Introduction
Colorectal cancer (CRC) is among the most common
malignancies and is becoming a leading cause of cancer-related
mortality worldwide (1). Although
the efficacy of multiple drug treatments, surgical treatments and
chemotherapy have extensively improved (2), CRC is still considered as a complex
and difficult disease to deal with (3). In recent years, studies have focused
on specific molecular targets, such as epidermal growth factor
receptor (EGFR) and vascular endothelial growth factor (VEGF) to
predict the progression of CRC (4,5).
However, due to the heterogeneous characteristics of CRC, there is
still controversy regarding the optimal treatment strategy for CRC.
Thus, in order to develop more effective targeting agents against
CRC, the identification of novel molecules is urgently required
(6).
Human DNA binding protein A (dbpA), a member of the
Y-box binding protein family, contains a highly conserved DNA
binding domain, named the cold shock domain (CSD) (7,8).
This family of proteins appears to play a critical role in cell
proliferation and growth, transcriptional and translational
regulation, DNA replication, drug resistance, the cell cycle and
malignancy (9,10). dbpA can bind to EGFR,
proliferating cell nuclear antigen, thymidine kinase and DNA
polymerase (11,12) to participate in cellular
activities. Previously, Tobita et al suggested that dbpA
induced carcinogenesis by regulating the expression of cellular
genes, such as insulin-like growth factor binding protein-1
(IGFBP-1) and carbonic anhydrase 3 (Car3) in dbpA-transgenic mice
(13). Furthermore, dbpA has been
reported as a prognostic marker for the advanced stages of and for
the poor prognosis of hepatocellular carcinoma by enhancing cell
proliferation and transformation (14,15). Our previous study demonstrated
that dbpA played a crucial role in the development of gastric
cancer by regulating the expression of E-cadherin, β-catinen,
adenomatous polyposis coli (APC) and cyclin D1 (16). These findings indicate the
significance of dbpA in the development of malignant diseases.
In the present study, to illustrate the role of dbpA
in CRC, the expression of dbpA in CRC tissues and cell lines was
examined. The effects of dbpA on CRC cells were investigated by
lentivirus-mediated short haripin RNA (shRNA) interference both
in vitro and in vivo. Our findings indicate that dbpA
is a vital driver of human CRC and that the knockdown of dbpA
markedly reduces cell proliferation in vitro and decreases
tumorigenesis in vivo. Our study may provide scientific
evidence for the further development of reliable molecular
biomarkers for CRC.
Materials and methods
Clinical specimen collection
Fresh colorectal tumor and adjacent normal tissues
were obtained from 44 patients who received surgery from May 2012
to July 2014 at the Department of General Surgery, the Third
Affiliated Hospital of Xi'an Jiaotong University, Xi'an, China. We
only collected the clinical data of patients that were complete and
from patients who had not received any radiotherapy or chemotherapy
prior to surgery. All specimens were classified according to the
TNM staging system enacted by International Union Against Cancer
(UICC) and American Joint Committee on Cancer (AJCC). The
correlations between dbpA expression and clinicopathological
parameters in CRC were analyzed by Pearson's Chi-square test.
Written informed consent was obtained from all the patients, and
this study was approved by the Human Ethics Committee of the Third
Affiliated Hospital of Xi'an Jiaotong University and all
experiments were performed in accordance with the 1964 Helsinki
declaration and its later amendments.
Immunohistochemical (IHC) staining
The colorectal tissues obtained from patients were
fixed immediately with 4% paraformaldehyde overnight at 4°C, and
were then embedded in paraffin and sectioned (4-µm-thick)
onto slides. The sections were baked at 60°C for 2 h,
deparaffinized by two changes of xylene, and rehydrated in graded
alcohol solutions. For antigen retrieval, the sections were heated
in 20 mmol/l sodium citrate (pH 6.0) at 95°C for 15 min. The slides
were treated then with 3% H2O2 to block
endogenous peroxidase activity. Subsequently, the slides were
incubated overnight at 4°C with the rabbit polyclonal anti-dbpA
antibody (ab48952; Boster Biological Technology, Ltd., Wuhan,
China) at a 1:500 dilutions. Subsequently, the sections were
incubated with HRP polymer (1:500; ab6721; Boster Biological
Technology, Ltd.) for 30 min at room temperature and DAB mix
(Tiangen Biotechnology, Beijing, China) was applied for staining.
Two independent pathologists blinded to the patient data evaluated
the scores of the IHC results. The scoring criteria was based on
the percentage of dpbA-positive cells in the tumor tissue as
follows: − (<10%) was considered as no staining; + (11–40%) was
considered weak staining; ++ (41–70%) was considered moderate
staining; and +++ (71–100%) was considered strong staining. If a
discrepancy was exited between the scores, the specimens would be
re-examined by both researchers together.
Cell lines and cell culture
The human CRC cell lines, LoVo, SW480, RKO, HT-29,
DLD-1, SW1463 and SW620, were obtained from the First Affiliated
Hospital of the Medical College of Xi'an Jiaotong University. The
normal colorectal mucosa cell line, FHC, and the 293T cell line
were purchased from the Animal Center of the Fourth Military
Medical University, Xi'an, China. All the cells were cultured in
Dulbecco's modified Eagle's medium (DMEM) supplemented with 10%
fetal bovine serum (FBS) at 37°C with 5% CO2.
dbpA RNAi lentivirus design and
packaging
The shRNA sequences were designed for dbpA as
follows: sense, 5′-AGACGUGGCUACUAUGGAATT-3′ and antisense,
5′-UUCCAUAGUAGCCACGUCUGT-3′ in accordance with our previous study
(16). The negative control shRNA
were randomly sequenced and homology with the dbpA sequence was
avoided using the Blast website (https://blast.ncbi.nlm.nih.gov/Blast.cgi). The shRNAs
were then cloned into the pGV115 entry vector (Invitrogen Life
Technologies, Carlsbad, CA, USA) and identified by PCR and DNA
sequencing. Lentiviruses were produced and packaged using the 293T
cells following co-transfection with pGV115 entry vector carrying
dbpA-shRNA (shRNA-dbpA-Lv) or scrambled shRNA (shNC-dbpA-Lv) and
pHelper plasmids, according to the lentivirus packaging protocol
(Genechem Co., Ltd., Shanghai, China). The lentivirus contained the
green fluorescent protein (GFP) and viral titers were then measured
by GFP-positive cell counts under the observation of a fluorescence
microscope (Olympus, Tokyo, Japan).
Infection of SW620 cells with
shRNA-dbpA-Lv
The SW620 cells were seeded at 5×105
cells/well in 6-well plates and incubated for 24 h at 37°C with 5%
CO2. The cells were infected with shRNA-dbpA-Lv
(GenePharma, Shanghai, China) at a multiplicity of infection (MOI)
of 10. The knockdown efficiency of dbpA was evaluated at 72 h by
the percentage of GFP-positive cells (>50%). Cells were also
infected with the shNC-dbpA-Lv plasmid as negative controls (NC).
Cells transfected with empty vector were used as controls (CON).
After infection at 120 h, the cells were harvested and prepared for
reverse transcription-quantitative PCR (RT-qPCR) and western blot
analysis.
RNA extraction and RT-qPCR
RNA from the cell cultures was extracted using
TRizol reagent (Gibco Life Technologies, Beijing, China) according
to the manufacturer's instructions. First strand cDNA was
synthesized from 5 µg of total RNA using SuperScript II RT
200 U/µl (Invitrogen). dbpA mRNA expression was evaluated by
qPCR on an ABI 7500 Real-Time PCR System (Applied Biosystems Life
Technologies, Beijing, China) with SYBR-Green PCR core reagents.
GAPDH was used as the input reference. The sequences of the primers
used were as follows: dbpA sense, 5′-CGTCGCTCACGGGTCTTA-3′ and
antisense, 5′-CCTGAAGTTGTGCTCCCTCT-3′; GAPDH sense,
5′-TGACTTCAACAGCGACACCCA-3′ and antisense,
5′-CACCCTGTTGCTGTAGCCAAA-3′. RT-qPCR was performed in triplicate
and the results are presented as the Ct values, defined as the
threshold PCR cycle number at which an amplified product is first
detected. The mean Ct value was calculated, and the ΔCt value was
determined as the mean Ct value for the target gene minus the mean
Ct value for GAPDH.
Western blot analysis
The cells were collected, washed with PBS and then
lysed in lysis buffer containing 100 mM Tris-HCl, pH 7.5, 0.5%
NP-40 and protease inhibitor cocktail. The supernatant was
collected after centrifugation (at 20,000 × g, for 15 min, at 4°C)
and the protein concentrations were determined using a BCA protein
assay kit (Pierce Biotechnology, Inc., Rockford, IL, USA). The cell
lysate samples (40 µg) were separated by 15% SDS-PAGE and
transferred onto a polyvinylidene difluoride filter (Immobilon;
Millipore, Bedford, MA, USA). After blocking with 5% milk, the
filter was incubated overnight with a primary rabbit polyclonal
anti-dbpA antibody (ab48952; Boster Biological Technology, Ltd.) at
1:500 dilutions for 1 h. The samples were then incubated with the
HRP-conjugated secondary antibodies (ab6721) at 1:1,000, and the
bands were detected by enhanced chemiluminescence (both from
Amersham Biosciences, Piscataway, NJ, USA), and then analyzed using
Quantity One software (Bio-Rad Laboratories, Inc., Hercules, CA,
USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The SW620 cells transfected with shRNA-dbpA-Lv or
shNC-dbpA-Lv for 1 to 5 days, respectively, followed by diluting
and seeding at a density of 2×103 cells/ml into a
96-well plate for 24 h. MTT solution was added to each well to a
final concentration of 5 mg/ml and culture was continued for 4 h at
37°C. The supernatant mixed with MTT was removed and DMSO was added
into each well. The OD data were analyzed once daily for 5 days
using an ELISA reader (Bio-Rad Laboratories) at a wavelength of 490
nm.
Colony formation assay
The SW620 cells transfected with shRNA-dbpA-Lv or
shNC-dbpA-Lv were plated into 6-well plates (2,000 cells/well) and
incubated for 14 days at 37°C, with the medium replaced every 3
days. After cultivating for 14 days, the cells were washed with
PBS, fixed with 4% paraformaldehyde for 30 min at room temperature
and stained with Giemsa (Tiangen Biotechnology) for 15 min. The
number of colonies containing >50 cells was counted under a
microscope (CKX53; Olympus).
Cell cycle analysis
The effect of shRNA-dbpA-Lv on the cell cycle
distribution was determined by flow cytometry. The cells were
suspended at the concentration of 1×106 and centrifuged
at 1,500 rmp for 5 min twice, then resuspended with 100 µl
PBS and fixed with 70% ice-cold ethanol at 4°C overnight. The cells
were washed with PBS and resuspended in 1 ml PBS containing 50
µg/ml PI and 100 µg/ml RNase A for 1 h in the dark at
4°C. The cell cycle was analyzed using a FACSCalibur flow cytometer
(Becton-Dickinson, San Jose, CA, USA) at 72 h after transduction.
The proportions of cells in the G2/M, S, and G0/G1 phases were
analyzed using special software FlowJo software (Tree Star, Inc.,
Ashland, OR, USA).
Apoptosis analysis
SW620 cells transfected shRNA-dbpA-Lv or
shNC-dbpA-Lv were collected and washed with PBS twice. Following
centrifugation (at 100 × g, for 5 min, at 4°C), the cells were
resuspended with 1X staining buffer at the concentration of
1×106 cells/ml, and the cells were then dyed with 5
µl Annexin V-APC in the dark at room temperature for 15 min.
Flow cytometry was performed using a FACSCalibur flow cytometer
(Becton-Dickinson) and analysis was performed with FlowJo software
(Tree Star, Inc.).
Tumorigenicity assay
BALB/C nude mice (4 weeks old) were purchased from
the Animal Center of the Fourth Military Medical University. Living
SW620 cells were detected and harvested after mixing with 4% trypan
blue, then washed and resuspended in PBS at 4°C. The animals were
randomly divided into 3 groups (n=10) as follows: the mice
subcutaneously injected with dbpA-shRNA (KD), those injected with
negative-shRNA (NC) and those injected with normal cells (empty
vector-transfected cells, CON). Each mouse was subcutaneously
injected with 1×106 cells on the right side of axilla.
The standard of tumorigenesis was based on a tumor diameter of ≥3
mm. Tumor sizes [volume (mm3) = width (mm2)/2
× length (mm)] were measured using calipers every 7 days.
Furthermore, the physical conditions of the mice were monitored as
follows: the thickness of subcutaneous fat of the mice was measured
by calipers after pinching with fingers; the body weight of the
mice was recorded using an electronic scale (Dongyi Biotechnology,
Beijing, China). All mice were euthanized by cervical dislocation
at 35 days post-inoculation, and the separated and complete tumors
were collected and disposed for further analysis. The animal
experiment was reviewed and approved by the Animal Care and Use
Committee of Xi'an Jiaotong University.
Statistical analysis
Statistical analyses were performed using SPSS 12.0
software. Each experiment was repeated 3 times, unless otherwise
indicated. All data are presented as the means ± standard deviation
(SD). Comparisons between groups were carried out using the
unpaired Student's t-test. A p-value of <0.05 was considered to
indicate a statistically significant difference for all
analyses.
Results
Expression of dbpA is increased in CRC
tumor samples and varies in CRC cell lines
To explore the role of dbpA in human colorectal
tumors, we began by analyzing the expression of dbpA in human CRC
tissues. Paired colorectal tumor samples were obtained from 44
patients and dbpA expression was examined by IHC staining.
Approximately 79.5% (35/44; p<0.001) of the CRC samples
exhibited a positive dbpA expression, while 20.5% (9/44) of the CRC
samples were negative for dbpA expression; only 9.1% (4/44) of the
adjacent normal tissue samples exhibited a positive dbpA expression
(Table I). We also found that
dbpA was predominantly expressed in the cytoplasm of the CRC cells
rather than in the nucleus or the cytomembrane region (Fig. 1A, top panels). Furthermore, IHC
analysis revealed that the level of dbpA expression correlated with
the depth of invasion in CRC. With the progression of the depth of
invasion of the CRC tumor tissues (T1 to T4), the level of dbpA
expression gradually increased (Fig.
1B, bottom panels). Collectively, the dbpA expression levels
were significantly higher in the CRC tissues than in the non-tumor
tissues, which implies that dbpA may facilitate tumorigenesis in
the colon.
 | Table IExpression of dbpA in colorectal
tumor and adjacent normal tissue samples. |
Table I
Expression of dbpA in colorectal
tumor and adjacent normal tissue samples.
| Total | DbpA (−) | DbpA (+) | P-value |
|---|
| Tumors | 44 | 9 (20.5%) | 35 (79.5%) | 0.001a |
| Normal tissues | 44 | 40 (90.9%) | 4 (9.1%) | |
In addition, the expression levels of dbpA were
assessed in different CRC cell lines, namely in the RKO, SW480,
LoVo, DLD-1, SW1463, HT-29, SW620 cells, and in the normal
colorectal mucosa cell line, FHC. The results of RT-qPCR revealed
that the mRNA expression levels of dbpA varied among the cell
lines. In the CRC cell lines, the expression levels from lowest to
highest were: LoVo, DLD-1, RKO, HT-29, SW1463, SW480 and SW620
cells (Fig. 1B); however, no dbpA
expression was detected in the normal colorectal mucosa cell line,
FHC, which was consistent with the results of western blot analysis
(Fig. 1C). The SW620 cells were
selected for use in futher experiments as they exhibited the
highest expression of dbpA. Our findings indicated that dbpA is
dominantly expressed in CRC tissues or cell lines rather than in
normal colon tissues or cells, and the expression profile differs
amongst CRC cell lines.
dbpA expression correlates with different
clinicopathological parameters
Considering that the level of dbpA in CRC tissues
was significantly higher than that in non-tumor tissues, the
correlations between dbpA expression and clinicopathological
parameters in CRC were analyzed by Pearson's Chi-square test. We
subdivided the CRC-positive cases into 3 groups according to the
criteria described in the Materials and methods. The percentage of
dbpA weak expression (+) was 28.6% (10/35), moderate expression
(++) was 31.4% (11/35), and strong expression was 40.0% (14/35).
Correlation analysis demonstrated that the strong expression of
dbpA was significantly associated with the degree of
differentiation (p<0.001), the depth of invasion (p<0.001),
lymphatic metastasis (p<0.001), vessel invasion (p<0.001) and
the TNM stage (p<0.05), but not with age, gender, tumor size,
stage, type, distant metastasis, or surgical method (Table II). Therefore, we hypothesized
that dbpA may be considered as a potential unfavorable prognostic
biomarker for patients with CRC.
| Table IIAssociation between dbpA expression
and clinicopathologic factors in patient with CRC. |
Table II
Association between dbpA expression
and clinicopathologic factors in patient with CRC.
| Parameters | The level dbpA
expression
| P-value |
|---|
| + | ++ | +++ |
|---|
| Age (years) | | | | |
| >60 | 8 | 6 | 9 | 0.431 |
| ≤60 | 2 | 5 | 5 | |
| Gender | | | | |
| Male | 7 | 7 | 8 | 0.708 |
| Female | 3 | 4 | 6 | |
| Tumor size
(cm) | | | | |
| >4 | 6 | 5 | 4 | 0.528 |
| ≤4 | 4 | 6 | 10 | |
| Degree of
differentiation | | | | |
| Well | 7 | 2 | 0 | <0.001b |
| Moderate | 3 | 9 | 5 | |
| Poor | 0 | 0 | 9 | |
| Invasion depth | | | | |
| T1 | 0 | 0 | 0 | <0.001b |
| T2 | 1 | 0 | 0 | |
| T3 | 5 | 5 | 2 | |
| T4 | 4 | 6 | 12 | |
| Tumor site | | | | |
| Colon | 5 | 2 | 6 | 0.900 |
| Rectum | 5 | 9 | 8 | |
| TNM stage | | | | |
| I+II | 5 | 4 | 0 | <0.05a |
| III+IV | 5 | 7 | 14 | |
| Lymphatic
metastasis | | | | |
| N0 | 7 | 5 | 1 | <0.001b |
| N1 | 3 | 4 | 10 | |
| N2 | 0 | 2 | 3 | |
| Distant
metastasis | | | | |
| M0 | 9 | 11 | 11 | 0.485 |
| M1 | 1 | 0 | 3 | |
| Tumor type | | | | |
| Infiltrate
type | 1 | 0 | 2 | 0.788 |
| Ulcerative
type | 9 | 9 | 12 | |
| Protrude type | 0 | 2 | 0 | |
| Vessel
invasion | | | | |
| 0 | 10 | 10 | 4 | <0.001b |
| 1 | 0 | 1 | 10 | |
| Surgical
method | | | | |
| Dixon | 5 | 7 | 7 | 0.670 |
| Mile's | 0 | 2 | 7 | |
| Hemicolectomy | 5 | 2 | 0 | |
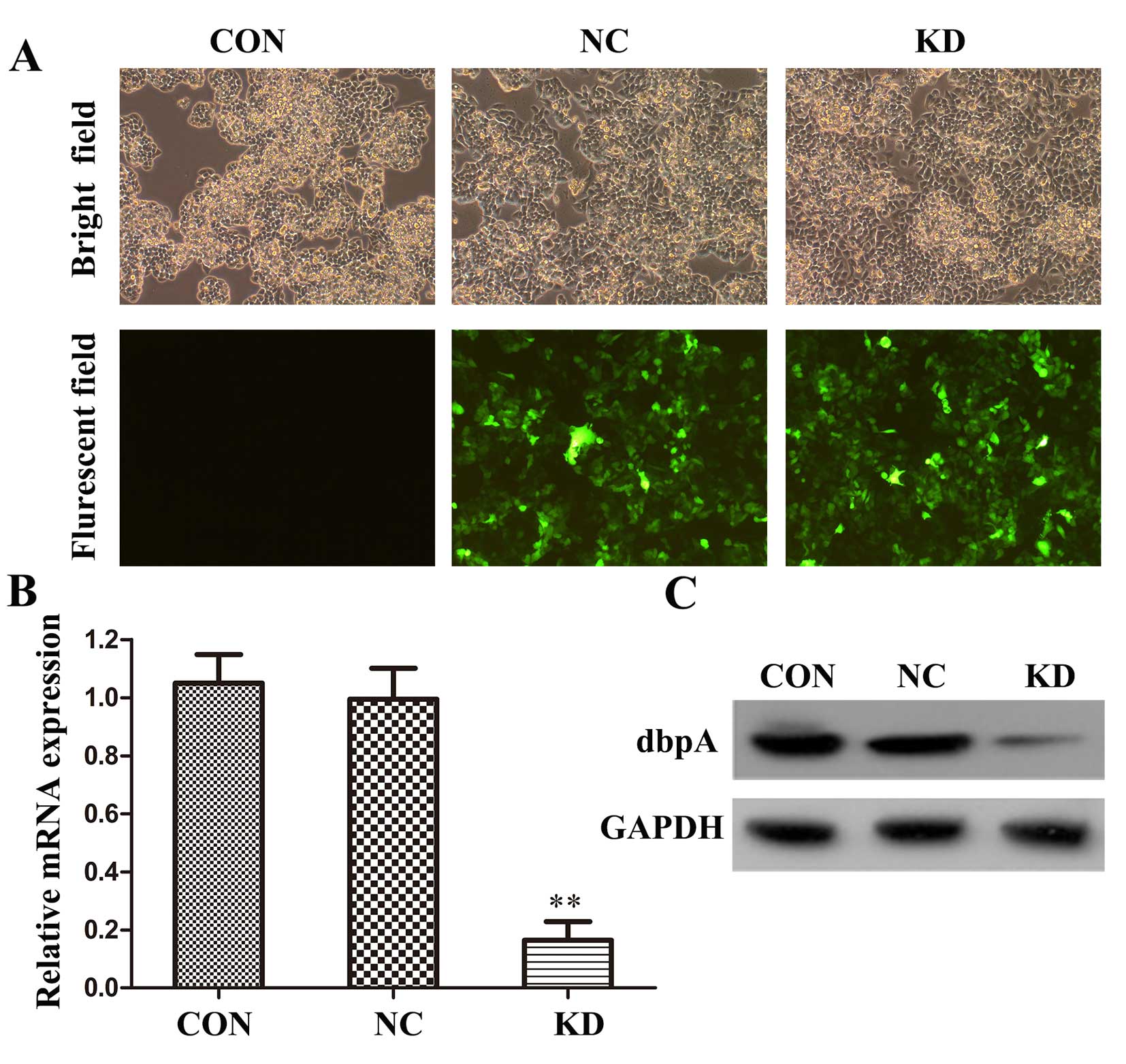
Effect of shRNA on dbpA expression in
SW620 cells
Lentivirus-mediated shRNA inferference was applied
to suppress the expression of dbpA in the SW620 cells. The
efficiency of shRNA-dbpA-Lv or shNC-dbpA-Lv was detected by
fluorescent microscopy in the infected cells. The results revealed
that a great proportion of cells was infected with the shRNA
(Fig. 2A); the infected
percentage was >90% (data not shown).
RT-qPCR and western blot analysis were performed to
further examine the silencing efficiency of the shRNA against dbpA.
At 72 h post-transfection, the mRNA level of dbpA in the
shRNA-dbpA-Lv group (KD) was significantly decreased when compared
to the shNC-dbpA-Lv (NC) or control (CON) group (p<0.01;
Fig. 2B). Western blot analysis
also verified that the protein expression of dbpA was markedly
decreased in the KD group in comparison to the CON or NC groups
(Fig. 2C); no significant
difference was observed between the NC and CON group. Therefore,
our results indicated that lentivirus-mediated dpbA RNAi
successfully and efficiently suppressed dbpA expression in the
SW620 cells.
Silencing of dbpA suppresses the
proliferation of SW620 cells
MTT assay was employed to assess the effects of the
silencing of dbpA expression on the proliferation of the SW620
cells in vitro. Cell proliferation was analyzed by MTT assay
once daily for 5 days. We found that the cells transfected with
shRNA-dbpA-Lv proliferated more slowly than the cells in the NC and
CON groups from 3 days onwards. In addition, on the 4th and 5th
day, the proliferation rates in the KD group were significantly
decreased (p<0.05 and p<0.01, respectively) when compared
with the NC group (Fig. 3A).
Downregulation of dbpA inhibits the
colony-forming ability of SW620 cells
Furthermore, when the SW620 cells were transfected
with dbpA-shRNA, negative-shRNA, or the empty vyector-transfected
cells were incubated for 14 days, the colony-forming capacity of
the SW620 cells was determined. The number of colonies was counted
following Giemsa staining. Our results revealed that the number
cell colonies in the KD group declined significantly (p<0.01) in
comparison with the control group (Fig. 3B and C). In addition, the size of
the colonies was markedly reduced in the KD group compared with the
control group (Fig. 3D). On the
whole, these findings indicated that the silencing of dbpA by
lentivirus-mediated RNAi efficiently suppressed SW620 cell
proliferation in vitro.
Silencing of dbpA expression induces
SW620 cell cycle arrest
Flow cytometry was adopted to detect cell cycle
progression in the dbpA-shRNA-transfected cells. The results
revealed that compared with the NC group, the proportion of
dbpA-shRNA-transfected cells in the S phase was significantly
decreased from 43.92 to 25.19 (p<0.01), while the ratio of cells
in the G0/G1 phase was significantly increased from 35.24 to 54.45
(p<0.01), indicating that dbpA-shRNA interfered with the
distribution of the cell cycle, leading to cell cycle arrest at the
G1 phase in the KD group (Fig.
4). However, no significant difference in the number of cells
in the S phase or the G0/G1 phase was observed between the cells in
the CON group and NC group. Thus, knocking down dbpA expression
suppressed SW620 cell proliferation by leading to cell cycle arrest
at the G0/G1 phase.
Knockdown of dbpA expression promotes the
apoptosis of SW620 cells
The balance between the cell cycle and cell
apoptosis is the key premise to maintain tumorigenesis in patients
(17). Hence, we considered it
necessary to examine the effects of dbpA silencing on SW620 cell
apoptosis in this study. Apoptosis was determined by FITC-labeled
Annexin V/PI double staining and flow cytometric analysis. The
results revealed that the silencing of the expression of dbpA in
the KD group significantly increased the apoptotic rate when
compared with the NC group (p<0.01; Fig. 5A and B). These results indicated
that the silencing of dbpA expression promoted cell apoptosis which
correlated with the inhibitory effects on cell proliferation.
Silencing dbpA decreases tumorigenesis in
CRC in vivo
Since the knocking down of dbpA exerted an
inhibitory effect on tumorigenesis in vitro, we considered
it crucial to assess its effects in vivo. A xenograft tumor
model was established by subcutaneously injecting normal cells
(empty vector-transfected cells), negative control- or
dbpA-shRNA-transfected SW620 cells into nude mice. After 14 days,
the average tumor volume in the mice in the KD group was markedly
smaller than that in the NC or CON groups (Fig. 6A). Furthermore, through monitoring
the physical conditions of the injected mice, at 30 days
post-treatment, the mice in the NC and CON groups reserved less
subcutaneous fat and exhibited more weight loss than the mice in
the KD group. At the 35th day, during the process of tumor
isolation, we found that in the NC and CON groups, the surfaces of
tumors appeared uneven with ulcerations and bleeding, while in the
KD group, the tumors appeared more complete and had smooth outer
members (Fig. 6C). The average
weight of the tumors in the KD group was significantly smaller than
that in the NC group, with the tumor inhibition rate at 93.97%
(p<0.05) (Fig. 6B and C).
However, no difference was observed either in tumor volume or tumor
weight between the CON group and NC group. In addition, western
blot analysis verified that the protein expression of dbpA was
downregulated in the KD group in comparison to the NC or CON groups
(Fig. 6D). Therefore, our results
demonstrated that the suppression of the expression of dbpA in the
SW620 cells significantly inhibited tumor growth in
vivo.
Discussion
dbpA as a member of the Y-box protein family, has
aroused great interest in recent years and has been reported to be
involved in the development of malignant tumors, such as
hepatocellular carcinoma (18)
and gastric cancer (16). dbpA
regulate the proliferation of epithelial cells, accelerates
inflammation-induced hepatocarcinogenesis, and plays a vital role
in the pathogenesis and development of gastric cancer (16). Although dbpA is considered as an
oncogene in several tumors, its pathogenic mechanisms of action in
CRC remain unknown. Hence, in this study, we examined dbpA
expression in tumor samples from patients with CRC, and clarified
that dbpA was overexpressed in CRC tumor tissues compared to paired
adjacent normal tissues. To the best of our knowledge, we provided
the first evidence to verify that dbpA plays an important role in
CRC tumorigenesis.
In this study, 35 CRC cases with various TNM stages
(I–IV) were selected to investigate the association between the
clinical characteristics of CRC tumors and dbpA expression levels.
Our findings illustrated that dbpA expression positively correlated
with the degree of differentiation, the depth of invasion, vessel
invasion and an advanced TNM stage, which are all the key features
to accelerate cancer development (19). Furthermore, IHC staining revealed
that dbpA was mainly expressed in the cytoplasm and was associated
with the progression of CRC in patients. Yasen et al had
emphasized that both the cytoplasmic expression and the nuclear
localization of dbpA, as a significant prognostic marker, was
responsible for the advanced stages of hepatocellular carcinoma
(14). However, during our study,
no dbpA expression was found in the nucleus in our CRC cases. These
exiting data indicate that the localization of dbpA may alternate,
depending on advanced cancer stages or different cancer types.
Further studies with larger sample sizes are warranted in order to
deeply investigate the variation of dbpA localization in CRC
progression.
In the present study, we found that dbpA expression
varied in different CRC cell lines. The SW620 cells derived from
colorectal adenocarcinoma had the highest expression of dbpA. Thus,
for this reason, we selected these cells for use in our subsequent
experiments. Small interference RNA and lentiviral vector-mediated
RNAi have been extensively used as efficient tools to investigate
the specific genes involved in abnormal cell proliferation and are
regarded as promising therapeutic methods to deal with malignant
tumors (20,21). Lentivirus-mediated shRNA
interference was conducted in this study to inhibit dbpA expression
in the SW620 cells. As a result, the knockdown of dbpA suppressed
SW620 cell proliferation by inducing cell cycle arrest in the G0/G1
phase in vitro. Furthermore, through establishing a
xenograft model using nude mice, we found that dbpA silencing
significantly inhibited tumor growth and tumorigenesis in
vivo. Therefore, modulating dbpA expression resulted in changes
in cell proliferation both in vitro and in vivo.
Abnormal cell proliferation is considered as the key element in the
progression of cancer (22). The
occurrence of cell proliferation in physiological conditions is
always regulated by specific molecular signaling pathways (23). For example, the duplicate
progression of a cell is mediated by a group of proteins known as
cyclins (24). Cyclins act in
part with the cyclin-dependent kinases (CDKs) to phosphorylate key
substrates that participate in each phase of the cell cycle
(25). Researchers have reported
that increased cyclin expression is frequently observed in human
malignancies (26). For example,
our previous study also proved that the silencing of dbpA
suppressed the transcription of cyclin D1 and resulted in the
inhibition of the proliferation of gastric cancer cells (16); this may also explain the similar
results obtained in our present study.
Apoptosis is a natural way of removing aged cells
from the living body (27),
whereas under cancer conditions, the uncontrolled regulation of
apoptotic signals assists cancer cells to escape from this
programmed death and leads to abnormal proliferation (28). Various molecular signaling
pathways have been found to be involved in this complex program,
such as the B-cell lymphoma 2 (Bcl-2) signaling pathway (29), the heat shock protein signaling
pathway (30) and the proteasome
pathway (31). In this study, we
confirmed that the silencing of dbpA significantly increased the
apoptosis of SW620 cells, suggesting that dbpA may regulate
apoptosis by activating or inactivating certain signaling pathways.
The exact underlying molecular mechanisms responsible for the
apoptosis observed by the silencing of dbpA in CRC requires further
investigation.
In conclusion, our study confirmed that dbpA was
overexpressed in CRC tissues and cell lines. The high expression
level of dbpA closely correlated with certain clinicopathological
parameters and tumor progression in CRC. Lentivirus-mediated RNAi
of dbpA inhibited SW620 cell growth in vitro and
tumorigenesis in vivo. Furthermore, the silencing of dbpA
induced cell cycle arrest and promoted cell apoptosis. Hence, our
findings illustrate the biological significance of dbpA in
tumorigenesis in CRC and provide scientific evidence to develop a
novel therapeutic target for the more effective treatment of
patients with CRC. Moreover, further investigations are required in
order to comprehensively reveal the intrinsic mechanisms of action
of dbpA in CRC.
Acknowledgments
The present study was funded by grants from the
National Natural Science Foundation of China (no. 81172363/H1617)
and the Natural Science Foundation of Shaanxi Province
(2014JM4089).
Reference
|
1
|
Siegel R, Desantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Shi Q, Mandrekar SJ and Sargent DJ:
Predictive biomarkers in colorectal cancer: usage, validation, and
design in clinical trials. Scand J Gastroenterol. 47:356–362. 2012.
View Article : Google Scholar
|
|
3
|
Compton CC: Colorectal carcinoma:
diagnostic, prognostic, and molecular features. Mod Pathol.
16:376–388. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Custodio A and Feliu J: Prognostic and
predictive biomarkers for epidermal growth factor receptor-targeted
therapy in colorectal cancer: beyond KRAS mutations. Crit Rev Oncol
Hematol. 85:45–81. 2013. View Article : Google Scholar
|
|
5
|
De Mattos-Arruda L, Dienstmann R and
Tabernero J: Development of molecular biomarkers in individualized
treatment of colorectal cancer. Clin Colorectal Cancer. 10:279–289.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Linnekamp JF, Wang X, Medema JP and
Vermeulen L: Colorectal cancer heterogeneity and targeted therapy:
a case for molecular disease subtypes. Cancer Res. 75:245–249.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kudo S, Mattei MG and Fukuda M:
Characterization of the gene for dbpA, a family member of the
nucleic-acid-binding proteins containing a cold-shock domain. Eur J
Biochem. 231:72–82. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Petruzzelli R, Gaudino S, Amendola G,
Sessa R, Puzone S, Di Concilio R, d'Urzo G, Amendolara M, Izzo P
and Grosso M: Role of the cold shock domain protein A in the
transcriptional regulation of HBG expression. Br J Haematol.
150:689–699. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hasegawa SL, Doetsch PW, Hamilton KK,
Martin AM, Okenquist SA, Lenz J and Boss JM: DNA binding properties
of YB-1 and dbpA: binding to double-stranded, single-stranded, and
abasic site containing DNAs. Nucleic Acids Res. 19:4915–4920. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kohno K, Izumi H, Uchiumi T, Ashizuka M
and Kuwano M: The pleiotropic functions of the Y-box-binding
protein, YB-1. BioEssays. 25:691–698. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wolffe AP: Structural and functional
properties of the evolutionarily ancient Y-box family of nucleic
acid binding proteins. BioEssays. 16:245–251. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Ladomery M and Sommerville J: A role for
Y-box proteins in cell proliferation. BioEssays. 17:9–11. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Tobita H, Kajino K, Inami K, Kano S, Yasen
M, Imamura O, Kinoshita Y and Hino O: Gene expression profile of
DNA binding protein A transgenic mice. Int J Oncol. 29:673–679.
2006.PubMed/NCBI
|
|
14
|
Yasen M, Kajino K, Kano S, Tobita H,
Yamamoto J, Uchiumi T, Kon S, Maeda M, Obulhasim G, Arii S, et al:
The up-regulation of Y-box binding proteins (DNA binding protein A
and Y-box binding protein-1) as prognostic markers of
hepatocellular carcinoma. Clin Cancer Res. 11:7354–7361. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Arakawa Y, Kajino K, Kano S, Tobita H,
Hayashi J, Yasen M, Moriyama M, Arakawa Y and Hino O: Transcription
of dbpA, a Y box binding protein, is positively regulated by E2F1:
implications in hepatocarcinogenesis. Biochem Biophys Res Commun.
322:297–302. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wang GR, Zheng Y, Che XM, Wang XY, Zhao
JH, Wu KJ, Zeng J, Pan CE and He DL: Upregulation of human DNA
binding protein A (dbpA) in gastric cancer cells. Acta Pharmacol
Sin. 30:1436–1442. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Evan GI and Vousden KH: Proliferation,
cell cycle and apoptosis in cancer. Nature. 411:342–348. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yasen M, Obulhasim G, Kajino K, Mogushi K,
Mizushima H, Tanaka S, Tanaka H, Hino O and Arii S: DNA binding
protein A expression and methylation status in hepatocellular
carcinoma and the adjacent tissue. Int J Oncol. 40:789–797.
2012.
|
|
19
|
Fallowfield LJ and Fleissig A: The value
of progression-free survival to patients with advanced-stage
cancer. Nat Rev Clin Oncol. 9:41–47. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Guo W, Chen W, Yu W, Huang W and Deng W:
Small interfering RNA-based molecular therapy of cancers. Chin J
Cancer. 32:488–493. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sumimoto H and Kawakami Y: Lentiviral
vector-mediated RNAi and its use for cancer research. Future Oncol.
3:655–664. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Von Wangenheim KH and Peterson HP: The
role of cell differentiation in controlling cell multiplication and
cancer. J Cancer Res Clin Oncol. 134:725–741. 2008. View Article : Google Scholar
|
|
23
|
Feitelson MA, Arzumanyan A, Kulathinal RJ,
Blain SW, Holcombe RF, Mahajna J, Marino M, Martinez-Chantar ML,
Nawroth R, Sanchez-Garcia I, et al: Sustained proliferation in
cancer: mechanisms and novel therapeutic targets. Semin Cancer
Biol. 35(Suppl): S25–S54. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Canavese M, Santo L and Raje N: Cyclin
dependent kinases in cancer: potential for therapeutic
intervention. Cancer Biol Ther. 13:451–457. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Węsierska-Gądek J and Maurer M: Promotion
of apoptosis in cancer cells by selective purine-derived
pharmacological CDK inhibitors: one outcome, many mechanisms. Curr
Pharm Des. 17:256–271. 2011. View Article : Google Scholar
|
|
26
|
Casimiro MC, Velasco-Velázquez M,
Aguirre-Alvarado C and Pestell RG: Overview of cyclins D1 function
in cancer and the CDK inhibitor landscape: past and present. Expert
Opin Investig Drugs. 23:295–304. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Elmore S: Apoptosis: a review of
programmed cell death. Toxicol Pathol. 35:495–516. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ouyang L, Shi Z, Zhao S, Wang FT, Zhou TT,
Liu B and Bao JK: Programmed cell death pathways in cancer: a
review of apoptosis, autophagy and programmed necrosis. Cell
Prolif. 45:487–498. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Yating Q, Yuan Y, Wei Z, Qing G, Xingwei
W, Qiu Q and Lili Y: Oxidized LDL induces apoptosis of human
retinal pigment epithelium through activation of ERK-Bax/Bcl-2
signaling pathways. Curr Eye Res. 40:415–422. 2015. View Article : Google Scholar
|
|
30
|
Qi Z, Shen L, Zhou H, Jiang Y, Lan L, Luo
L and Yin Z: Phosphorylation of heat shock protein 27 antagonizes
TNF-α induced HeLa cell apoptosis via regulating TAK1
ubiquitination and activation of p38 and ERK signaling. Cell
Signal. 26:1616–1625. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Mohammad RM, Muqbil I, Lowe L, Yedjou C,
Hsu HY, Lin LT, Siegelin MD, Fimognari C, Kumar NB, Dou QP, et al:
Broad targeting of resistance to apoptosis in cancer. Semin Cancer
Biol. 35(Suppl): S78–S103. 2015. View Article : Google Scholar : PubMed/NCBI
|