1.387778635 |
| GO:0045761 -
regulation of adenylate cyclase activity | 25 | 8.13E-04 | 1.504562886 |
| GO:0022037 -
metencephalon development | 13 | 9.00E-04 | 1.663443567 |
| GO:0048568 -
embryonic organ development | 38 | 9.18E-04 | 1.697487359 |
| GO:0021766 -
hippocampus development | 10 | 9.31E-04 | 1.721218707 |
| GO:0016481 -
negative regulation of transcription | 83 | 9.44E-04 | 1.744715102 |
| GO:0030808 -
regulation of nucleotide biosynthetic process | 27 | 0.001237 | 2.280679952 |
| GO:0030802 -
regulation of cyclic nucleotide biosynthetic process | 27 | 0.001237 | 2.280679952 |
| GO:0010629 -
negative regulation of gene expression | 89 | 0.001288 | 2.373367738 |
| GO:0031279 -
regulation of cyclase activity | 25 | 0.001292 | 2.380392647 |
| GO:0035107 -
appendage morphogenesis | 25 | 0.001292 | 2.380392647 |
| GO:0035108 - limb
morphogenesis | 25 | 0.001292 | 2.380392647 |
| GO:0021543 -
pallium development | 15 | 0.00132 | 2.432197518 |
| GO:0014031 -
mesenchymal cell development | 16 | 0.001327 | 2.444625723 |
| GO:0048762 -
mesenchymal cell differentiation | 16 | 0.001327 | 2.444625723 |
| GO:0060562 -
epithelial tube morphogenesis | 19 | 0.001468 | 2.701431115 |
| GO:0010941 -
regulation of cell death | 134 | 0.001518 | 2.791424456 |
| GO:0035295 - tube
development | 45 | 0.001562 | 2.870764354 |
| GO:0048705 -
skeletal system morphogenesis | 27 | 0.001632 | 2.999117627 |
| GO:0060485 -
mesenchyme development | 16 | 0.001648 | 3.027543666 |
| GO:0030799 -
regulation of cyclic nucleotide metabolic process | 27 | 0.001868 | 3.424511611 |
| GO:0001709 - cell
fate determination | 12 | 0.001898 | 3.478414435 |
| GO:0043009 -
chordate embryonic development | 62 | 0.001904 | 3.49042025 |
| GO:0016331 -
morphogenesis of embryonic epithelium | 17 | 0.001959 | 3.588514741 |
| GO:0042127 -
regulation of cell proliferation | 129 | 0.002101 | 3.843962583 |
| GO:0048736 -
appendage development | 25 | 0.002293 | 4.187971146 |
| GO:0060173 - limb
development | 25 | 0.002293 | 4.187971146 |
| GO:0051349 -
positive regulation of lyase activity | 17 | 0.002374 | 4.334185806 |
| GO:0035239 - tube
morphogenesis | 29 | 0.002504 | 4.564895517 |
| GO:0043067 -
regulation of programmed cell death | 132 | 0.002508 | 4.573381853 |
| GO:0017145 - stem
cell division | 6 | 0.002742 | 4.98879198 |
In addition, we obtained 3 KEGG pathways for genes
in patients with SLE (Table IV),
although no significant enrichment was found for immune-correlated
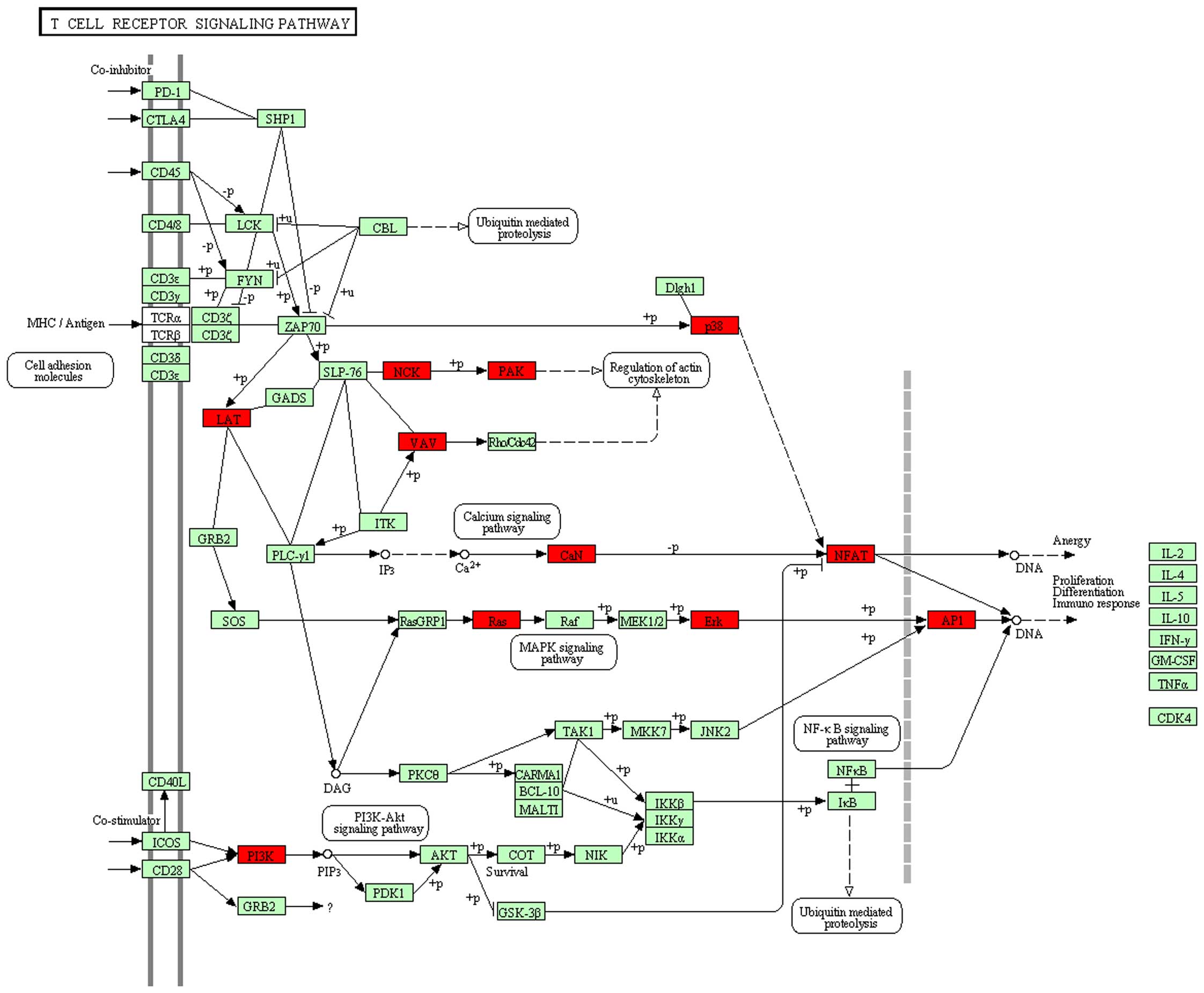
process KEGG pathways, such as hsa04660:T cell receptor signaling
pathway (Fig. 1; 15 genes:
PIK3CG, HRAS, VAV1, LOC407835, MAPK1, LAT, MAPK12, NCK1, PAK4,
JUN, NFAT5, PPP3CB, CHP, PIK3R3, NFATC1, P=0.406339, FDR
>0.05).
| Table IVKEGG pathway annotation of
corresponding CpG-methylated genes in patients with SLE. |
Table IV
KEGG pathway annotation of
corresponding CpG-methylated genes in patients with SLE.
| Pathways | Gene count | P-value | FDR |
|---|
| hsa05200: pathways
in cancer | 60 | 5.25E-04 | 0.6471034 |
| hsa04916:
melanogenesis | 23 | 0.002387 | 2.9086362 |
| hsa05217: basal
cell carcinoma | 15 | 0.003991 | 4.8195711 |
Effect of T-UCR expression levels on
immunological processes
We also annotated T-UCR-corresponding genes with GO
schemes using the DAVID gene annotation tool. The genes produced
total 43 GO terms in patients with SLE (Table V); however, no significant
enrichment was found for immune-correlated process GO terms. In
addition, we did not obtain immune-correlated process KEGG pathways
for the genes in patients with SLE, i.e., there was no significant
enrichment.
| Table VGO term annotation of T-UCR
corresponding genes in SLE patients. |
Table V
GO term annotation of T-UCR
corresponding genes in SLE patients.
| GO term | Gene count | P-value | FDR |
|---|
| GO:0008380 - RNA
splicing | 35 | 1.24E-11 | 2.15E-08 |
| GO:0006397 - mRNA
processing | 37 | 2.04E-11 | 3.55E-08 |
| GO:0016071 - mRNA
metabolic process | 38 | 3.00E-10 | 5.22E-07 |
| GO:0006396 - RNA
processing | 47 | 6.45E-10 | 1.12E-06 |
| GO:0006357 -
regulation of transcription from RNA polymerase II promoter | 55 | 1.75E-09 | 3.04E-06 |
| GO:0045449 -
regulation of transcription | 129 | 1.15E-08 | 1.99E-05 |
| GO:0051252 -
regulation of RNA metabolic process | 97 | 5.09E-08 | 8.85E-05 |
| GO:0045935 -
positive regulation of nucleobase, nucleoside, | 44 | 7.25E-07 | 0.001261 |
| nucleotide and
nucleic acid metabolic process | | | |
| GO:0051254 -
positive regulation of RNA metabolic process | 37 | 9.25E-07 | 0.00161 |
| GO:0006355 -
regulation of transcription, DNA-dependent | 91 | 1.01E-06 | 0.00175 |
| GO:0010558 -
negative regulation of macromolecule biosynthetic process | 40 | 1.07E-06 | 0.001865 |
| GO:0000398 -
nuclear mRNA splicing, via spliceosome | 19 | 1.20E-06 | 0.002082 |
| GO:0000377 - RNA
splicing, via transesterification reactions with bulged adenosine
as nucleophile | 19 | 1.20E-06 | 0.002082 |
| GO:0000375 - RNA
splicing, via transesterification reactions | 19 | 1.20E-06 | 0.002082 |
| GO:0045944 -
positive regulation of transcription from RNA polymerase II
promoter | 31 | 1.60E-06 | 0.002782 |
| GO:0051173 -
positive regulation of nitrogen compound metabolic process | 44 | 1.68E-06 | 0.002914 |
| GO:0031327 -
negative regulation of cellular biosynthetic process | 40 | 1.97E-06 | 0.003434 |
| GO:0045893 -
positive regulation of transcription, DNA-dependent | 36 | 2.06E-06 | 0.003575 |
| GO:0045941 -
positive regulation of transcription | 40 | 2.26E-06 | 0.00393 |
| GO:0009890 -
negative regulation of biosynthetic process | 40 | 3.34E-06 | 0.005812 |
| GO:0048598 -
embryonic morphogenesis | 27 | 3.46E-06 | 0.006015 |
| GO:0045934 -
negative regulation of nucleobase, nucleoside, nucleotide and
nucleic acid metabolic process | 37 | 3.86E-06 | 0.00672 |
| GO:0006350 -
transcription | 101 | 4.00E-06 | 0.006958 |
| GO:0010628 -
positive regulation of gene expression | 40 | 4.62E-06 | 0.008041 |
| GO:0051172 -
negative regulation of nitrogen compound metabolic process | 37 | 5.24E-06 | 0.009119 |
| GO:0031328 -
positive regulation of cellular biosynthetic process | 44 | 8.06E-06 | 0.014026 |
| GO:0051253 -
negative regulation of RNA metabolic process | 29 | 8.35E-06 | 0.014517 |
| GO:0010605 -
negative regulation of macromolecule metabolic process | 46 | 8.87E-06 | 0.015419 |
| GO:0010604 -
positive regulation of macromolecule metabolic process | 51 | 1.09E-05 | 0.018888 |
| GO:0009891 -
positive regulation of biosynthetic process | 44 | 1.15E-05 | 0.020058 |
| GO:0016481 -
negative regulation of transcription | 33 | 1.61E-05 | 0.027966 |
| GO:0045892 -
negative regulation of transcription, DNA-dependent | 28 | 1.72E-05 | 0.029862 |
| GO:0010557 -
positive regulation of macromolecule biosynthetic process | 41 | 3.08E-05 | 0.053493 |
| GO:0010629 -
negative regulation of gene expression | 34 | 4.16E-05 | 0.072327 |
| GO:0000122 -
negative regulation of transcription from RNA polymerase II
promoter | 21 | 2.37E-04 | 0.412261 |
| GO:0048568 -
embryonic organ development | 16 | 2.97E-04 | 0.515968 |
| GO:0016055 - Wnt
receptor signaling pathway | 13 | 8.69E-04 | 1.501407 |
| GO:0030900 -
forebrain development | 14 | 8.89E-04 | 1.535269 |
| GO:0043009 -
chordate embryonic development | 22 | 0.001544 | 2.651627 |
| GO:0009792 -
embryonic development ending in birth or egg hatching | 22 | 0.001718 | 2.946609 |
| GO:0046907 -
intracellular transport | 35 | 0.002355 | 4.018647 |
| GO:0015931 -
nucleobase, nucleoside, nucleotide and nucleic acid transport | 11 | 0.002648 | 4.507588 |
| GO:0048562 -
embryonic organ morphogenesis | 12 | 0.002821 | 4.795051 |
Correlation between MHC mutation and CpG
methylation
In this study, we found 4 SNPs in the CpG promoter
(chr6:31323946-31325211) of HLA-B and 2 SNPs in
chr6:29521110-29521833 in the control patients (Table VI).
| Table VISix CpG-methylated SNPs of the MHC
segment in patients with SLE. |
Table VI
Six CpG-methylated SNPs of the MHC
segment in patients with SLE.
| Chromosome
segment | SNP | Gene | Location |
|---|
| chr6:31324019 | rs1050683 | HLA-B | Exonic |
| chr6:31324057 | rs12697943 | HLA-B | Exonic |
| chr6:31324448 | rs17881210 | HLA-B | Intronic |
| chr6:31324633 | rs1065378 | HLA-B | Exonic |
| chr6:29521289 | rs17184255 | | Intergenic |
| chr6:29521557 | rs16895070 | | Intergenic |
Discussion
The first genetic factors to be identified as
important in the pathogenesis of SLE were those of the MHC on
chromosome 6. It is now widely accepted that MHC genes constitute a
part of the genetic susceptibility to SLE (12). However, previous studies on SLE
have lacked statistical power and the genetic resolution to fully
define the influences of the MHC (13,14). In this study, we attempted to
identify MHC, CpG methylation and T-UCR to reveal the potential
mechanisms responsible for the development of SLE using a novel and
combinatorial approach involving MHC gene capture technology,
hMeDIP-chip, T-UCR microarray and bioinformatics analysis. A total
of 27,066 SNPs were detected and thus these may be involved in SLE.
Moreover, we integrated the datasets and identified 6 of the most
important SNPs in SLE. Our next step is to perform research on the
function of these SNPs.
HLA antigens and genes have long been reported to be
associated with SLE susceptibility in a number of populations
(15). With advances in
technologies, such as genome-wide association studies, a number of
newly discovered SLE-associated SNPs have been reported in recent
years. These include HLA-DRB1/HLA-DQA1 rs9271366 and
HLA-DQB1/HLA-DQA2 rs9275328 (15). Previously, a meta-analysis of the
MHC region in patients with SLE was performed to determine
associations with both SNPs and classical HLA alleles. The results
of a conditional analysis and model choice with the use of the
Bayesian information criterion indicated that the best model for
SLE association includes both classical loci
(HLA-DRB1*03:01, HLA-DRB1*08:01
and HLA-DQA1*01:02) and 2 SNPs, rs8192591 (in
class III and upstream of NOTCH4) and rs2246618 (MICB in class I)
(16). Single-marker analyses
have revealed strong signals for SNPs within several MHC regions,
as well as for HLA-DRB1. The most strongly associated DRB1
alleles are *0301, *1401 and
*1501, and the MHC region SNP demonstrating the
strongest evidence of an association with SLE is rs3117103
(3). These results delineate with
high resolution several MHC regions which contribute independently
to the risk of developing SLE. In the present study, we integrated
the MHC and CpG-methylated datasets, and the results indicated CpG
methylation enrichment at 6 sites: RNF39, HLA-B,
DDAH2, LY6G6C, MSH5 and BRD2 in the MHC
regions of SLE. These genes play important roles in various immune
diseases, including SLE. SNPs in the region of the RNF39
gene have been found to be associated with the disease course of
HIV-1 (17,18). Behcet's disease is a chronic
inflammatory autoimmune disease that is strongly associated with
HLA-B51 and -A26. It has previously been suggested
that RNF39 is involved in the etiology of Behcet's disease
(19). The MHC region is
suspected to host susceptibility loci for HIV-related Kaposi's
sarcoma, involving the rs1065356 (LY6G6C) and rs3749953
(MSH5-SAPCD1) (20).
MSH5 has been found to be mutated in patients with common
variable immunodeficiency (21).
In a previous study, a significant increase in the frequency of
HLA-A*01, A*03, A*11,
A*23, A*26 A*69,
HLA-B*27, B*40, B*49,
B*51, B*52, B*53, B*54,
B*95, HLA-DRBI*01, DRBI*03,
DRBI*11 and DRBI*14 was observed
in SLE patients, indicating a positive association of these alleles
with SLE. By contrast, HLA-A*24, A*29,
A*31, A*34, A*68, A*92,
HLA-B*18 and HLA-DRB1*12 were
found to be decreased in the patient group compared to the
controls, indicating a negative association of these alleles with
SLE. Thus, it was concluded that SLE is associated with certain MHC
alleles, such as HLA-B, in the Pakistani population
(12). As the results of the
present study indicated the enrichment of one CpG methylation site
located in the HLA-B promoter region, HLA-B may
indeed play an important role in the pathogenesis of SLE. Moreover,
we found 4 SNPs (rs1050683, rs12697943, rs17881210 and rs1065378)
in the CpG region of the HLA-B promoter and 2 SNPs
(rs17184255 and rs16895070) in MHC regions. These SNPs were
significantly associated with an increased risk of developing
SLE.
SLE is an autoimmune disease with known genetic,
epigenetic, and environmental risk factors. Epigenetic events play
a central role in the priming, differentiation and subset
determination of T lymphocytes. CpG-DNA methylation and
post-translational modifications to histone tails are the two most
well-accepted epigenetic mechanisms. Furthermore, the involvement
of epigenetic mechanisms in the pathogenesis of SLE has been
suggested by the development of lupus-like symptoms in individuals
who are treated with procainamide or hydralazine, resulting in a
reduction in CpG-DNA methylation (22). In SLE, global CpG-DNA
hypomethylation correlates with disease activity. A number of
cytokine genes are overexpressed in CD4+ T lymphocytes
from patients with SLE in a chromatin-dependent manner, including
IL-6 (23). Region-specific
histone acetylation in certain tissues is associated with increased
disease activity, whereas histone acetylation in other regions has
protective effects. In SLE, acetylation of the TNF promoter in
monocytes is associated with increased monocyte maturation and
cytokine expression (24). Thus,
a better understanding of the molecular events that contribute to
epigenetic alterations and subsequent immune imbalance is essential
for the establishment of disease biomarkers and the identification
of potential therapeutic targets (22). To assess the role of DNA
methylation in SLE, researchers collected CD4+ T-cells,
CD19+ B-cells, and CD14+ monocytes, and
performed a genome-wide DNA methylation analysis with the use of
IlluminaMethylation 450 microarrays. Interferon hypersensitivity
was apparent in memory, naïve and regulatory T-cells, suggesting
that this epigenetic state in lupus patients is established in
progenitor cell populations. These cell type-specific effects are
consistent with the disease-specific changes in the composition of
the CD4+ population and suggest that shifts in the
proportion of CD4+ subtypes can be monitored at CpGs
with subtype-specific DNA methylation patterns (25). In the present study, we annotated
the corresponding CpG-methylated genes using the DAVID gene
annotation tool. However, immune-correlated process GO terms, such
as GO:0006955-immune response, and KEGG pathways, such as
hsa04660-T-cell receptor (TCR) signaling pathway exhibited no
significant enrichment. The GO analysis did reveal that the 'theme'
immune response (GO:0006955), which is known to be affected by
anti-TNF treatment in the inflammatory tissue of rheumatoid
arthritis patients, was significantly over-represented (26). Regardless, it is relevant to note
in our context that our GO analysis identified immune functions as
potentially relevant mechanisms. The activation of T lymphocytes is
a key event for an efficient response of the immune system
(hsa04660-TCR signaling pathway) and requires the involvement of
the TCR as well as costimulatory molecules, such as CD28. The
engagement of these receptors through interaction with a foreign
antigen is associated with MHC molecules (27), and our findings may thus
facilitate the selection of better target molecules for further
studies. The findings of the present study may also aid future
research by providing details of new pathways to be studied using a
more focused approach, confirmation at the protein level and
emphasis of the clinical significance.
lncRNAs are transcripts longer than ~200 nucleotides
with little or no protein-coding capacity (28). Research has shown that lncRNAs
play important roles in disease development and are associated with
a number of human diseases, such as cancer, Alzheimer's disease and
heart disease (29). T-UCR
transcripts are a novel class of lncRNAs transcribed from UCRs, a
class of 481 non-coding sequences located in both intra- and
intergenic regions of the genome. UCRs are absolutely conserved
(100%) between the orthologous regions of the human, rat, and mouse
genomes and are actively transcribed (30,31). It has recently been proven in
cancer systems that differentially expressed T-UCRs alter the
functional characteristics of malignant cells. Indeed, recent data
suggest that T-UCRs are altered at the transcriptional level in
human tumorigenesis and that the aberrant T-UCR expression profiles
can be used to differentiate human cancer types (31,32). Researchers observed that DNA
hypomethylation induces T-UCR silencing in cancer cells, and the
analysis of a large set of primary human tumors demonstrated that
the hypermethylation of the described T-UCR CpG islands is a common
event in the various tumor types (33). In the present study, we integrated
the MHC and T-UCR datasets and examined the expression levels of
T-UCR in the MHC segment by T-UCR microarray. We annotated the
T-UCR corresponding genes using the DAVID gene annotation tool.
However, no significant enrichment was found for immune-correlated
process GO terms and KEGG pathways. Thus, T-UCR expression levels
did not correlate with the commonly used clinicopathological
features of the patients with SLE.
Taken together, in the present study, we identified
6 of the most important SNPs (rs1050683, rs12697943, rs17881210,
rs1065378, rs17184255 and rs16895070) in patients with SLE. The
present study indicates that SNPs in the MHC segment are potential
biomarkers and are likely factors which are involved in the
pathogenesis of SLE. However, further studies are required to
investigate the mechanisms through which polymorphisms in this
region lead to the development of SLE. A major advantage of
combining multiple levels of measurement is the ability to dissect
mechanisms not apparent in a single dimension. The integration of
MHC, CpG methylation, and T-UCR datasets is a powerful strategy for
understanding SLE biology. Our findings provide insight into the
potential contribution of anomalously regulated SNPs to the
abnormalities in SLE and may aid in the structuring of antenatal
diagnostic biomarkers of SLE, as well as in obtaining novel
therapeutic targets which can be used in the treatment of patients
with SLE. Moreover, our study of SNPs may aid in the development of
novel methods which may prove to be useful for treating and
preventing other diseases.
Acknowledgments
The authors of this study would like to thank the
patients with SLE and the healthy volunteers who participated in
this study. Bioinformatics analysis was performed by Shanghai
Biotree Biotech Co., Ltd., Shanghai, China. The present study was
supported financially by the Key Project of Guangxi Natural Science
Foundation (no. 2012GXNSFDA053017), the Construction Project
Planning Assignment of Guangxi Key Laboratory (no. 13-051-31) and
the Scientific Problem Tackling of Guilin Science and Technology
Program (no. 20130120-20), China.
References
|
1
|
Mirkazemi S, Akbarian M, Jamshidi AR,
Mansouri R, Ghoroghi S, Salimi Y, Tahmasebi Z and Mahmoudi M:
Association of STAT4 rs7574865 with susceptibility to systemic
lupus erythematosus in Iranian population. Inflammation.
36:1548–1552. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Zhang J, Zhang Y, Yang J, Zhang L, Sun L,
Pan HF, Hirankarn N, Ying D, Zeng S, Lee TL, et al: Three SNPs in
chromosome 11q23.3 are independently associated with systemic lupus
erythematosus in Asians. Hum Mol Genet. 23:524–533. 2014.
View Article : Google Scholar
|
|
3
|
Barcellos LF, May SL, Ramsay PP, Quach HL,
Lane JA, Nititham J, Noble JA, Taylor KE, Quach DL, Chung SA, et
al: High-density SNP screening of the major histocompatibility
complex in systemic lupus erythematosus demonstrates strong
evidence for independent susceptibility regions. PLoS Genet.
5:e10006962009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fernando MM, Freudenberg J, Lee A, Morris
DL, Boteva L, Rhodes B, Gonzalez-Escribano MF, Lopez-Nevot MA,
Navarra SV, Gregersen PK, Martin J; IMAGEN; Vyse TJ: Transancestral
mapping of the MHC region in systemic lupus erythematosus
identifies new independent and interacting loci at MSH5, HLA-DPB1
and HLA-G. Ann Rheum Dis. 71:777–784. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Al-Motwee S, Jawdat D, Jehani GS, Anazi H,
Shubaili A, Sutton P, Uyar AF, Hajeer AH and AI-Motwee S:
Association of HLA-DRB1*15 and HLADQB1*06
with SLE in Saudis. Ann Saudi Med. 33:229–234. 2013.PubMed/NCBI
|
|
6
|
Ruiz-Narvaez EA, Fraser PA, Palmer JR,
Cupples LA, Reich D, Wang YA, Rioux JD and Rosenberg L: MHC region
and risk of systemic lupus erythematosus in African American women.
Hum Genet. 130:807–815. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Morris DL, Fernando MM, Taylor KE, Chung
SA, Nititham J, Alarcón-Riquelme ME, Barcellos LF, Behrens TW,
Cotsapas C, Gaffney PM, et al: Systemic Lupus Erythematosus
Genetics Consortium: MHC associations with clinical and
autoantibody manifestations in European SLE. Genes Immun.
15:210–217. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mantila Roosa SM, Turner CH and Liu Y:
Regulatory mechanisms in bone following mechanical loading. Gene
Regul Syst Bio. 6:43–53. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tan EM, Cohen AS, Fries JF, Masi AT,
McShane DJ, Rothfield NF, Schaller JG, Talal N and Winchester RJ:
The 1982 revised criteria for the classification of systemic lupus
erythematosus. Arthritis Rheum. 25:1271–1277. 1982. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hochberg MC: Updating the American College
of Rheumatology revised criteria for the classification of systemic
lupus erythematosus. Arthritis Rheum. 40:17251997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sui W, Tan Q, Yang M, Yan Q, Lin H, Ou M,
Xue W, Chen J, Zou T, Jing H, et al: Genome-wide analysis of 5-hmC
in the peripheral blood of systemic lupus erythematosus patients
using an hMeDIP-chip. Int J Mol Med. 35:1467–1479. 2015.PubMed/NCBI
|
|
12
|
Hussain N, Jaffery G, Sabri AN and Hasnain
S: HLA association in SLE patients from Lahore-Pakistan. Bosn J
Basic Med Sci. 11:20–26. 2011.PubMed/NCBI
|
|
13
|
International Consortium for Systemic
Lupus Erythematosus Genetics (SLEGEN); Harley JB, Alarcón-Riquelme
ME, Criswell LA, Jacob CO, Kimberly RP, Moser KL, Tsao BP, Vyse TJ,
Langefeld CD, et al: Genome-wide association scan in women with
systemic lupus erythematosus identifies susceptibility variants in
ITGAM, PXK, KIAA1542 and other loci. Nat Genet. 40:204–210. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Graham RR, Cotsapas C, Davies L, Hackett
R, Lessard CJ, Leon JM, Burtt NP, Guiducci C, Parkin M, Gates C, et
al: Genetic variants near TNFAIP3 on 6q23 are associated with
systemic lupus erythematosus. Nat Genet. 40:1059–1061. 2008.
View Article : Google Scholar
|
|
15
|
Chai HC, Phipps ME, Othman I, Tan LP and
Chua KH: HLA variants rs9271366 and rs9275328 are associated with
systemic lupus erythematosus susceptibility in Malays and Chinese.
Lupus. 22:198–204. 2013. View Article : Google Scholar
|
|
16
|
Morris DL, Taylor KE, Fernando MM,
Nititham J, Alarcón-Riquelme ME, Barcellos LF, Behrens TW, Cotsapas
C, Gaffney PM, Graham RR, et al International MHC and Autoimmunity
Genetics Network: Systemic Lupus Erythematosus Genetics Consortium:
Unraveling multiple MHC gene associations with systemic lupus
erythematosus: model choice indicates a role for HLA alleles and
non-HLA genes in Europeans. Am J Hum Genet. 91:778–793. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
van Manen D, Kootstra NA, Boeser-Nunnink
B, Handulle MA, van't Wout AB and Schuitemaker H: Association of
HLA-C and HCP5 gene regions with the clinical course of HIV-1
infection. AIDS. 23:19–28. 2009. View Article : Google Scholar
|
|
18
|
Trachtenberg E, Bhattacharya T, Ladner M,
Phair J, Erlich H and Wolinsky S: The HLA-B/-C haplotype block
contains major determinants for host control of HIV. Genes Immun.
10:673–677. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kurata R, Nakaoka H, Tajima A, Hosomichi
K, Shiina T, Meguro A, Mizuki N, Ohono S, Inoue I and Inoko H:
TRIM39 and RNF39 are associated with Behçet's disease independently
of HLA-B-51 and -A-26. Biochem Biophys Res Commun. 401:533–537.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Aissani B, Boehme AK, Wiener HW, Shrestha
S, Jacobson LP and Kaslow RA: SNP screening of central
MHC-identified HLA-DMB as a candidate susceptibility gene for
HIV-related Kaposi's sarcoma. Genes Immun. 15:424–429. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Glocker E, Ehl S and Grimbacher B: Common
variable immunodeficiency in children. Curr Opin Pediatr.
19:685–692. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hedrich CM, Crispin JC and Tsokos GC:
Epigenetic regulation of cytokine expression in systemic lupus
erythematosus with special focus on T cells. Autoimmunity.
47:234–241. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Lal G, Zhang N, van der Touw W, Ding Y, Ju
W, Bottinger EP, Reid SP, Levy DE and Bromberg JS: Epigenetic
regulation of Foxp3 expression in regulatory T cells by DNA
methylation. J Immunol. 182:259–273. 2009. View Article : Google Scholar
|
|
24
|
Sullivan KE, Suriano A, Dietzmann K, Lin
J, Goldman D and Petri MA: The TNF alpha locus is altered in
monocytes from patients with systemic lupus erythematosus. Clin
Immunol. 123:74–81. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Absher DM, Li X, Waite LL, Gibson A,
Roberts K, Edberg J, Chatham WW and Kimberly RP: Genome-wide DNA
methylation analysis of systemic lupus erythematosus reveals
persistent hypomethylation of interferon genes and compositional
changes to CD4+ T-cell populations. PLoS Genet.
9:e10036782013. View Article : Google Scholar
|
|
26
|
Lindberg J, af Klint E, Catrina AI,
Nilsson P, Klareskog L, Ulfgren AK and Lundeberg J: Effect of
infliximab on mRNA expression profiles in synovial tissue of
rheumatoid arthritis patients. Arthritis Res Ther. 8:R1792006.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Diehn M, Alizadeh AA, Rando OJ, Liu CL,
Stankunas K, Botstein D, Crabtree GR and Brown PO: Genomic
expression programs and the integration of the CD28 costimulatory
signal in T cell activation. Proc Natl Acad Sci USA.
99:11796–11801. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yan B, Tao ZF, Li XM, Zhang H, Yao J and
Jiang Q: Aberrant expression of long noncoding RNAs in early
diabetic retinopathy. Invest Ophthalmol Vis Sci. 55:941–951. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Haemmerle M and Gutschner T: Long
non-coding RNAs in cancer and development: where do we go from
here? Int J Mol Sci. 16:1395–1405. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Scaruffi P, Stigliani S, Coco S, Valdora
F, De Vecchi C, Bonassi S and Tonini GP: Transcribed-ultra
conserved region expression profiling from low-input total RNA. BMC
Genomics. 11:1492010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Peng JC, Shen J and Ran ZH: Transcribed
ultraconserved region in human cancers. RNA Biol. 10:1771–1777.
2013. View Article : Google Scholar
|
|
32
|
Sana J, Hankeova S, Svoboda M, Kiss I,
Vyzula R and Slaby O: Expression levels of transcribed
ultraconserved regions uc.73 and uc.388 are altered in colorectal
cancer. Oncology. 82:114–118. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lujambio A, Portela A, Liz J, Melo SA,
Rossi S, Spizzo R, Croce CM, Calin GA and Esteller M: CpG island
hypermethylation-associated silencing of non-coding RNAs
transcribed from ultraconserved regions in human cancer. Oncogene.
29:6390–6401. 2010. View Article : Google Scholar : PubMed/NCBI
|