Introduction
Vascular endothelial injury is the pathological and
physiological basis for cardiovascular diseases, including
atherosclerosis and hypertension (1,2).
Recently, the enhancement of re-endothelialization has been
recognized as a therapeutic option to repair injured vessels
(3). During the process of
re-endothelialization, it was previously considered that the
adjacent endothelial cells (ECs) of injured vessels were the major
contributors to the regeneration of the injured endothelium.
However, their low proliferative potential limits their application
in substituting the damaged endothelium following damage. Recently,
emerging evidence has confirmed the important roles of endothelial
progenitor cells (EPCs) in vascular injury due to their capacity to
home to injury sites, and to differentiate into mature ECs to
participate in re-endothelialization and angiogenesis following
vascular injury (4–6). It has been reported that bone marrow
(BM)-derived EPCs can control the angiogenic switch to induce tumor
angiogenesis (7). Mounting
evidence suggests that EPCs are the endogenous repair mechanism for
maintaining the integrity of the endothelial monolayer (8). However, the underlying mechanisms
involved in the regulation of EPC properties remain undefined.
Inhibitor of DNA-binding (ID) proteins are a
helix-loop-helix (HLH) family of transcription factors that play
pivotal roles in various developmental processes, such as cell
differentiation, development, migration and angiogenesis (9). As an important member of this
family, ID1 has drawn increasing attention due to its important
function in cell growth, migration and differentiation. ID1 lacks a
DNA-binding domain, and can act as a dominant-negative regulator of
the basic HLH (bHLH) transcription factors, which have been shown
to be implicated in endothelial cell angiogenic activities
(10). A previous study
demonstrated the abundant expression of ID1 during blood vessel
formation (11). ID1-knockout
mice have been shown to exhibit abnormal angiogenesis (12). Moreover, the ectopic expression of
ID1 has been shown to enhance EC proliferation and migration
(13,14). Recently, ID1 has been shown to
positively regulate the proliferation and migration of
spleen-derived EPCs (15).
Furthermore, the silencing of ID1 reduces human ovarian cancer EPC
angiogenesis (16). Although
several studies have demonstrated the crucial role of ID1 in EPC
angiogenesis, the mechanisms through which ID1 triggers
angiogenesis remain poorly understood.
In this study, the ID1 interaction partners were
analyzed using immunoprecipitation analysis, and the basic HLH
transcription factor, E2-2, was identified. We investigated the
mechanisms through which ID1 regulates E2-2 expression, as well as
its role in ID1-indcued EPC proliferation and migration.
Materials and methods
Reagents and antibodies
Unless otherwise specified, all reagents used were
from Sigma-Aldrich (St. Louis, MO, USA). The Dil-labeled acetylated
low-density lipoprotein (Dil-Ac-LDL; #BT-902) was obtained from
Biomedical Technologies, Inc. (Stoughton, MA, USA). Fluorescein
isothiocyanate-Ulex europaeus lectin-1 (FITC-UEA1; #L9006)
was obtained from Sigma-Aldrich. Fluorescein isothiocyanate
(FITC)-conjugated antibodies against mouse stem cell antigen-1
(Sca-1; #553335), vascular endothelial growth factor receptor 2
(VEGFR2; #560680) and isotype control (#553929) were purchased from
BD Biosciences (San Diego, CA, USA). Rabbit polyclonal antibodies
against mouse ID1 (ab52998) and E2-2 (ab185736) were obtained from
Abcam (Cambridge, MA, USA). Antibodies against mouse Ki67 (sc-7846)
and cyclin D1 (sc-753) were from Santa Cruz Biotechnology, Inc.
(Santa Cruz, CA, USA). Rabbit monoclonals against
p27Kip1 (ab62364), β-actin (ab6276), matrix
metalloproteinase (MMP)-2 (ab92536) and MMP-9 (ab58803) were from
Abcam.
Preparation and identification of
EPCs
Spleen-derived EPCs were isolated as previously
described (15). Briefly, male
Kunming mice (n=5, weighing 25–30 g; Kunming General Hospital of
Chengdu Military Command, Kunming, China) were employed to prepare
the spleens. All mice were allowed to acclimatize for at least 2
weeks under normal husbandry conditions. The mice were then
euthanized by an overdose of anesthesia with pentobarbital sodium
(150 mg/kg, ip) and the tissues of the spleens were harvested
aseptically by surgery. The animal experimental procedures were
carried out after obtaining approval from the Institution Animal
Care and Use Committee of Kunming General Hospital of Chengdu
Military Command, Kunming, China. Total mononuclear cells from the
spleens were isolated by density gradient centrifugation with
Histopaque-1077 at 400 × g for 20 min. Approximately 24 h later,
the unattached cells were discarded. The cells were then cultured
in low-glucose Dulbecco's modified Eagle's medium (DMEM)
supplemented with 10% FCS and 10 ng/ml VEGF at 37°C with 5%
CO2. The medium was replaced every 2 days thereafter.
After 4 days of culture, colony-forming cells were recognized as
adherent cells, and were cultured continuously for further
experiments. To identify the EPCs, the adherent cells were
incubated with Dil-Ac-LDL (10 mg/ml) for 4 h. Following fixation
with 4% paraformaldehyde, the cells were then incubated with
FITC-labeled lectin (UEA-1, 10 mg/ml) for 1 h. Dual-stained cells
(positive for Dil-Ac-LDL and UEA-1) were identified as EPCs.
Cellular nuclei were stained with DAPI (Sigma-Aldrich). The
fluorescent images were recorded under a fluorescence microscope
(IX51; Olympus, Tokyo, Japan).
Flow cytometric analysis
For further characterization of the EPCs, the
dual-stained cells positive for Dil-Ac-LDL and UEA-1 were incubated
with antibodies against mouse Sca-1 and VEGFR2 at 37°C with 5%
CO2. Approximately 30 min later, the cells were
evaluated by flow cytometry using a FACSAria flow cytometer (BD
Immunocytometry Systems, Franklin Lakes, NJ, USA).
Adenoviruses and plasmids
Adenoviral vectors expressing Flag-IDl were prepared
by inserting the coding sequence of ID1 into the pShuttle-CMV
vector using the AdEasy system as previously described (17). After recombination of
pShuttle-CMV-flag-ID1 with pADEasy-1, the obtained plasmids were
transfected into 293T cells (ATCC, Manassas, VA, USA) to amplify
the adenoviruses. EPCs were infected with recombinant ID1
adenovirus (AD-ID1) and empty vector (Ad). Cells without any
treatment were defined as the control group. The adenoviralvector
expressing Myc-E2-2 was also generated using the AdEasy system, as
previously described (15).
The full-length cDNA of E2-2 was obtained by PCR, as
previously described (18) and
then cloned into the pMD19-T vector, followed by subcloning into
pAdTrack-CMV to generate pAdTrack-E2-2. The resulting plasmids were
transfected into 293T cells.
ID1ΔHLH was generated by Pfx DNA polymerase
(Invitrogen, Carlsbad, CA, USA) using mouse ID1 as a template with
previously published methods (18).
Immunoprecipitation assay
To analyze the interaction between ID1 and E2-2, the
prepared plasmids were transfected into COS7 cells (ATCC).
Approximately 40 h later, the cells were lysed with lysis buffer.
Following centrifugation, the supernatants were pre-cleared with
protein G-Sepharose beads (GE Healthcare, Little Chalfont,
Buckinghamshire, UK) for 30 min at 4°C. The antibodies against Flag
(Sigma-Aldrich) were then added for a further 2-h incubation. The
protein complexes were immunoprecipitated by incubating with
G-Sepharose Fast Flow beads, which had been pre-equilibrated in
lysis buffer, for 30 min. Beads collected by centrifugation were
washed and resuspended in an equal volume of 5X sodium dodecyl
sulfate (SDS) loading buffer. Immunoprecipitated proteins were
separated by 10% SDS-PAGE, followed by transfer to Hybond-C Extra
membranes. The membranes were incubated with anti-Myc 9E10 antibody
(ab32; Abcam). For the detection of the endogenous interaction
between ID1 and E2-2 in EPCs, cells were stimulated with bone
morphogenetic protein 6 (BMP6; P20722; R&D Systems;
Minneapolis, MN, USA)) for 4 h, and the immunoprecipitation was
carried out with antibodies against E2-2 followed by western blot
analysis with an anti-ID1 antibody.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
The transfected EPCs were incubated for 48 h prior
to RNA extraction. Total RNA was extracted using TRIzol reagent
according to the manufacturer's instructions (Biostar, Shanghai,
China). Subsequently, 5 μg of total RNA of each sample was
reverse transcribed into first-strand cDNA using the Promega
Reverse Transcription System (Promega, Southampton, UK). The cDNA
was used as the template for qPCR in a final volume of 20 μl
with the SYBR Premix Ex Taq II kit (Takara, Otsu, Japan). The
following specific primers were used: ID1 sense,
5′-AGTGGTGCTTGGTCTGTCG-3′ and antisense, 5′-GCAGGTCCCTGATGTAGTCG-3′
and E2-2 sense, 5′-ATGGCTGCCTTAGGGACGGACA-3′ and antisense,
5′-AGGACCCTGAGCTACTTCTG-3′. β-actin was used as the endogenous
control, and all results were calculated using the
2−ΔΔCt method, as previously described (19).
Western blot analysis
Following lysis with lysis buffer (Beyotime,
Nantong, China), the protein concentrations were determined using
the micro-BCA protein assay (Pierce, Rockford, IL, USA). The
protein was separated by SDS-PAGE using a 12% polyacrylamide gel
followed by electroblotting onto polyvinylidene difluoride (PVDF)
membranes. After blocking the non-specific binding with 5% non-fat
milk, the membranes were incubated with the primary antibodies
against ID1, E2-2, Ki67, p27Kip1, cyclin D1, MMP-9 and
MMP-2. Horseradish peroxidase (HRP)-conjugated secondary antibodies
were added for a further incubation for 1 h. The binding signals
were determined by electrogenerated chemilummescence (ECL)
detection reagent (Beyotime). Blots against β-actin served as the
loading control, and the results were normalized according to
β-actin.
Transcriptional reporter assay
To examine the effects of ID1 on E2-2-induced
transcription, the MCKpfos-luc reporter construct consisting of 4
E-box elements system was used as previously described (20). Briefly, the EPCs were seeded at a
density of 5×104 cells/well in 24-well plates 1 day
prior to transfection. The cells were then transfected with
MCKpfos-luc, E2-2, and either Ad-ID1 or ID1ΔHLH using Lipofectamine
(Life Technologies, Carlsbad, CA, USA). Approximately 40 h later,
the lysates were collected and were subsequently analyzed for
luciferase activity using a luciferase assay system (Promega,
Madison, WI, USA).
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The EPCs were collected and seeded into 96-well
plates at a density of 2×106 cells/well. After
preconditioning with the indicated treatments, the cells were
cultured with fresh medium containing 15 μl MTT reagent for
a further 5 h at 37°C. The supernatant was then discarded, and 200
μl of DMSO were added to each well to dissolve the formazan.
The absorbance at 490 nm was measured to analyze cell proliferation
using a micro-enzyme-linked immunosorbent assay (ELISA) reader
(Bio-Rad, Hercules, CA, USA).
Cell migration assay
The migration of EPCs was analyzed by a modified
Boyden's chamber assay as previously described (15). Briefly, the cells
(1×106/well in 200 μl serum-free medium) were
seeded into the upper chamber. DMEM medium containing 10% FCS and
50 ng/ml VEGF was added to the lower chamber as the
chemoattractant. Approximately 8 h later, the non-migrating cells
on the upper surface of the 8-μm filters were removed with a
cotton swab. Cells that had penetrated to the lower surface were
fixed with 4% paraformaldehyde and stained with 0.1% crystal
violet. The cell migration ability was evaluated by counting cells
in 5 randomly selected visual fields with an inverted microscope at
×100 magnification.
Statistical analysis
All results are presented as the means ± SD of at
least 3 independent measurements in triplicate. SPSS 13.0 software
was used to analyze all data. Statistically significant differences
between different groups were determined based on the Student's
t-test and ANOVA (analysis of variance). A value of P<0.05 was
considered to indicate a statistically significant difference.
Results
Characterization of spleen-derived
EPCs
After 5–7 days of culture, the attached EPCs derived
from the spleens of mice exhibited a spindle-shaped morphology.
EPCs are characterized by being double positive for Dil-Ac-LDL
uptake and lectin binding (Fig.
1A). FACS analysis revealed expression of the stem cell marker,
Sca-1, and the endothelial marker, VEGFR2. The Sca-1-positive cells
accounted for 84.2±3.6% of the cells, while the VEGFR2-positive
cells accounted for 58.3±4.1% of the cells (Fig. 1B).
Identification of E2-2 as an interaction
partner of ID1
To determine the mechanisms through which ID1
regulates EPC proliferation and migration, we investigated the
protein of E2-2, which has been proven to interact with human ID1
using a yeast two-hybrid system in a previous study (18). To demonstrate the interaction
between endogenous ID1 and E2-2, the EPCs were stimulated with BMP6
for 4 h to induce the expression of ID1. Following lysis and
immunoprecipitation, western blot analysis revealed that endogenous
ID1 formed a complex with endogenous E2-2 in the EPCs (Fig. 2A). Moreover, following
transfection with Myc-E2-2 and Flag-ID1 into the COS7 cells, the
complex between ID1 and E2-2 was also observed (Fig. 2C). It is known that ID HLH
proteins often act as dominant-negative regulators of bHLH
transcriptional regulators. To further elucidate the mechanisms
through which ID1 interacts with E2-2, we constructed ID1 mutants
lacking the HLH domains (Fig.
2B). An immunoprecipitation assay revealed that E2-2 did not
interact with ID1ΔHLH. Taken together, these results suggest that
ID1 heterodimerizes with E2-2 via its HLH domain.
ID1 suppresses the expression of E2-2 and
E2-2-mediated transcription
To examine the effects of the ID1-E2-2 interaction
on the E2-2 levels, an adenoviral vector carrying ID1 (Ad-ID1) was
constructed. Following the transfection of Ad-ID1 into EPCs, the
mRNA levels of ID1 were upregulated (P<0.05; Fig. 3A), accompanied by a similar
upregulation in its protein levels (P<0.05; Fig. 3B). The results of RT-qPCR revealed
that the overexpression of ID1 markedly suppressed the mRNA levels
of E2-2 (P<0.05; Fig. 3C), as
well as its protein levels (P<0.05; Fig. 3D). To further examine the effects
of ID1 on E2-2, the MCKpfos-luc reporter construct containing 4
E-box elements was transfected into the EPCs. As one of the
E-proteins, E2-2 transfection enhanced the reporter activity
(P<0.05; Fig. 3E). However,
the upregulation of ID1 significantly blocked E2-2-induced
MCKpfos-luc reporter activity (P<0.05; Fig. 3E). The above-mentioned data
verified that ID1 inhibited the expression and transcriptional
activity of E2-2.
HLH-dependent inhibition of E2-2 by
ID1
In order to investigate the mechanisms responsible
for the regulation of E2-2 by ID1, the functional role of its HLH
motif was analyzed. As shown in Fig.
2B, the deletion mutants of ID1 were constructed and
transfected into the EPCs. The results of RT-qPCR revealed that the
upregulation of ID1 inhibited the mRNA levels of E2-2 (P<0.05);
however, this did not occur in the cells transfected with ID1ΔHLH
(Fig. 3C). Similarly, the
corresponding decrease in the E2-2 protein levels triggered by the
overexpression of ID1 was attenuated when the cells were
transfected with ID1ΔHLH (P<0.05). The deletion of the HLH
domain in ID1 did not suppress the MCKpfos-luc reporter activity
induced by E2-2 (Fig. 3E),
suggesting that ID1 inhibited E2-2 expression and transcriptional
regulation via the HLH motif.
E2-2 is responsible for ID1-induced EPC
proliferation
EPC-based gene therapy can contribute to
re-endothelialization and can inhibit intimal hyperplasia following
vascular injury (5). Our previous
study demonstrated the important role of ID1 in regulating EPC
proliferation and migration (15). In this study, to investigate the
underlying mechanisms, we examined the effect of E2-2 during this
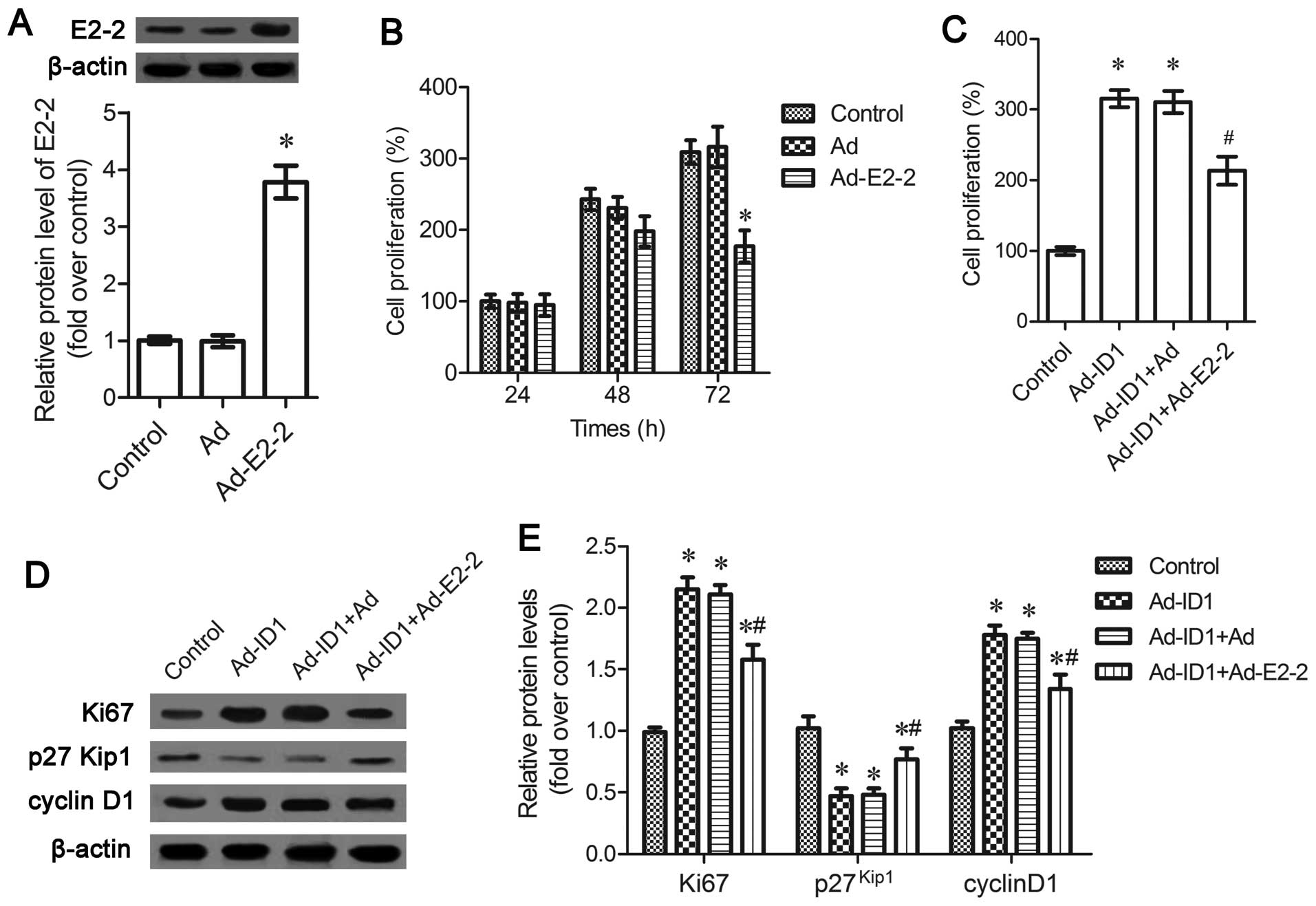
processs. Following infection with Ad-E2-2, the expression levels
of E2-2 were significantly upregulated (P<0.05; Fig. 4A). The upregulation of E2-2
antagonized EPC proliferation and induced a time-dependent
inhibition of cell proliferation (Fig. 4B). To further clarify the role of
E2-2 in ID1-induced cell proliferation, MTT assay was carried out
and the results corroborated that ID1 enhanced EPC proliferation,
which was attenuated by E2-2 upregulation (P<0.05; Fig. 4C). Simultaneously, the expression
of the cell proliferation marker, Ki67, indcued by ID1 was
downregulated in the cells transfected with Ad-E2-2 (P<0.05;
Fig. 4D and E). Moreover, the
inhibitory effects of ID1 on the expression of
proliferation-related cyclin-dependent kinase (Cdk) inhibitor
p27Kip1 were also abated in the Ad-E2-2-transfected
cells, concomitant with a decrease in cyclin D1 expression
(P<0.05; Fig. 4D and E).
ID1 induces EPC migration by suppressing
E2-2
As shown in Fig.
5A, the upregulation of E2-2 suppressed EPC migration
(P<0.05), and the overespression of ID1 significantly increased
the number of migrated EPC cells (P<0.05; Fig. 5B). However, this increase in cell
migration was significantly attenuated by the overexpression of
E2-2 (P<0.05; Fig. 5B).
Both MMP-2 and MMP-9 are associated with cell
migration (16,21). The results of western blot
analysis revealed that the enhanced expression of MMP-2 and MMP-9
induced by ID1 were also decreased in the E2-2-overexpressing cells
(P<0.05; Fig. 5C and D).
Overall, the above-mentioned results confirm that ID1 enhances EPC
migration in an E2-2-dependent manner.
The HLH domain is critical for
ID1-induced EPC proliferation and migration
Our above-mentioned findings demonstrated the
important role of the HLH motif in E2-2 expression. However, its
role in ID1 function remains unclear. To further clarify the
function of the HLH domain, we constructed ID1 mutants lacking the
respective HLH domains. The reslts of MTT assay revealed that ID1
overexpression induced EPC proliferation; however, this increase
was significantly inhibited in the ID1ΔHLH-transfected cells
(P<0.05; Fig. 6A). Moreover,
similar changes in proliferation-related proteins were also
observed, including Ki67 and cyclin D1. The expression of
p27Kip1 was inhibited by the overexpression of ID1 and
it was increased in the ID1ΔHLH-transfected cells (P<0.05;
Fig. 6C and D). The enhanced
number of cells that had migrated due to ID1 upregulation was also
blocked when ID1 lacked the HLH motif (P<0.05; Fig. 6B), which was accompanied by
corresponding changes in the expression of MMP-2 and MMP-9
(P<0.05; Fig. 6C and D). On
the whole, these results suggest that the HLH domain is pivotal in
ID1-induced EPC proliferation and migration.
Discussion
Endothelial damage is a major contributor to
atherosclerosis and other cardiovascular diseases (22). Recently, the therapeutic
significance of EPCs has drawn increasing attention due to their
critical function in re-endothelialization (4,5).
EPCs are recognized as the major source of cells participating in
endothelial repair and subsequently, re-endothelialization
following vascular injury. Their number and function are inversely
correlated with the risk factors for coronary artery disease
(23,24). Increasing evidence suggests that
the proliferation and migration of EPCs is the key mechanism in
re-endothelialization following vascular injury (25–28). Therefore, understanding the
mechanisms involved in re-endothelialization by EPCs will lead to
novel strategies for the treatment of vascular endothelial
injury-related diseases.
ID1 is a critical subfamily member of the HLH
proteins, and plays a pivotal role in angiogenesis (11). The gigh expression of ID1 has been
observed during vascular formation (11), and it can enhance EC proliferation
and migration (14). Compared
with healthy patients, ID1 expression is increased in ovarian
cancer patients, and its silencing substantially reduces EPC
angiogenesis (16). Our previous
study demonstrated that ID1 overexpression promoted EPC
proliferation and migration, indicating a vital role for ID1 in
re-endothelialization (15).
However, its underlying mechanisms of action remain unclear. In
this study, we searched for ID1 interaction partners using
immunoprecipitation analysis and found that ID1 formed a complex
with E2-2. Moreover, ID1 upregulation inhibited the expression of
E2-2 and the E2-2-induced activity of an artificial
E-box-containing reporter (MCKpfos-luc). Therefore, these results
suggest that ID1 interacts with E2-2. However, whether E2-2 plays
an important role in ID1-induced EPC function needs to be explored
further.
E2-2, which is also known as TCF4, belongs to the
E-protein family or the class-A type of bHLH transcription factors
that are involved in various physiological processes, such as
cellular growth, differentiation, and neural development (29–31). Previous studies have demonstrated
that ID proteins lack the basic DNA-binding domain, and can act as
a dominant-negative regulator of bHLH to regulate cell commitment,
differentiation and embryogenesis by forming inactive hererodimers
(9,10). Recently, E2-2 was shown to repress
VEGFR2 reporter activity, endothelial cell activation and
subsequent angiogenesis (10,20). In this study, we demonstrated the
inhibitory effect of ID1 on E2-2 levels. To further clarify the
underlying mechanisms involved in ID1-regulated EPC proliferation
and migration, we investigated the function of E2-2. E2-2
overexpression inhibited EPC proliferation, accompanied by a
corresponding decrease in the expression of the cell proliferation
marker, Ki67, and cell cycle-related proteins (cyclin D1), and an
increase in p27 expression. Moreover, E2-2 upregulation also
attenuated cell migration, concomitant with a downregulation in
MMP-2 and MMP-9 levels. Importantly, the enhanced effects of ID1 on
EPC proliferation and migration were attenuated when the cells were
transfected with Ad-E2-2. These data indicate that ID1 may trigger
EPC angiogenesis by enhancing cell proliferation and migration
through E2-2.
ID proteins often bind to E-proteins through the HLH
motif to inhibit the corresponding transcription (9). In this study, we demonstrated the
critical role of the HLH domain in the interaction between ID1 and
E2-2. Furthermore, the lack of HLH exhibited little effect on
E2-2-induced MCKpfos-luc transcriptional activity. Importantly, the
inhibitory effect of ID1 on E2-2 expression was abolished when the
cells were transfected with ID1 that lacked the HLH domain. It has
been demonstrated that blocking ID1 function by dominant
interfering HLH dimerization mutant 13I represses angiogenic factor
vascular endothelial growth factor (32). To further investigate the
underlying mechanisms involved in ID1-induced EPC proliferation and
migration by E2-2, we analyzed the function of HLH during these
processes. As expected, the enhanced effect of ID1 on EPC
proliferation was notably attenuated when the HLH domain in ID1 was
deleted. A similar effect on cell migration was also observed.
These results allow us to speculate that the HLH domain is critical
for ID1-induced EPC proliferation and migration via E2-2.
In conclusion, this study demonstrates that ID1
interacts with E2-2 in EPCs. Importantly, ID1 exerted its positive
regulatory effect on EPC proliferation and migration via E2-2 and
its HLH domain. Therefore, this study indicates a potential role
for ID1 in the development of the re-endothelialization processes
based on EPCs, and supports a promising therapeutic option for
repairing injured blood vessels.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (no. 81270224) and the Chengdu military
region's 12th five foundation (no. C12053).
References
|
1
|
Sena CM, Pereira AM and Seiça R:
Endothelial dysfunction - a major mediator of diabetic vascular
disease. Biochim Biophys Acta. 1832:2216–2231. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hirase T and Node K: Endothelial
dysfunction as a cellular mechanism for vascular failure. Am J
Physiol Heart Circ Physiol. 302:H499–H505. 2012. View Article : Google Scholar
|
|
3
|
Hu CH, Ke X, Chen K, Yang DY, Du ZM and Wu
GF: Transplantation of human umbilical cord-derived endothelial
progenitor cells promotes re-endothelialization of the injured
carotid artery after balloon injury in New Zealand white rabbits.
Chin Med J (Engl). 126:1480–1485. 2013.
|
|
4
|
Balaji S, King A, Crombleholme TM and
Keswani SG: The role of endothelial progenitor cells in postnatal
vasculogenesis: Implications for therapeutic neovascularization and
wound healing. Adv Wound Care (New Rochelle). 2:283–295. 2013.
View Article : Google Scholar
|
|
5
|
Zhang M, Malik AB and Rehman J:
Endothelial progenitor cells and vascular repair. Curr Opin
Hematol. 21:224–228. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yu J, Wang Q, Wang H, Lu W, Li W, Qin Z
and Huang L: Activation of liver X receptor enhances the
proliferation and migration of endothelial progenitor cells and
promotes vascular repair through PI3K/Akt/eNOS signaling pathway
activation. Vascul Pharmacol. 62:150–161. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dudley AC, Cloer EW and Melero-Martin JM:
The role of bone marrow-derived progenitor cells in tumor growth
and angiogenesis. Stem Cells and Cancer Stem Cells. 8. Springer;
pp. 45–54. 2012, View Article : Google Scholar
|
|
8
|
Hibbert B, Ma X, Pourdjabbar A, Holm E,
Rayner K, Chen YX, Sun J, Filion L and O'Brien ER: Inhibition of
endothelial progenitor cell glycogen synthase kinase-3β results in
attenuated neointima formation and enhanced re-endothelialization
after arterial injury. Cardiovasc Res. 83:16–23. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Norton JD: ID helix-loop-helix proteins in
cell growth, differentiation and tumorigenesis. J Cell Sci.
113:3897–3905. 2000.PubMed/NCBI
|
|
10
|
Tanaka A, Itoh F, Itoh S and Kato M:
TAL1/SCL relieves the E2-2-mediated repression of VEGFR2 promoter
activity. J Biochem. 145:129–135. 2009. View Article : Google Scholar
|
|
11
|
Benezra R, Rafii S and Lyden D: The Id
proteins and angiogenesis. Oncogene. 20:8334–8341. 2001. View Article : Google Scholar
|
|
12
|
Lyden D, Young AZ, Zagzag D, Yan W, Gerald
W, O'Reilly R, Bader BL, Hynes RO, Zhuang Y, Manova K and Benezra
R: Id1 and Id3 are required for neurogenesis, angiogenesis and
vascularization of tumour xenografts. Nature. 401:670–677. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Valdimarsdottir G, Goumans M-J, Rosendahl
A, Brugman M, Itoh S, Lebrin F, Sideras P and ten Dijke P:
Stimulation of Id1 expression by bone morphogenetic protein is
sufficient and necessary for bone morphogenetic protein-induced
activation of endothelial cells. Circulation. 106:2263–2270. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ling M-T, Lau TC, Zhou C, Chua CW, Kwok
WK, Wang Q, Wang X and Wong YC: Overexpression of Id-1 in prostate
cancer cells promotes angiogenesis through the activation of
vascular endothelial growth factor (VEGF). Carcinogenesis.
26:1668–1676. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wang H, Yu Y, Guo RW, Shi YK, Song MB,
Chen JF, Yu SY, Yin YG, Gao P and Huang L: Inhibitor of DNA
binding-1 promotes the migration and proliferation of endothelial
progenitor cells in vitro. Mol Cell Biochem. 335:19–27. 2010.
View Article : Google Scholar
|
|
16
|
Su Y, Gao L, Teng L, Wang Y, Cui J, Peng S
and Fu S: Id1 enhances human ovarian cancer endothelial progenitor
cell angiogenesis via PI3K/Akt and NF-κB/MMP-2 signaling pathways.
J Transl Med. 11:1322013. View Article : Google Scholar
|
|
17
|
Itoh F, Itoh S, Goumans MJ,
Valdimarsdottir G, Iso T, Dotto GP, Hamamori Y, Kedes L, Kato M and
ten Dijke Pt P: Synergy and antagonism between Notch and BMP
receptor signaling pathways in endothelial cells. EMBO J.
23:541–551. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Tanaka A, Itoh F, Nishiyama K, Takezawa T,
Kurihara H, Itoh S and Kato M: Inhibition of endothelial cell
activation by bHLH protein E2-2 and its impairment of angiogenesis.
Blood. 115:4138–4147. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar
|
|
20
|
Yang W, Itoh F, Ohya H, Kishimoto F,
Tanaka A, Nakano N, Itoh S and Kato M: Interference of
E2-2-mediated effect in endothelial cells by FAM96B through its
limited expression of E2-2. Cancer Sci. 102:1808–1814. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Von Offenberg Sweeney N, Cummins PM,
Cotter EJ, Fitzpatrick PA, Birney YA, Redmond EM and Cahill PA:
Cyclic strain-mediated regulation of vascular endothelial cell
migration and tube formation. Biochem Biophys Res Commun.
329:573–582. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cai H and Harrison DG: Endothelial
dysfunction in cardiovascular diseases: The role of oxidant stress.
Circ Res. 87:840–844. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Pearson JD: Endothelial progenitor cells -
hype or hope? J Thromb Haemost. 7:255–262. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Vasa M, Fichtlscherer S, Aicher A, Adler
K, Urbich C, Martin H, Zeiher AM and Dimmeler S: Number and
migratory activity of circulating endothelial progenitor cells
inversely correlate with risk factors for coronary artery disease.
Circ Res. 89:E1–E7. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Asahara T, Masuda H, Takahashi T, Kalka C,
Pastore C, Silver M, Kearne M, Magner M and Isner JM: Bone marrow
origin of endothelial progenitor cells responsible for postnatal
vasculogenesis in physiological and pathological
neovascularization. Circ Res. 85:221–228. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Takahashi T, Kalka C, Masuda H, Chen D,
Silver M, Kearney M, Magner M, Isner JM and Asahara T: Ischemia-
and cytokine-induced mobilization of bone marrow-derived
endothelial progenitor cells for neovascularization. Nat Med.
5:434–438. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
He T, Smith LA, Harrington S, Nath KA,
Caplice NM and Katusic ZS: Transplantation of circulating
endothelial progenitor cells restores endothelial function of
denuded rabbit carotid arteries. Stroke. 35:2378–2384. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Walter DH, Rittig K, Bahlmann FH,
Kirchmair R, Silver M, Murayama T, Nishimura H, Losordo DW, Asahara
T and Isner JM: Statin therapy accelerates reendothelialization: A
novel effect involving mobilization and incorporation of bone
marrow-derived endothelial progenitor cells. Circulation.
105:3017–3024. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Flora A, Garcia JJ, Thaller C and Zoghbi
HY: The E-protein Tcf4 interacts with Math1 to regulate
differentiation of a specific subset of neuronal progenitors. Proc
Natl Acad Sci USA. 104:15382–15387. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang Q, Lu PH, Shi ZF, Xu YJ, Xiang J,
Wang YX, Deng LX, Xie P, Yin Y and Zhang B: Glucocorticoid receptor
β acts as a co-activator of T-cell factor 4 and enhances glioma
cell proliferation. Mol Neurobiol. 52:1–13. 2014.
|
|
31
|
Forrest MP, Hill MJ, Quantock AJ,
Martin-Rendon E and Blake DJ: The emerging roles of TCF4 in disease
and development. Trends Mol Med. 20:322–331. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ciarapica R, Annibali D, Raimondi L,
Savino M, Nasi S and Rota R: Targeting Id protein interactions by
an engineered HLH domain induces human neuroblastoma cell
differentiation. Oncogene. 28:1881–1891. 2009. View Article : Google Scholar : PubMed/NCBI
|