Introduction
Obesity is defined by the excessive formation of
adipose tissue, and thus an understanding of the molecular
mechanism behind adipose tissue formation, i.e., adipogenesis, is
necessary to find ways of preventing and treating obesity and
obesity-related diseases including type 2 diabetes, hyperlipidemia
and cardiovascular disease (1).
Adipogenesis is mediated by the coordinated expression of the genes
involved in adipocyte differentiation and intracellular fat
accumulation (2–4). The 3T3-L1 cell line has been widely
used as an in vitro model of adipogenesis, and the treating
these cells with 3-isobutyl-1-methylxanthine, dexamethasone and
insulin is known to upregulate the major transcription factors of
adipogenesis, peroxisome proliferator-activated receptor γ (PPARγ)
and CCAAT/enhancer-binding protein α (C/EBPα), which directly
transcribe various adipocyte marker genes, including fatty
acid-binding protein (FABP)4 (2–4).
AMP-activated protein kinase (AMPK) is a major
regulator of cellular energy homeostasis (5), and it regulates carbohydrate and fat
metabolism in order to maintain the cellular energy balance
(6–8). AMPK is activated by the increased
ratio of AMP/ATP, and it is known to phosphorylate various target
proteins involved in cell growth and metabolism (9). Dysregulation of AMPK is involved in
various disorders including metabolic diseases, cardiovascular
disease, cancer and dementia (10). Recently, Li et al reported
that sterol regulatory element binding protein (SREBP)1c was one of
the target proteins directly phosphorylated by AMPK (11). AMPK was found to phosphorylate the
Ser372 residue of SREBP-1c, which inhibited the proteolytic
cleavage of the precursor form of SREBP-1c (precursor SREBP-1c)
into mature SREBP-1c, resulting in the inhibition of hepatic
steatosis in insulin-resistant mice. SREBP-1c has also been
identified as one of the transcription factors involved in
adipogenesis (12). However, to
the best of our knowledge, AMPK-mediated modulation of SREBP-1c has
never been suggested to be the anti-adipogenic mechanism of any
compound which inhibits adipogenesis.
Shikonin is a major chemical ingredients of
Lithospermum erythrorhizon, a therapeutic plant used for the
treatment of macular eruption, measles, sore-throat, carbuncles and
burns (13). Lithospermum
erythrorhizon contains various shikonin compounds, including
shikonin, acetylshikonin, isobutyrylshikonin and
β-hydroxyisovalerylshikonin (β-HIVS) (14). In the present study, we
demonstrated the anti-adipogenic effect of β-HIVS and elucidated
its molecular mechanisms. It was found that β-HIVS activated AMPK,
resulting in the increased phosphorylation of SREBP-1c, preventing
its proteolytic maturation. This was followed by the reduced
expression of fat-forming enzymes, including acetyl-CoA carboxylase
(ACC)1, fatty acid synthase (FAS) and stearoyl-CoA desaturase
(SCD)1, which resulted in reduced intracellular fat
accumulation.
Materials and methods
Chemicals and reagents
The cell culture reagents, Dulbecco's modified
Eagle's medium (DMEM), fetal bovine serum (FBS) and
penicillin/streptomycin, were all obtained from Life Technologies
(Grand Island, NY, USA). Anti-PPARγ (#2430), anti-FABP4 (#2120),
anti-p-AMPK (#2535), anti-AMPK (#2603), anti-FABP4 (#2120) and
anti-p-SREBP1c (#9847) antibodies, and anti-mouse
(#7076S)/anti-rabbit (#7074) secondary antibody were all purchased
from Cell Signaling Technology (Beverly, MA, USA). Anti-C/EBPα
(sc-61) and anti-β-actin (sc-47778) antibodies were all purchased
from Santa Cruz Biotechnology, Inc (Santa Cruz, CA, USA).
Anti-SREBP1c (557036) was purchased from BD science (Franklin
Lakes, NJ, USA). AMPK small interfering (si)RNA and control siRNA
were both purchased from Santa Cruz Biotechnology, Inc.
Lipofectamine RNAiMAX transfection reagent was purchased from
Invitrogen (Carlsbad, CA, USA). Shikonin, acetylshikonin,
isobutyrylshikonin and β-HIVS were all purchased from Wako Pure
Chemical Industries, Ltd. (Osaka, Japan). All other chemicals were
from Sigma-Aldrich (St. Louis, MO, USA).
Cell culture
The 3T3-L1 preadipocytes were purchased from the
American Type Culture Collection (ATCC; Manassas, VA, USA) and
subcultured every 2 days. The cells were seeded in 6-well plates at
a density of 1.5×105 cells/well. Two days after reaching
confluence (day 0), the 3T3-L1 cells were differentiated in a
differentiation-induction medium containing DMEM, supplemented with
1 µg/ml insulin, 0.25 µM dexamethasone, 0.5 mM
3-isobutyl-1-methylxanthine and 10% FBS, for 2 days. The cells were
then maintained in a differentiation-maintenance medium containing
DMEM, supplemented with 1 µg/ml insulin and 10% FBS. The
differentiation-maintenance medium was replaced every 2 days until
the cells were harvested.
Oil Red O staining of intracellular
fat
The 3T3-L1 cells were seeded in 12-well plates at a
density of 7.5×104 cells/well. After differentiation for
7 days, the cells were washed with phosphate-buffered saline (PBS),
and incubated for 4 h in 4% paraformaldehyde (Sigma-Aldrich). The
cells were then washed with distilled water and stained with 0.3%
Oil Red O (Sigma-Aldrich) solution for 1 h. After washing them 3
times with distilled water, the cells were completely dried and
observed under an inverted microscope (Olympus, Tokyo, Japan).
Photographic images were captured and then 100% isopropanol
(Sigma-Aldrich) was added to each well to extract the stained Oil
Red O dye. The contents of the extracted dye were measured at 540
nm using a spectrophotometer (BioTek Instruments, Inc., Winooski,
VT, USA).
Cell viability assay
Cell viability was determined using a CellCountEZ™
Cell Survival assay kit (purchased from Rockland Immunochemicals,
Limerick, PA, USA), as previously described (15); viability was determined based on
the ability of viable mammalian cells to convert hydroxyethyl
disulfide into mercaptoethanol. The amount of mercaptoethanol
produced from hydroxyethyl disulfide can be measured in the
extracellular culture media since mercaptoethanol produced inside
cells is extruded quickly by cells through an active transport
mechanism.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
The 3T3-L1 cells were seeded into 12-well plates at
a density of 7.5×104 cells/well, and, after
differentiation, total RNA was extracted using an RNeasy kit
(Qiagen, Hilden, Germany). One microgram of total RNA was reverse
transcribed at 37°C using the cDNA reverse transcription kit
(Applied Biosystems, Inc., Foster City, CA, USA). qPCR was
performed using the 7000 Real-Time PCR System (Applied Biosystems)
in a final volume of 20 µl, which included the TaqMan Gene
Expression Master Mix, 250 nM of TaqMan probe, an optimized
concentration of each primer and 1 µl of the reverse
transcription product containing cDNA. The reaction mixtures were
preheated at 95°C for 10 min to activate the enzyme and then
subjected to 40 cycles of melting at 95°C for 15 sec and
annealing/extension at 60°C for 1 min. The efficiency of RT-qPCR
was approximately 100%. The Assays-on-Demand gene expression
products (Applied Biosystems) were used to evaluate the mRNA levels
of PPARγ (Mm00440945_m1), C/EBPα (Mm01265914_s1), SREBP-1c
(Mm00550338_m1), FABP4 (Mm00445880_m1), FAS (Mm01253292_m1), ACC1
(Mm01304257_m1), SCD1 (Mm00772290_m1) as well as the level of 18S
rRNA (Hs99999901_s1). 18S rRNA was used as an internal control, as
previously described (16). For
each sample, the mRNA level was normalized against the level of 18S
rRNA, and the ratio of normalized mRNA in each sample to that in
the control sample was determined using the comparative Ct method
(17).
Protein extraction and western blot
analysis
Cells were harvested using a cell scraper in
ice-cold PBS, and lysed with RIPA buffer containing 25 mM Tris-HCl
(pH 7.6), 150 mM NaCl, 1% Nonidet P-40, 1% sodium deoxycholate,
0.1% SDS and a protease inhibitor cocktail (Sigma-Aldrich) for 30
min at 4°C. The total cell lysates were then obtained after
centrifuging at 20,000 × g for 20 min at 4°C to remove the
insoluble materials. The protein concentrations were determined
using a BCA protein assay kit (Pierce, Rockford, IL, USA). Twenty
micrograms of protein were separated using 10% polyacrylamide gel
electrophoresis and electrotransferred onto nitrocellulose
membranes at 180 mA for 1 h. The membranes were then blocked for 2
h at room temperature with Tris-buffered saline (TBS) containing 5%
skim milk. Blocked nitrocellulose membranes were incubated with TBS
containing 0.1% Tween-20 and a 1:1,000-dilution of primary antibody
overnight at 4°C followed by a 1:1,000-dilution of horseradish
peroxidase-conjugated secondary antibody for 1 h at room
temperature. Peroxidase activity was visualized using an ECL kit
(Pierce).
Transfection with siRNA
Two days after reaching confluence, 3T3-L1 cells
were incubated in serum-free medium for 1 h and then transfected
with 60 nM of AMPK siRNA or 60 nM of control siRNA using
Lipofectamine RNAiMAX transfection reagent. Six hours later, the
transfected cells were differentiated by replacing the medium with
differentiation-induction medium. Total RNA and protein were
extracted for RT-qPCR and western blot analysis, respectively.
Analysis of nuclear SREBP-1c levels
In the present study, the cells were harvested using
cell scrapers, and the nuclear extracts were prepared using a
nuclear extract kit (Active Motif, Carlsbad, CA, USA). Protein
concentrations in the nuclear extracts were determined using a BCA
protein assay kit (Pierce). Twenty micrograms of nuclear protein
were separated using 10% polyacrylamide gel electrophoresis and
analyzed by western blot analysis using an anti-SREBP-1c antibody
followed by secondary antibody.
Statistical analyses
All data are represented as the means ± standard
deviation (SD) of at least three replicated experiments.
Statistically significant differences between treated and untreated
samples were detected using unpaired t-tests. All analyses were
performed using SPSS v. 14 (SPSS, Inc., Chicago, IL, USA). A
P-value <0.05 was considered to indicate a statistically
significant difference.
Results
Comparison of the anti-adipogenic effect
of various shikonin compounds
It has previously been reported that Lithospermum
erythrorhizon contains various shikonin compounds, including
shikonin, acetylshikonin, isobutyrylshikonin and
β-hydroxy-isovalerylshikonin (β-HIVS) (14). These shikonin compounds have the
common basic structure of naphthoquinone, while the attached
chemical groups are slightly different (Fig. 1A). To determine which compound
exerted the greatest anti-adipogenic effect, the 3T3-L1 cells were
treated with the four shikonin compounds at various concentrations
for 7 days. Analysis of the effects on intracellular fat content
revealed that isobutyrylshikonin and β-HIVS exerted a greater
anti-adipogenic effect than shikonin and acetylshikonin (Fig. 1B). However, the intracellular fat
content of 3T3-L1 cells treated with isobutyrylshikonin or β-HIVS
for 7 days were similar. Thus, in the subsequent experiment, 3T3-L1
cells were treated with isobutyrylshikonin or β-HIVS on days 0–2,
3–4 and 4–7 of adipogenic differentiation (Fig. 2). The anti-adipogenic effect of
β-HIVS was found to be slightly greater than that of
isobutyrylshikonin (Fig. 2C and
D). These data indicate that β-HIVS exerted the greatest
anti-adipogenic effect among the shikonin compounds, and further
studies were therefore conducted using β-HIVS.
Anti-adipogenic effect of β-HIVS
In order to determine the most effective
concentration of β-HIVS required for anti-adipogenic activity, the
3T3-L1 cells were treated with β-HIVS at various concentrations
(0.25, 0.5, 1, 1.5 and 2 µM) for 7 days. The Oil Red O
staining revealed that intracellular fat droplet formation was
inhibited by β-HIVS in a dose-dependent manner, and that β-HIVS
markedly inhibited fat droplet formation at a concentration of 2
µM, almost to the control level (Fig. 3A and B). Cytotoxicity was not
observed when the 3T3-L1 cells were treated with β-HIVS at
concentrations of up to 2 µM for 7 days, and cell viability
was maintained at >90% of the untreated control cells (Fig. 3C), indicating that the inhibitory
effects of β-HIVS on fat accumulation were not mediated by
cytotoxicity. Further experiments were conducted with β-HIVS at a
concentration of 2 µM.
During adipogenesis of the 3T3-L1 cells, the mRNA
expression of the fat-forming enzymes, ACC1, FAS and SCD1, was
upregulated, and this effect was significantly reduced by β-HIVS
treatment (Fig. 4A–C). The major
transcription factors of adipogenesis, PPARγ and C/EBPα, which are
markedly upregulated during adipogenesis and are involved in the
transcription of adipocyte marker genes including FABP4, were also
significantly reduced by β-HIVS treatment (Fig. 4D–F).
 | Figure 4Effects of β-hydroxyisovalerylshikonin
(β-HIVS) on the expression of fat-forming enzymes and
adipocyte-specific genes. (A–C) Effects of 2 µM of β-HIVS on
the mRNA expression of fat-forming enzymes, acetyl-CoA carboxylase
(ACC)1, fatty acid synthase (FAS) and stearoyl-CoA desaturase
(SCD)1, in 3T3-L1 cells differentiated for 0, 2, 4 and 7 days.
(D–F) Effects of 2 µM of β-HIVS on the mRNA and protein
expression of adipocyte-specific genes such as major adipogenic
transcription factors, peroxisome proliferator-activated receptor γ
(PPARγ) and CCAAT/enhancer-binding protein α (C/EBPα), and an
adipocyte marker gene, fatty acid-binding protein (FABP)4, in
3T3-L1 cells differentiated for 0, 2, 4 and 7 days.
*P<0.05, **P<0.01,
***P<0.001 compared with the untreated adipocytes on
the same differentiation day. |
It is noteworthy that ACC1, FAS and SCD1 have been
reported to be transcribed by a transcription factor, SREBP-1c. It
has also been reported that the mRNA expression of SCD1 is induced
by the binding of nuclear SREBP-1c to its promoter (18), and SREBP-1c is the transcription
factor that binds to a regulatory element in the enhancer of the
FAS gene (19). The level of ACC1
mRNA is also controlled by the binding of SREBP-1c to the ACC1
promoter (19,20). It has also been reported that
SREBP-1c induces the expression of PPARγ, which collaborates with
C/EBPα to transcribe adipocyte marker genes such as FABP4 (21).
Effects of β-HIVS on AMPK activation and
SREBP-1c downregulation
AMPK is known to suppress adipogenesis when it is
activated by phosphorylation (22). To elucidate the association
between β-HIVS and the activatation of AMPK, we determined whether
β-HIVS induces AMPK phosphorylation during adipogenesis, and it was
found that the p-AMPK level was markedly upregulated in the
β-HIVS-treated cells compared with the untreated cells. The total
AMPK protein level was almost unchanged, demonstrating that β-HIVS
induced the phosphorylation of AMPK without affecting its total
protein level (Fig. 5A).
5-Aminoimidazole-4-carbox-amide-1-β-dribofuranoside (AICAR) is the
most well-known activator of AMPK, which activates AMPK by
increasing its phosphorylation, and it was thus necessary to
compare the AMPK-activating effect of β-HIVS with AICAR (23). The levels of p-AMPK and
p-precursor-SREBP-1c, which is one of the products made by p-AMPK,
were measured in order to compare the AMPK-activating effect of
AICAR and β-HIVS at various concentrations (AICAR: 25, 150, 250 and
500 mM vs. β-HIVS: 0.1, 0.5, 1 and 2 µM). The results
revealed that the levels of p-AMPK and p-precursor-SREBP-1c were
increased by AICAR and β-HIVS (Fig.
5B). However, β-HIVS increased the levels of p-AMPK and
p-precursor-SREBP-1c at a much lower concentration, <1/100,000,
compared with AICAR, demonstrating that β-HIVS is a much more
effective AMPK activator compared with AICAR.
Li et al previously reported that
phosphorylated/activated AMPK directly phosphorylates precursor
SREBP-1c at the Ser372 residue, which prevents the proteolytic
maturation of precuser SREBP-1c into mature SREBP-1c and the
translocation of mature SREBP-1c into the nucleus (11). When the 3T3-L1 cells were treated
with β-HIVS for 6, 12, 24 and 48 h, the level of
p-precursor-SREBP-1c was increased similarly to the p-AMPK level.
As a result, the level of precursor SREBP-1c (unphosphorylated) and
the proteolytic cleavage of precurser SREBP-1c (unphosphorylated)
to mature-SREBP-1c was decreased by β-HIVS treatment. Furthermore,
the level of nuclear mature SREBP-1c was also decreased, as the
phosphorylation of Ser372 prevented nuclear translocation of
SREBP-1c (Fig. 5C). When the
3T3-L1 cells were treated with β-HIVS for longer periods of time,
of 2, 4 and 7 days, similar patterns of change were observed in the
phosphorylation, proteolytic maturation and nuclear translocation
of SREBP-1c. The level of precursor SREBP-1c (unphosphorylated) and
its proteolytic cleavage into mature SREBP-1c, followed by its
nuclear translocation, were reduced by the increase of p-precursor
SREBP-1c in the β-HIVS-treated cells compared with the untreated
cells (Fig. 5D).
Effects of AMPK knockdown on the
anti-adipogenic effect of β-HIVS
To confirm the essential role which AMPK plays in
the anti-adipogenic mechanism of β-HIVS, we performed
siRNA-mediated knockdown of AMPK in 3T3-L1 cells in the presence or
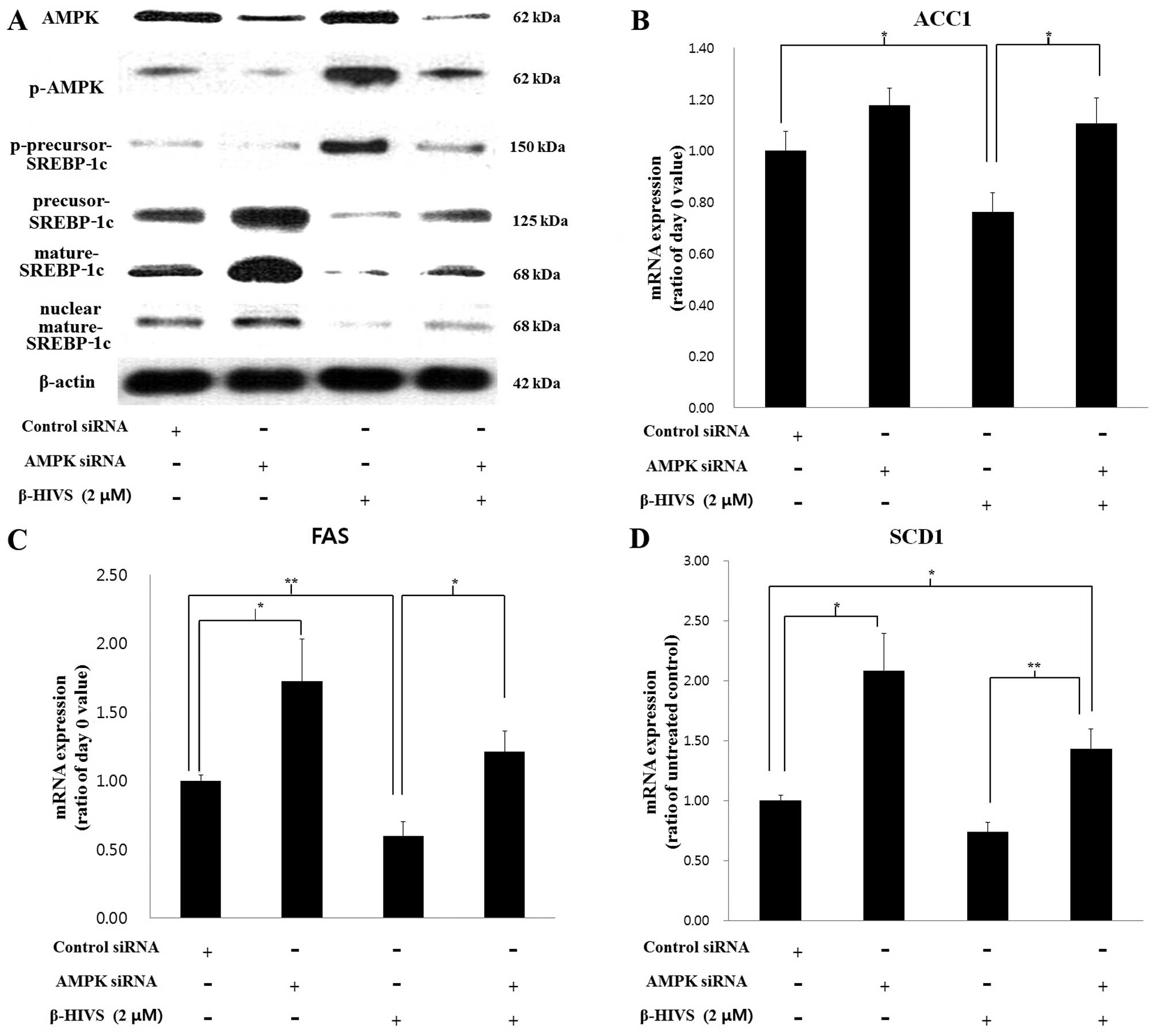
absence of β-HIVS. The siRNA-mediated knockdown of AMPK reduced the
levels of AMPK and p-AMPK. We noted that p-precursor-SREBP-1c,
which is a product made by p-AMPK, was also reduced by AMPK
knockdown. As a result, levels of precursor SREBP-1c
(unphosphorylated), mature SREBP-1c and nuclear mature SREBP-1c
were increased by AMPK knockdown (Fig. 6A). Mature SREBP-1c is a
transcription factor required for the expression of the fat-forming
enzymes ACC1, FAS and SCD1, the levels of which were all
significantly increased by AMPK siRNA compared to transfection with
control siRNA (Fig. 6B–D).
SREBP-1c is also known to induce self mRNA transcription by binding
to its own promoter (24,25). In the present study, it was also
found that the SREBP-1c mRNA level was increased by AMPK knockdown
in β-HIVS-treated cells, possibly through the reduction in SREBP-1c
phosphorylation and increase in mature SREBP-1c level (data not
shown).
We noted that as a result of the effects of AMPK
knockdown on the expression of the fat-forming enzymes,
intracellular fat accumulation, which had been suppressed by the
anti-adipogenic activity of β-HIVS, was significantly recovered by
AMPK siRNA compared to transfection with the control siRNA
(Fig. 7A and B). AMPK knockdown
also reduced the effect of β-HIVS on the expression level of the
major adipogenic transcription factors, PPARγ and C/EBPα, and an
adipocyte marker gene, FABP4, transcribed by them. The mRNA and
protein levels of PPARγ, C/EBPα and FABP4, which had been
suppressed by β-HIVS, were significantly increased by AMPK
knockdown (Fig. 7C–E).
The results of the present study demonstrated that
AMPK is involved in the anti-adipogenic mechanism of β-HIVS through
the modulation of SREBP-1c phosphorylation, maturation and nuclear
translocation as well as the transcription of downstream
fat-forming enzymes and adipogenic transcription factors. A
possible molecular mechanism for the anti-adipogenic activity of
β-HIVS and the effects of AMPK knockdown are described in Fig. 8.
 | Figure 8Molecular mechanisms for the
anti-adipogenic effect of β-hydroxyisovalerylshikonin (β-HIVS) and
effect of AMP-activated protein kinase (AMPK) knockdown. (A) β-HIVS
activates AMPK by increasing its phosphorylation. Phosphorylated
(p-)AMPK directly phosphorylates precursor-sterol regulatory
element binding protein (SREBP)-1c into p-precursor SREBP-1c, which
cannot be cleaved into mature SREBP-1c, and the level of mature
SREBP-1c level decreases. A decreased level of mature SREBP-1c,
which is a transcription factor required for the expression of
acetyl-CoA carboxylase (ACC)1, fatty acid synthase (FAS) and
stearoyl-CoA desaturase (SCD)1 and peroxisome
proliferator-activated receptor γ (PPARγ), leads to reduced
intracellular fat accumulation. (B) AMPK siRNA reduces the levels
of AMPK and p-AMPK, which leads to the reduced phosphorylation of
precursor SREBP-1c and an increased level of unphosphorylated
precursor SREBP-1c. Unphosphorylated precursor SREBP-1c is cleaved
into mature SREBP-1c, which is a transcription factor required for
the expression of ACC1, FAS, SCD1 and PPARγ, leads to increased
intracellular fat accumulation. |
Discussion
A number of natural compounds have the potential to
exert anti-obesity effects through the inhibition of adipogenesis
(26). As previously noted,
β-HIVS is one of the natural shikonin compounds contained in a
therapeutic plant, Lithospermum erythrorhizon (14). In the present study, modulations
of AMPK and SREBP-1c were analyzed in relation to the
anti-adipogenic mechanisms of β-HIVS, resulting in novel findings.
Firstly, we noted that β-HIVS exerted the greatest anti-adipogenic
effect of the four shikonin compounds. Secondly, β-HIVS effectively
activated AMPK by increasing its phosphorylation at a much lower
concentration, <1/100,000, compared with AICAR, a well-known
AMPK activator. Thirdly, β-HIVS increased the phosphorylation of
precursor SREBP-1c through the activation of AMPK, which prevented
the cleavage of precursor SREBP-1c into mature SREBP-1c and its
nuclear translocation. Accordingly, the expression of genes
transcribed by mature SREBP-1c, which included fat-forming enzymes
and an adipogenic transcription factor, were decreased, resulting
in reduced intracellular fat accumulation. Finally, knockdown of
AMPK attenuated the anti-adipogenic activity of β-HIVS, including
the expression of fat-forming enzymes and an adipogenic
transcription factor as well as intracellular fat accumulation,
through the modulation of SREBP-1c.
AMPK is a serine/threonine kinase expressed in
various tissues such as the skeletal muscle, liver and adipose
tissue (27), and it regulates
metabolic processes by upregulating catabolism and downregulating
anabolism of lipid and carbohydrate (6–8).
SREBPs are a family of transcription factors that regulate lipid
homeostasis by controlling the expression of enzymes required for
the synthesis of endogenous cholesterol, fatty acids,
triacylglycerols and phospholipids. Accordingly, SREBPs have been
identified as a novel therapeutic target for metabolic diseases
such as insulin resistance, type 2 diabetes, fatty liver and
atherosclerosis (28). The three
SREBP isoforms, SREBP1a, SREBP-1c and SREBP2, play different roles
in lipid metabolism. SREBP-1c is involved in fatty acid synthesis
and lipogenesis, whereas SREBP2 and SREBP1a are mainly involved in
cholesterol synthesis. SREBPs are synthesized as inactive
precursors, and upon activation, the precursors undergo proteolytic
cleavage to release the mature SREBPs into the nucleus (29). A previous study demonstrated
AMPK-SREBP-1c modulation by revealing that activated AMPK directly
phosphorylates precursor SREBP-1c at the Ser372 residue, which
suppresses the proteolytic cleavage of precursor SREBP-1c into
mature SREBP-1c and inhibits its nuclear translocation (11).
Many phytochemicals, including resveratrol,
epigallocatechin gallate, berberine, quercetin, rutaecarpine
analogues and ursolic acid, have previously been reported to
inhibit adipogenesis by activating AMPK but the molecular
mechanisms downstream of AMPK have not been fully elucidated
(30–32). It is noteworthy that until the
present study, AMPK-SREBP-1c modulation has never been suggested as
the anti-adipogenic mechanism of any compound which inhibits
adipogenesis, to the best of our knowledge. In the present study,
the detailed molecular mechanism for the anti-adipogenic activity
of β-HIVS, which was found to be an efficient AMPK activator, was
elucidated. β-HIVS activated AMPK to increase the phosphorylation
of precursor SREBP-1c, which inhibited the formation of mature
SREBP-1c, necessary for the transcription of fat-forming enzymes as
well as a major transcription factor of adipogenesis.
Acknowledgments
The present study was supported by the Biomedical
Science, Department of Medicine Research Scholarship Grants, of
Chung-Ang University in 2014.
Abbreviations:
|
ACC
|
acetyl-CoA carboxylase
|
|
AMPK
|
AMP-activated protein kinase
|
|
C/EBPα
|
CCAAT/enhancer-binding protein α
|
|
FABP
|
fatty acid-binding protein
|
|
FAS
|
fatty acid synthase
|
|
β-HIVS
|
β-hydroxyisovalerylshikonin
|
|
PPARγ
|
peroxisome proliferator-activated
receptor γ
|
|
SCD
|
stearoyl-CoA desaturase
|
|
SREBP
|
sterol regulatory element binding
protein
|
References
|
1
|
Grundy SM: Obesity, metabolic syndrome,
and cardiovascular disease. J Clin Endocrinol Metab. 89:2595–2600.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Farmer SR: Transcriptional control of
adipocyte formation. Cell Metab. 4:263–273. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rosen ED and MacDougald OA: Adipocyte
differentiation from the inside out. Nat Rev Mol Cell Biol.
7:885–896. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rosen ED, Walkey CJ, Puigserver P and
Spiegelman BM: Transcriptional regulation of adipogenesis. Genes
Dev. 14:1293–1307. 2000.PubMed/NCBI
|
|
5
|
Hardie DG, Ross FA and Hawley SA: AMPK: a
nutrient and energy sensor that maintains energy homeostasis. Nat
Rev Mol Cell Biol. 13:251–262. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Makinde AO, Gamble J and Lopaschuk GD:
Upregulation of 5′-AMP-activated protein kinase is responsible for
the increase in myocardial fatty acid oxidation rates following
birth in the newborn rabbit. Circ Res. 80:482–489. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ojuka EO, Jones TE, Nolte LA, Chen M,
Wamhoff BR, Sturek M and Holloszy JO: Regulation of GLUT4
biogenesis in muscle: evidence for involvement of AMPK and
Ca2+. Am J Physiol Endocrinol Metab. 282:E1008–E1013.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fazakerley DJ, Holman GD, Marley A, James
DE, Stöckli J and Coster ACF: Kinetic evidence for unique
regulation of GLUT4 trafficking by insulin and AMP-activated
protein kinase activators in L6 myotubes. J Biol Chem.
285:1653–1660. 2010. View Article : Google Scholar :
|
|
9
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth, autophagy and
metabolism. Nat Cell Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Steinberg GR and Kemp BE: AMPK in health
and disease. Physiol Rev. 89:1025–1078. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Li Y, Xu S, Mihaylova MM, Zheng B, Hou X,
Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al: AMPK
phosphorylates and inhibits SREBP activity to attenuate hepatic
steatosis and atherosclerosis in diet-induced insulin-resistant
mice. Cell Metab. 13:376–388. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kim JB, Wright HM, Wright M and Spiegelman
BM: ADD1/SREBP1 activates PPARgamma through the production of
endogenous ligand. Proc Natl Acad Sci USA. 95:4333–4337. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen X, Yang L, Oppenheim JJ and Howard
MZ: Cellular pharmacology studies of shikonin derivatives.
Phytother Res. 16:199–209. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ito Y, Onobori K, Yamazaki T and Kawamura
Y: Tigloylshikonin, a new minor Shikonin derivative, from the roots
and the commercial root extract of Lithospermum erythrorhizon. Chem
Pharm Bull (Tokyo). 59:117–119. 2011. View Article : Google Scholar
|
|
15
|
Li J, Zhang D, Ward KM, Prendergast GC and
Ayene IS: Hydroxyethyl disulfide as an efficient metabolic assay
for cell viability in vitro. Toxicol In Vitro. 26:603–612. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gustafson B and Smith U: Cytokines promote
Wnt signaling and inflammation and impair the normal
differentiation and lipid accumulation in 3T3-L1 preadipocytes. J
Biol Chem. 281:9507–9516. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
18
|
Paton CM and Ntambi JM: Biochemical and
physiological function of stearoyl-CoA desaturase. Am J Physiol
Endocrinol Metab. 297:E28–E37. 2009. View Article : Google Scholar :
|
|
19
|
Shimomura I, Bashmakov Y, Ikemoto S,
Horton JD, Brown MS and Goldstein JL: Insulin selectively increases
SREBP-1c mRNA in the livers of rats with streptozotocin-induced
diabetes. Proc Natl Acad Sci USA. 96:13656–13661. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Magaña MM, Lin SS, Dooley KA and Osborne
TF: Sterol regulation of acetyl coenzyme A carboxylase promoter
requires two interdependent binding sites for sterol regulatory
element binding proteins. J Lipid Res. 38:1630–1638.
1997.PubMed/NCBI
|
|
21
|
Fajas L, Schoonjans K, Gelman L, Kim JB,
Najib J, Martin G, Fruchart JC, Briggs M, Spiegelman BM and Auwerx
J: Regulation of peroxisome proliferator-activated receptor gamma
expression by adipocyte differentiation and determination factor
1/sterol regulatory element binding protein 1: implications for
adipocyte differentiation and metabolism. Mol Cell Biol.
19:5495–5503. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bijland S, Mancini SJ and Salt IP: Role of
AMP-activated protein kinase in adipose tissue metabolism and
inflammation. Clin Sci (Lond). 124:491–507. 2013. View Article : Google Scholar
|
|
23
|
Lee H, Kang R, Bae S and Yoon Y: AICAR, an
activator of AMPK, inhibits adipogenesis via the WNT/β-catenin
pathway in 3T3-L1 adipocytes. Int J Mol Med. 28:65–71.
2011.PubMed/NCBI
|
|
24
|
Zhang C, Shin DJ and Osborne TF: A simple
promoter containing two Sp1 sites controls the expression of
sterol-regulatory-element-binding protein 1a (SREBP-1a). Biochem J.
386:161–168. 2005. View Article : Google Scholar :
|
|
25
|
Amemiya-Kudo M, Shimano H, Yoshikawa T,
Yahagi N, Hasty AH, Okazaki H, Tamura Y, Shionoiri F, Iizuka Y,
Ohashi K, et al: Promoter analysis of the mouse sterol regulatory
element-binding protein-1c gene. J Biol Chem. 275:31078–31085.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rayalam S, Della-Fera MA and Baile CA:
Phytochemicals and regulation of the adipocyte life cycle. J Nutr
Biochem. 19:717–726. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Shirwany NA and Zou MH: AMPK in
cardiovascular health and disease. Acta Pharmacol Sin.
31:1075–1084. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xiao X and Song BL: SREBP: a novel
therapeutic target. Acta Biochim Biophys Sin (Shanghai). 45:2–10.
2013. View Article : Google Scholar
|
|
29
|
Eberlé D, Hegarty B, Bossard P, Ferré P
and Foufelle F: SREBP transcription factors: master regulators of
lipid homeostasis. Biochimie. 86:839–848. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Hwang JT, Kwon DY and Yoon SH:
AMP-activated protein kinase: A potential target for the diseases
prevention by natural occurring polyphenols. N Biotechnol.
26:17–22. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Chen YC, Zeng XY, He Y, Liu H, Wang B,
Zhou H, Chen JW, Liu PQ, Gu LQ, Ye JM and Huang ZS: Rutaecarpine
analogues reduce lipid accumulation in adipocytes via inhibiting
adipogenesis/lipogenesis with AMPK activation and UPR suppression.
ACS Chem Biol. 8:2301–2311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
He Y, Li Y, Zhao T, Wang Y and Sun C:
Ursolic acid inhibits adipogenesis in 3T3-L1 adipocytes through
LKB1/AMPK pathway. PLoS One. 8:e701352013. View Article : Google Scholar : PubMed/NCBI
|