Introduction
Vitamin D and its active metabolites play a major
role in bone mineral metabolism. The prevalence of vitamin D
deficiency is high worldwide, with latitude and socioeconomic
status as major determinants (1).
Subclinical vitamin D deficiency is associated with the development
of osteoporosis and an increased risk of falls and fractures in
older patients (2,3). In addition, vitamin D deficiency is
associated with an increased risk of cardiovascular disease,
diabetes, colon and breast cancer, infectious diseases and
allergies (4). Meta-analyses of
data have demonstrated that vitamin D supplementation may have
beneficial effects on bone health in older individuals and on
mortality in adults; however, evidence for the effectiveness of
vitamin D supplementation in cancer prevention is contradictory
(5,6). In animal studies, vitamin D therapy
has been shown to prevent progressive cardiac hypertrophy and the
development of heart failure (7,8).
However, results regarding the benefits of vitamin D
supplementation in cardiovascular disease in humans remain
controversial (9). Therefore,
further investigation is warranted into the effects of vitamin D on
the cardiovascular system.
Vascular alterations in sepsis are an important
pathophysiological mechanism that is characterized by the
dysregulation of vasoreactive mediators, increased vascular
permeability, leukocyte recruitment, unregulated expression of cell
adhesion molecules and abnormal hemostasis (10). Sepsis also induces myocardial
depression through microcirculatory alterations, autonomic
dysregulation, metabolic and mitochondrial dysfunction, cell death,
and myocardial inflammation (11). These vascular alterations in
combination with myocardial dysfunction lead to high mortality and
morbidity in patients with sepsis (11).
Pharmacological intervention for the prevention and
treatment of sepsis is challenging due to limitations to the
development of novel drugs, and a need for multidisciplinary
approaches to managing patients with sepsis. Targeting vascular
alterations would provide novel therapeutic strategies for the
treatment of sepsis. Previously, we reported that the blockade of
Janus kinase 3 (JAK3) by JANEX-1 (a JAK3 inhibitor) attenuated the
tumor necrosis factor (TNF)-α-induced increase in cell adhesion
molecule expression in endothelial cells, and improved myocardial
vascular permeability in endotoxemic mice (12).
Paricalcitol, 19-nor-1-α-25-dihydroxyvitamin D2, is
a vitamin D2 analogue that activates the vitamin D receptor (VDR),
resulting in the reduced intestinal absorption of calcium and
phosphorus, and calcium mobilization from bone (13). Paricalcitol is well tolerated in
patients with secondary hyperparathyroidism and it is available in
two formulations, intravenous and oral (14–16). Studies have suggested that
paricalcitol exerts beneficial effects on renal inflammation
through the modulation of the nuclear factor (NF)-κB pathway in a
model of cyclosporine-induced kidney injury and a model of renal
fibrosis (17,18). However, the protective effects of
paricalcitol on endotoxemia-induced myocardial inflammation remain
unknown. Thus, in the present study, we aimed to investigate the
effects of paricalcitol on lipopolysaccharide (LPS)-induced
myocardial inflammation, and to elucidate the mechanisms
responsible for these effects.
Materials and methods
Cell culture and immunocytochemistry
Human umbilical vein endothelial cells (HUVECs) were
obtained from Lonza Walkersville, Inc. (Walkersville, MD, USA) and
cultured in endothelial basal medium (EBM)-2 complete medium
supplemented with 2% (v/v) heat-inactivated fetal bovine serum at
37°C in 5% CO2. To examine the effects of paricalcitol
on the LPS-induced increase in cell adhesion molecule expression in
HUVECs, subconfluent endothelial cells were incubated with
paricalcitol (Tocris Bioscience, Minneapolis, MN, USA; 0.01, 0.1 or
1 µM) for 30 min and then stimulated with 10 ng/ml TNF-α
(R&D Systems, Minneapolis, MN, USA) for 6 h.
For p65 immunocytochemistry, the HUVECs were
cultured in 6-well plates and treated with 1 µM paricalcitol
for 30 min and then stimulated with 10 ng/ml TNF-α for 6 h.
Following 4% paraformaldehyde fixation, the cells were washed with
phosphate-buffered saline (PBS), permeabilized in 0.25% Triton
X-100 and blocked with 1% goat serum. The HUVECs were then
incubated with primary anti-p65 antibody (#4764; Cell Signaling
Technology, Beverly, MA, USA), followed by staining with
Cy3-conjugated secondary antibody (AP182C; Chemicon, Temecula, CA,
USA). For nuclear staining, the HUVECs were incubated with
4′,6-diamidino-2-phenylinole (DAPI; Molecular Probes, Eugene, OR,
USA).
Western blot analysis
Western blot analysis was performed as described
previously (12). Primary
antibodies against intercellular adhesion molecule-1 (ICAM-1;
sc-1511), vascular cell adhesion molecule-1 (VCAM-1; sc-1504) (both
from Santa Cruz Biotechnology, Santa Cruz, CA, USA), fractalkine
(TP-213; Torrey Pines BioLabs, Houston, TX, USA), phosphorylated
(p)-p65 (#3033) and p65 (#4764; both from Cell Signaling
Technology) were used. The membranes were reblotted with anti-actin
antibody to verify the equal loading of protein in each lane. All
signals were visualized using chemiluminescent reagents (Amersham
Pharmacia Biotech, London, UK) and analyzed using a densitometric
scanner (LAS-3000; FujiFilm, Tokyo, Japan).
Electrophoretic mobility shift assays
(EMSAs)
EMSAs for NF-κB p65 were performed as described
previously (12). The HUVECs were
incubated with control buffer or 1 µM paricalcitol for 30
min and then stimulated with 10 ng/ml TNF-α for 4 h and nuclear
extracts were prepared. PBS was used as a control buffer. Signals
were detected using chemiluminescent EMSA imaging kits according to
the manufacturer's instructions (Panomics, Inc., Fremont, CA,
USA).
Animal experiments
The animal experimental protocol was reviewed and
approved by the Institutional Animal Care and Use Committee of
Chonbuk National University, Jeonju, Korea (CBU 2014-00060). Male
C57BL/6 mice (7 weeks old, weighing 20–23 g) were obtained from
Orient Bio Inc. (Seoul, Korea) and maintained in an environmentally
controlled room (temperatue, 23±1°C; humidity, 50±10%), with
lighting on a 12-h light/12-h dark cycle and free access to water
and standard chow. The mice were divided into the following 4
groups: i) control buffer-treated (n=10); ii) paricalcitol-treated
(n=10); iii) LPS administration (n=10); and iv) LPS administration
plus paricalcitol treatment (n=10). PBS was used as the control
buffer. Pre-treatment with 0.2 µg/kg paricalcitol was
administered by daily intraperitoneal injections for 5 days.
Endotoxemia was induced by an intraperitoneal injection of 15 mg/kg
LPS (Sigma-Aldrich, St. Louis, MO, USA). The doses of LPS and
paricalcitol were selected based on previous studies (12,17). At 16 h after the induction of
endotoxemia, the mice were anesthetized with 100 mg/kg ketamine
(Huons, Seoul, Korea) and 10 mg/kg xylazine (Bayer Korea, Seoul,
Korea). The hearts were harvested for western blot analysis and
histological examination.
Immunohistochemical analysis of
myocardial ICAM-1 expression
Immunohistochemical staining for ICAM-1 was
performed as previously described (19). The isolated heart tissues were
fixed by immersion in 4% paraformaldehyde and embedded in paraffin.
The tissue sections were deparaffinized with xylene and rehydrated
in graded ethanol baths. Following treatment with blocking buffer,
the slides were incubated overnight at 4°C with either hamster
anti-mouse ICAM-1 or hamster IgG (both from BD
Biosciences-Pharmingen, San Jose, CA, USA) as a control. The heart
sections were exposed to Dako Chromogen (DakoCytomation, Glostrup,
Denmark) to visualize immunocomplexes, and counterstained with
hematoxylin (Sigma-Aldrich). For morphometric analysis, the slides
were evaluated using a Zeiss Z1 microscope (Carl Zeiss, Göttingen,
Germany) by two observers who were unaware of the slide origins.
The extent of ICAM-1 immunostaining in the heart tissue was
expressed as a percentage of the area of 10 random, non-overlapping
fields per slide at x400 magnification using ImageJ software
(http://rsb.info.nih.gov/ij).
Measurement of myocardial tumor necrosis
factor-α levels
The TNF-α levels were determined using enzyme-linked
immunosorbent assay (ELISA) kits (R&D Systems) according to the
manufacturer's instructions. Briefly, 50 µl of assay
diluent, standard dilutions and protein samples were added to each
well and mixed by gently tapping the plate. The plate was covered
and incubated for 2 h at room temperature. After washing each well
with washing buffer, mouse TNF-α conjugate was added to each well
and incubated for 2 h at room temperature. After washing, substrate
solutions were added to each well followed by incubation for 30 min
and stop solutions were then added. The optical density of each
well was determined using a microplate reader set at 450 nm.
Measurement of vascular permeability
using Evans blue dye
To measure myocardial permeability, Evans blue dye
assays were performed as previously described (12). Evans blue dye (50 mg/kg;
Sigma-Aldrich) dissolved in 200 µl PBS was injected into the
tail veins of the mice following the induction of endotoxemia
induction. At 16 h after the induction of endotoxemia, the mice
were sacrificed by CO2 asphyxiation. At sacrifice, the
mice were perfused with PBS through the left ventricle until all
blood was eliminated. The hearts were excised, weighed and
homogenized in 1 ml formamide. The samples were incubated at 55°C
for 18 h and centrifuged at 16,000 × g for 30 min at 4°C. The
amount of Evans blue dye in the supernatant was determined by
measuring the absorbance at 620 nm using a SmartSpec Plus
spectrophotometer (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Data are expressed as the means ± SD. Mean
comparisons between 2 groups were examined for significant
differences using ANOVA, followed by individual comparisons with a
Tukey's post hoc test, with a P-value <0.05 considered to
indicate a statistically significant difference.
Results
Paricalcitol suppresses the TNF-α-induced
increase in the expression of cell adhesion molecules in
HUVECs
To examine the effects of paricalcitol on the
TNF-α-induced increase in the expression of ICAM-1, VCAM-1 and
fractalkine, the HUVECs were treated with paricalcitol for 30 min
and then stimulated with TNF-α for 6 h. Western blot analysis
revealed that TNF-α significantly increased the protein levels of
ICAM-1, VCAM-1 and fractalkine compared with the controls.
Treatment with paricalcitol suppressed the TNF-α-induced increase
in the expression of ICAM-1, VCAM-1 and fractalkine in a
dose-dependent manner. Treatment with paricalcitol alone did not
alter the expression of the cell adhesion molecules (Fig. 1).
Paricalcitol suppresses the TNF-α-induced
activation of NF-κB in HUVECs
The NF-κB signaling pathway is a major signaling
pathway involved in the TNF-α-induced expression of cell adhesion
molecules. Therefore, we examined the effects of paricalcitol on
the TNF-α-induced activation of the NF-κB signaling pathway using
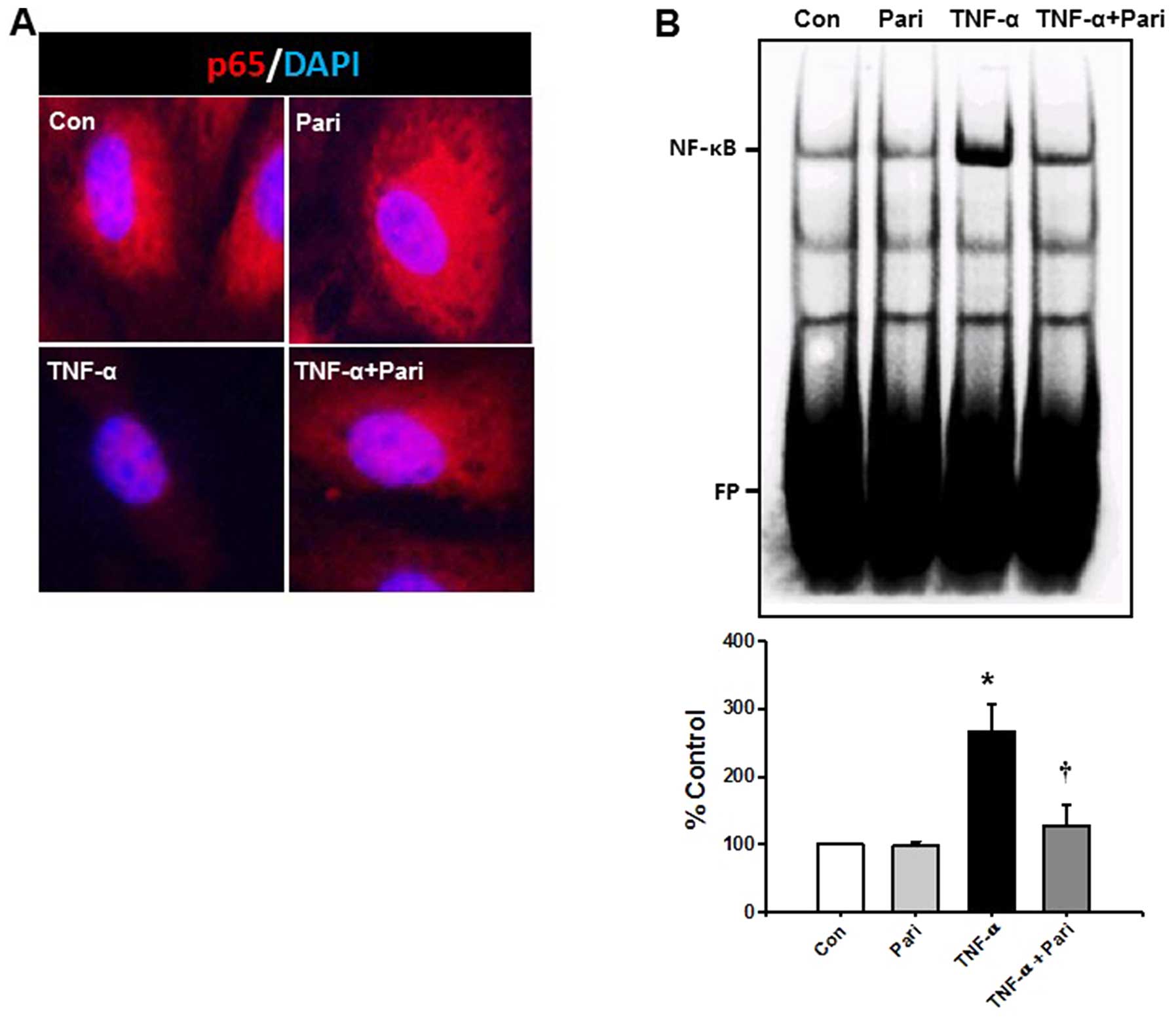
immunocytochemistry and EMSA. p65 protein was abundant in the
cytoplasm of the control- and paricalcitol-treated cells. However,
TNF-α decreased the cytoplasmic expression of p65 in the HUVECs.
Pre-treatment with paricalcitol prevented the TNF-α-induced
depletion of cytoplasmic p65 in the HUVECs (Fig. 2A).
To examine the effects of paricalcitol on the NF-κB
DNA binding activity induced by TNF-α, nuclear extracts from the
TNF-α-stimulated HUVECs were examined using EMSA. TNF-α
significantly increased the NF-κB DNA binding activity in the
nuclear extracts by 2.6-fold compared with that observed in the
HUVECs treated with control buffer. The increase in the
TNF-α-induced DNA binding activity of NF-κB in the HUVECs was
significantly decreased by 51.8% following treatment with
paricalcitol (Fig. 2B). We did
not observe any increase in the DNA binding activity of NF-κB in
the cells treated only with control buffer or paricalcitol. These
data suggested that paricalcitol prevented the TNF-α-induced
increase in the DNA binding activity of NF-κB in the HUVECs.
Paricalcitol attenuates LPS-induced
myocardial inflammation in mice
We wished to determine whether paricalcitol
suppresses LPS-induced myocardial inflammation. At 16 h after the
LPS injection, myocardial ICAM-1 expression had increased by
3.2-fold and VCAM-1 expression had increased by 2.1-fold compared
with the controls. Pre-treatment with paricalcitol suppressed the
LPS-induced increase in myocardial ICAM-1 and VCAM-1 expression by
approximately 43.8 and 37.1%, respectively, compared with the
LPS-treated group (Fig. 3A).
Treatment with paricalcitol alone did not induce ICAM-1 and VCAM-1
expression in the mouse hearts. To evaluate the localization of
LPS-induced ICAM-1 expression in the myocardium, we examined the
heart sections using ICAM-1 immunohistochemistry. After the
injection of LPS, myocardial ICAM-1 expression increased in the
arteriolar and capillary endothelial cells compared with the
control samples. Pre-treatment with paricalcitol suppressed the
increase in myocardial ICAM-1 expression (Fig. 3B).
 | Figure 3Paricalcitol decreases
endotoxemia-induced myocardial inflammation. (A) Myocardial
intercellular adhesion molecule-1 (ICAM-1) and vascular cell
adhesion molecule-1 (VCAM-1) expression were evaluated by western
blot analysis. Hearts from the mice treated with control buffer
(Con), paricalcitol (Pari), lipopolysaccharide (LPS), and LPS plus
Pari were harvested 16 h following the induction of endotoxemia.
Data from densitometric analysis are presented as the relative
ratio of each protein to actin. The relative ratio measured in
hearts from control buffer-treated mice was arbitrarily set to 1.
Data are expressed as the means ± SD. **P<0.01 vs.
Con or Pari; †P<0.05 vs. LPS. (B)
Immunohistochemistry of ICAM-1 in hearts from the mice treated with
Con, Pari, LPS or LPS plus Pari were harvested 16 h following the
induction of endotoxemia. Ten randomly selected, non-overlapping
fields at ×200 magnification were quantified (n=10/group). Data are
expressed as the means ± SD. **P<0.01 vs. Con or
Pari; †P<0.05 vs. LPS. (C) Heart tumor necrosis
factor-α (TNF-α) levels from Con, Pari, LPS or LPS plus Pari were
measured using ELISA, normalized to 1 mg heart protein. Data are
expressed as the means ± SD. (n=10/group). *P<0.05
vs. Con or Pari; †P<0.05 vs. LPS. (D) Western blot
analysis of myocardial phosphorylated (p-)p65. Hearts from mice
treated with Con, Pari, LPS or LPS plus Pari were harvested 16 h
following the induction of endotoxemia. Data from densitometric
analysis are presented as the relative ratio of each protein to
actin. The relative ratio measured in the heart from control
buffer-treated mice is arbitrarily presented as 1. Data are
expressed as the means ± SD. **P<0.01 vs. Con or
Pari; †P<0.05 vs. LPS. |
To further examine the anti-inflammatory effects of
paricalcitol, we measured the myocardial TNF-α levels using ELISA.
The myocardial TNF-α levels increased significantly following the
induction of endotoxemia by LPS (7.29±1.02 pg/mg protein) compared
with that in the controls (4.66±0.11 pg/mg protein) or in the
samples from the mice treated with paricalcitol (4.74±0.59 pg/mg
protein) (Fig. 3C). Treatment
with paricalcitol reduced the LPS-induced increase in TNF-α levels
(5.42±0.11 pg/mg protein). These data suggested that paricalcitol
attenuated LPS-induced myocardial inflammation.
Our in vitro data suggested that paricalcitol
exerted anti-inflammatory effects through the modulation of the
NF-κB signaling pathway. Therefore, we wished to determine whether
paricalcitol can prevent the LPS-induced myocardial activation of
p65 in vivo. The level of p-p65 in the myocardium after the
injection of LPS increased 1.66-fold compared with that in the
control samples. Treatment with paricalcitol suppressed the
LPS-induced increase in p-p65 levels (Fig. 3D). These data suggested that
paricalcitol inhibited the LPS-induced activation of the myocardial
NF-κB signaling pathway.
Paricalcitol alleviates myocardial
vascular leakage in mice with LPS-induced endotoxemia
Endotoxemia is characterized by an increase in
vascular leakage (12); thus, in
this study, we examined the effects of paricalcitol on LPS-induced
myocardial permeability using Evans blue dye, as previoulsy
described (12). At 16 h after
the LPS injection, myocardial vascular leakage was significantly
increased. Treatment with paricalcitol significantly suppressed the
LPS-induced increase in vascular leakage (Fig. 4). These data suggested that
paricalcitol exerted protective effects against LPS-induced
myocardial vascular leakage.
Discussion
The present study demonstrated that paricalcitol
suppressed the TNF-α-induced expression of cell adhesion molecules
in endothelial cells by modulating the NF-κB signaling pathway.
Paricalcitol also attenuated endotoxemia-induced myocardial
inflammation and vascular leakage.
Vascular endothelial cells constitute a continuous
and semipermeable barrier in the vascular bed (20). During septic shock, this
endothelial barrier function is impaired. The breakdown of the
endothelial barrier is initiated by the activation of the
coagulation system and impaired fibrinolysis, which leads to tissue
hypoperfusion. Leukocyte adhesion to endothelial cells is mediated
by soluble cytokines or cell adhesion molecules. Finally,
endothelial cell-cell junctions, such as vascular endothelial
cadherin and zona occludens are broken, resulting in the leakage of
proteins and solutes that induces interstitial edema (21). Therefore, preserving endothelial
barrier function is an important approach in the treatment of
patients with sepsis.
The NF-κB signaling pathway is central to
TNF-α-induced endothelial cell activation. Tan et al
reported that paricalcitol decreased TNF-α-induced tubular RANTES
induction through VDR-mediated NF-κB sequestration (18). In our in vitro experiments,
we demonstrated that paricalcitol effectively suppressed the
TNF-α-induced increase in ICAM-1, VCAM-1 and fractalkine expression
in endothelial cells. For the activation of the NF-κB signaling
pathway, nuclear translocation of the NF-κB p65 subunit is
important (22). Our
immunocyto-chemical data revealed that paricalcitol effectively
decreased the TNF-α-induced NF-κB p65 nuclear translocation. Our
EMSA data demonstrated that paricalcitol decreased the
TNF-α-induced DNA binding activity of NF-κB in endothelial cells.
These data suggest that paricalcitol may exert protective effects
against TNF-α-induced endothelial cell activation and inflammation
by regulating NF-κB nuclear translocation and DNA binding.
Impaired vascular permeability is important in the
pathophysiology of sepsis (12).
Increased vascular permeability is characterized by the
extravasation of interstitial fluid and the failure of vascular
responses (23). In our animal
model, myocardial permeability was increased following LPS-induced
endotoxemia. Pre-treatment with paricalcitol significantly
decreased the increase in vascular permeability induced by
endotoxemia. In addition, paricalcitol effectively decreased the
LPS-induced increase in myocardial ICAM-1 and VCAM-1 expression.
These data suggested that paricalcitol affected both cell adhesion
molecule expression and vascular permeability in mice with
endotoxemia.
VDR is ubiquitously distributed in human tissue and
exerts pleiotropic effects on the regulation of oxidative stress,
immune modulation and inflammation (24,25). The co-stimulation of human
macrophages by Toll-like receptor ligands and 25(OH) vitamin D has
been shown to upregulate the VDR and 1α-hydroxylase genes, which
enhances the antimicrobial effects of cathelicidin (26,27). In ischemic kidney injury,
pre-treatment with paricalcitol has been shown to decrease renal
inflammation by suppressing the TLR4-NF-κB signaling pathway
(28). Therefore, vitamin D may
be important in the innate immune system. Jeng et al found
that patients in the intensive care unit with sepsis had lower
25(OH) vitamin D levels than the healthy controls (29). This study demonstrated that
paricalcitol decreased the LPS-induced increase in myocardial TNF-α
levels, and suppressed the activation of myocardial NF-κB in mice
with endotoxemia.
In conclusion, the findings of our study
demonstrated that paricalcitol suppressed the TNF-α-induced
increase in the expression of cell adhesion molecules in
endothelial cells and thus it may exert protective effects against
endotoxemia-induced vascular inflammation by modulating the NF-κB
signaling pathway.
Acknowledgments
The authors would like to thank Kieu Thi Thu Trang
for providing excellent technical assistance. This study was
supported by the National Research Foundation of Korea, funded by
the Korean government (2008-0062279, to W.K. and
NRF-2014R1A1A4A01003832, to S.K.P.) and by the Fund of Biomedical
Research Institute, Chonbuk National University Hospital in 2012
(to S.K.P.).
References
|
1
|
Lips P: Worldwide status of vitamin D
nutrition. J Steroid Biochem Mol Biol. 121:297–300. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Faulkner KA1, Cauley JA, Zmuda JM,
Landsittel DP, Newman AB, Studenski SA, Redfern MS, Ensrud KE, Fink
HA, Lane NE and Nevitt MC: Higher 1,25-dihydroxyvitamin D3
concentrations associated with lower fall rates in older
community-dwelling women. Osteoporos Int. 17:1318–1328. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bischoff-Ferrari HA, Dietrich T, Orav EJ
and Dawson-Hughes B: Positive association between 25-hydroxy
vitamin D levels and bone mineral density: a population-based study
of younger and older adults. Am J Med. 116:634–639. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Thacher TD and Clarke BL: Vitamin D
insufficiency. Mayo Clinic proceedings. Mayo Clin. 86:50–60. 2011.
View Article : Google Scholar
|
|
5
|
Bjelakovic G, Gluud LL, Nikolova D,
Whitfield K, Krstic G, Wetterslev J and Gluud C: Vitamin D
supplementation for prevention of cancer in adults. Cochrane
Database Syst Rev. 6:CD0074692014.PubMed/NCBI
|
|
6
|
Bjelakovic G, Gluud LL, Nikolova D,
Whitfield K, Wetterslev J, Simonetti RG, Bjelakovic M and Gluud C:
Vitamin D supplementation for prevention of mortality in adults.
Cochrane Database Syst Rev. 1:CD0074702014.PubMed/NCBI
|
|
7
|
Bae S, Yalamarti B, Ke Q, Choudhury S, Yu
H, Karumanchi SA, Kroeger P, Thadhani R and Kang PM: Preventing
progression of cardiac hypertrophy and development of heart failure
by paricalcitol therapy in rats. Cardiovasc Res. 91:632–639. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bodyak N, Ayus JC, Achinger S,
Shivalingappa V, Ke Q, Chen YS, Rigor DL, Stillman I, Tamez H,
Kroeger PE, et al: Activated vitamin D attenuates left ventricular
abnormalities induced by dietary sodium in Dahl salt-sensitive
animals. Proc Natl Acad Sci USA. 104:16810–16815. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zittermann A: Vitamin D and cardiovascular
disease. Anticancer Res. 34:4641–4648. 2014.PubMed/NCBI
|
|
10
|
Dauphinee SM and Karsan A:
Lipopolysaccharide signaling in endothelial cells. Lab Invest.
86:9–22. 2006. View Article : Google Scholar
|
|
11
|
Rudiger A and Singer M: Mechanisms of
sepsis-induced cardiac dysfunction. Crit Care Med. 35:1599–1608.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Lee JE, Lee AS, Kim DH, Jung YJ, Lee S,
Park BH, Lee SH, Park SK, Kim W and Kang KP: Janex-1, a JAK3
inhibitor, ameliorates tumor necrosis factor-α-induced expression
of cell adhesion molecules and improves myocardial vascular
permeability in endotoxemic mice. Int J Mol Med. 29:864–870.
2012.PubMed/NCBI
|
|
13
|
Dyer CA: Safety and tolerability of
paricalcitol in patients with chronic kidney disease. Expert Opin
Drug Saf. 12:717–728. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lindberg J, Martin KJ, González EA,
Acchiardo SR, Valdin JR and Soltanek C: A long-term, multicenter
study of the efficacy and safety of paricalcitol in end-stage renal
disease. Clin Nephrol. 56:315–323. 2001.PubMed/NCBI
|
|
15
|
Llach F, Keshav G, Goldblat MV, Lindberg
JS, Sadler R, Delmez J, Arruda J, Lau A and Slatopolsky E:
Suppression of parathyroid hormone secretion in hemodialysis
patients by a novel vitamin D analogue:
19-nor-1,25-dihydroxyvitamin D2. Am J Kidney Dis. 32(Suppl 2):
S48–S54. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Coyne D, Acharya M, Qiu P, Abboud H,
Batlle D, Rosansky S, Fadem S, Levine B, Williams L, Andress DL and
Sprague SM: Paricalcitol capsule for the treatment of secondary
hyperparathyroidism in stages 3 and 4 CKD. Am J Kidney Dis.
47:263–276. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Park JW, Bae EH, Kim IJ, Ma SK, Choi C,
Lee J and Kim SW: Paricalcitol attenuates cyclosporine-induced
kidney injury in rats. Kidney Int. 77:1076–1085. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tan X, Wen X and Liu Y: Paricalcitol
inhibits renal inflammation by promoting vitamin D
receptor-mediated sequestration of NF-kappaB signaling. J Am Soc
Nephrol. 19:1741–1752. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kang KP, Kim DH, Jung YJ, Lee AS, Lee S,
Lee SY, Jang KY, Sung MJ, Park SK and Kim W: Alpha-lipoic acid
attenuates cisplatin-induced acute kidney injury in mice by
suppressing renal inflammation. Nephrol Dial Transplant.
24:3012–3020. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Matsuda N and Hattori Y: Vascular biology
in sepsis: pathophysiological and therapeutic significance of
vascular dysfunction. J Smooth Muscle Res. 43:117–137. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Opal SM and van der Poll T: Endothelial
barrier dysfunction in septic shock. J Intern Med. 277:277–293.
2015. View Article : Google Scholar
|
|
22
|
Sung MJ, Kim W, Ahn SY, Cho CH, Koh GY,
Moon SO, Kim DH, Lee S, Kang KP, Jang KY and Park SK: Protective
effect of alpha-lipoic acid in lipopolysaccharide-induced
endothelial fractalkine expression. Circ Res. 97:880–890. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Aird WC: The role of the endothelium in
severe sepsis and multiple organ dysfunction syndrome. Blood.
101:3765–3777. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Donate-Correa J, Domínguez-Pimentel V,
Méndez-Pérez ML, Muros-de-Fuentes M, Mora-Fernández C, Martín-Núñez
E, Cazaña-Pérez V and Navarro-González JF: Selective vitamin D
receptor activation as anti-inflammatory target in chronic kidney
disease. Mediators Inflamm. 2014(670475)2014. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Andress D: Nonclassical aspects of
differential vitamin D receptor activation: implications for
survival in patients with chronic kidney disease. Drugs.
67:1999–2012. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Watkins RR, Yamshchikov AV, Lemonovich TL
and Salata RA: The role of vitamin D deficiency in sepsis and
potential therapeutic implications. J Infect. 63:321–326. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Liu PT, Stenger S, Li H, Wenzel L, Tan BH,
Krutzik SR, Ochoa MT, Schauber J, Wu K, Meinken C, et al: Toll-like
receptor triggering of a vitamin D-mediated human antimicrobial
response. Science. 311:1770–1773. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lee JW, Kim SC, Ko YS, Lee HY, Cho E, Kim
MG, Jo SK, Cho WY and Kim HK: Renoprotective effect of paricalcitol
via a modulation of the TLR4-NF-κB pathway in
ischemia/reperfusion-induced acute kidney injury. Biochem Biophys
Res Commun. 444:121–127. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Jeng L, Yamshchikov AV, Judd SE, Blumberg
HM, Martin GS, Ziegler TR and Tangpricha V: Alterations in vitamin
D status and anti-microbial peptide levels in patients in the
intensive care unit with sepsis. J Transl Med. 7(28)2009.
View Article : Google Scholar : PubMed/NCBI
|