Introduction
Sudden cardiac arrest (CA) is a major burden on
health care in the US, with estimates ranging from 200,000 to
500,000 patients who experience out-of-hospital CA each year
(1). The median rate of survival
in adults following hospital discharge is approximately 6.7% in the
US (2). It has been widely
accepted that cardiopulmonary resuscitation (CPR) is an important
first aid technique for CA. A 9-year retrospective cohort study of
5,958 patients receiving emergency medical service (EMS)-initiated
resuscitation demonstrated that 16.8% survived at hospital
discharge. Of those individuals discharged, 5-year survival was
better in those who had received percutaneous coronary intervention
(3). Notably, a recent randomized
controlled trial has demonstrated that mild therapeutic hypothermia
for 12–24 h improves neurologic recovery and survival after CA
(4). However, even after
successful CPR and therapeutic hypothermia treatment, myocardial
tissue may still suffer from severe ischemia/reperfusion (I/R)
injury. In many patients who initially achieve return of
spontaneous circulation (ROSC) after CA, subsequent morbidity and
mortality is caused by cardiac ischemia that accompanies prolonged
cardiac dysfunction (5). This
disease is known as post-cardiac arrest syndrome (PCAS) (5), which is a serious threat to patient
health. A better understanding of the cellular and molecular
mechanisms responsible for myocardial ischemia may assist in the
development of more efficient treatment strategies for PCAS.
The renin-angiotensin-aldosterone system (RAAS) is
an endogenous hormonal cascade that functions in the homeostatic
control of arterial pressure, extracellular volume and tissue
perfusion (6). Currently,
blockade of the RAAS with angiotensin-converting enzyme inhibitors
(ACEIs) or angiotensin II type 1 receptor (AT1R) blockers (ARBs)
has been widely used as a treatment strategy in the management of
patients with hypertension, acute myocardial infarction, chronic
systolic heart failure, stroke and diabetic renal disease (6). However, these drugs are also
associated with adverse reactions, such as hypotension (6). Following myocardial I/R injury, the
cardioprotective effects of ACEIs have been well established
(7). ACEIs are capable of acutely
limiting myocardial injury and necrosis in models of permanent
coronary artery occlusion. The mechanisms responsible for this
cardioprotection include reductions in myocardial oxygen
supply/demand, inhibition of bradykinin metabolism, increased
prostaglandin synthesis and attenuated inflammatory responses
(8). However, there is limited
data regarding the application of ACEIs or ARBs in a model of CA
and resuscitation, which features complex pathophysiological
changes such as brain ischemia and renal ischemia (9,10).
Furthermore, there is concern that blockade of the RAAS reduces
perfusion pressure, which may attenuate the beneficial effects of
resuscitation.
Thus, we performed this study in order to examine
the effects of enalapril pre-treatment on myocardial injury in a
swine model of CA and resuscitation. We examined the effects of
enalapril infusion on myocardial ultrastructure, hemodynamics and
RAAS system activation, including the plasma levels of angiotensin
(Ang) II and Ang-(1–7). Finally, the effects of enalapril
pre-treatment on the mRNA and protein levels of
angiotensin-converting enzyme (ACE), AT1R, ACE2 and MAS were
evaluated in swine myocardial tissue.
Materials and methods
Animals
Inbred, male, Landrace miniature piglets aged 11–13
months, with an average weight of 30±2 kg, were used as previously
described (11). All procedures
were performed in accordance with the Animal Care Guidelines of the
Institutional Animal Care and Use Committee of Capital Medical
University (Beijing, China) as well as the Utstein-style guidelines
(12). The experimental protocol
was also approved by the Committee on the Ethics of Animal
Experiments of Capital Medical University. All animals were housed
in a dedicated, pathogen-free environment in the Capital Medical
University animal facility. Animals were fed standard chow and had
free access to water.
Groups, anesthesia and ventilation
The pigs were randomly assigned to three groups: the
sham-operated group (sham group, n=8), the experimental
CA-resuscitation + saline (0.9% NaCl) group (saline group, n=12)
and the experimental CA-resuscitation + enalapril group (enalapril
group, n=12). On the experimental day, anesthesia was induced by an
intramuscular injection of midazolam (0.5 mg/kg) followed by an
injection of propofol (1.0 mg/kg via an ear vein). The animals were
anesthetized with an intravenous infusion of sodium pentobarbital
(8 mg/kg/h) to maintain favorable surgical anesthesia. The chest
and back were thoroughly shaven to allow for direct contact of the
paddles used for defibrillation during CPR immediately following
the induction of anaesthesia.
An endotracheal tube (6.5-mm internal diameter) was
used as an ordinary endotracheal tube. The tube was connected to a
Servo Ventilator (Servo 900C; Siemens, Munich, Germany) with an
FiO2 of 0.21. All the animals were intubated and
mechanically ventilated with room air using a tidal volume of 12
ml/kg and a respiratory frequency of 12 breaths/min. Respiratory
frequency was adjusted to maintain end-tidal pCO2
between 35 and 40 mmHg, which was continuously monitored using an
infrared CO2 analyzer (CO2SMO Plus monitor;
Respirometric Inc., Murrysville, PA, USA).
Preparation and monitoring
Acetated Ringer's solution (30 ml/kg) was infused
via an ear vein to compensate for fluid loss during the first hour
of preparation. During the procedure, 2.5% glucose-electrolyte
solution (8 ml/kg/h) was continuously infused. A Swan-Ganz catheter
(7 Fr; Edwards Life Sciences, Irvine, CA, USA) was inserted into
the pulmonary artery via the right external femoral vein for
cardiac output (CO) measurement (13). Electrocardiogram and hemodynamic
parameters were monitored using the M1165 monitoring system
(Hewlett-Packard, Palo Alto, CA, USA). Self-adhesive defibrillation
electrodes were positioned on the chest wall. Coronary perfusion
pressure (CPP) was calculated as the difference, measured at the
end of each minute of precordial compression, between decompression
diastolic aortic and time-coincident right-atrial pressure.
Experimental procedure
Experimental ventricular fibrillation (VF) in pigs
was induced in the saline and enalapril groups, but not in the sham
group, as described previously (11). Pure enalapril (E6888) was
purchased from Sigma-Aldrich (St. Louis, MO, USA). Following the
surgical preparations described above, baseline measurements and
blood samples were obtained. Then, the animals were intravenously
infused with enalapril solution (0.2 mg/kg) or an equal volume of
saline 30 min prior to VF induction. This dose was selected based
on data from a previous study (14).
Successful VF was induced using a method described
in previous study from our laboratory (15). A temporary pacemaker conductor was
inserted into the right ventricle through the right sheathing canal
and connected to an electrical stimulator (GY-600A; Kaifeng Huanan
Equipment Co., Ltd., Kaifeng, China). The electrical stimulator was
programmed in the S1S2 mode (300/200 msec, 40 V, 8:1 proportion and
10 msec step length) to provide a continuous electrical stimulus
until the occurrence of VF. Ventricular fibrillation was defined as
an electrocardiogram showing waveforms corresponding to VF and a
rapid decline in mean arterial pressure (MAP) toward zero (16).
Mechanical ventilation was stopped after successful
VF induction. At 8 min post VF induction, monophasic electrical
shock was attempted, beginning at 3 J/kg. If the initial
defibrillation shock failed, energy was increased by 1 J/kg
increments for subsequent defibrillation shocks. If the first
defibrillation was unsuccessful, CPR was performed for 2 min and
the second defibrillation was induced. Manual CPR was carried out
at a frequency of 100 compressions/min with mechanical ventilation
at an FiO2 of 100% and a compression-to-ventilation
ratio of 30:2. The swine chest was compressed to approximately one
third of the anteroposterior diameter. Notably, the same CPR
technician performed CPR throughout the entire study. The quality
of chest compressions was controlled by a HeartStart MRx
Monitor/Defibrillator with Q-CPR (Philips Medical Systems, Best,
The Netherlands). The compression-to-ventilation ratio was 30:2
(17). If spontaneous circulation
was not achieved after the second defibrillation attempt, CPR was
continued for an additional 2 min, and defibrillation was attempted
a third time. In this manner, CPR was continued for up to 30 min.
Only the principal investigator, who did not take part in any
resuscitation efforts, knew the treatment group assignment of each
animal.
ROSC was defined as 10 consecutive min of systolic
blood pressure at 50 mmHg. At 30 min after CPR, those animals
remaining in VF, pulseless electrical activity or asystole were
considered resuscitation failures (18). After successful resuscitation, the
animals were mechanically ventilated with 100% oxygen. During the
next 6 h, the animals were maintained in a surgical plane of
anesthesia with an intravenous infusion of sodium pentobarbital (8
mg/kg/h). Heart rate (HR), CO, CPP and MAP were monitored in these
animals hourly. Blood samples were obtained and blood plasma was
prepared at 0, 30 min, 1, 2, 4 and 6 h post-ROSC from the sham,
saline- and enalapril-treated groups. During this period, two pigs
in the saline-treated group and one pig in the enalapril-treated
group died due to sudden cardiac arrest. At the end of the
experiments, the animals were euthanized with intravenous potassium
chloride (15%, 10 ml). The cardiac tissue was isolated, immediately
frozen in liquid nitrogen, and stored at −80°C.
Hemodynamic measurements
Hemodynamic parameters were measured at baseline and
0.5, 1, 2, 4 and 6 h after ROSC. Measured parameters included HR,
CO, CPP and MAP. MAP was monitored via a right femoral arterial
catheter.
Enzyme-linked immunosorbent assay
(ELISA)
Plasma levels of Ang II and Ang-(1–7)
were measured. Plasma Ang II levels were determined using a
commercial ELISA kit purchased from R&D Systems (Minneapolis,
MN, USA) and plasma Ang-(1–7)
plasma levels were determined using a commercial ELISA kit
purchased from BlueGene Biotech Co., Ltd. (Shanghai, China). The
ELISA samples consisted of 50 µl blood plasma and 50
µl deionized water in a 96-well plate, according to the
manufacturer's instructions. Optical density (OD) values (450 nm)
were determined using a microplate reader (Tefcan M200; Tecan,
Crailsheim, Germany) as previously described (19).
Measurement of plasma cardiac enzyme
levels and a terminal deoxynucleotidyl transferase-mediated dUTP
nick-end labeling (TUNEL) assay
According to the manufacturer's instructions, the
plasma cardiac troponin I (cTNI) level was determined using a
high-sensitive assay (Abbott Diagnostics, Lake Forest, IL, USA),
and TUNEL assay was performed using a commercial kit (In
Situ Cell Death Detection kit; Roche, Mannheim, Germany)
(20).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
RT-qPCR was performed as previously described and
according to the manufacturer's protocol (21,22). Total RNA was extracted from the
myocardial tissue using TRIzol reagent (Invitrogen, Carlsbad, CA,
USA) and then reverse transcribed into cDNA (23). For all experiments, 5 ng cDNA was
subjected to RT-qPCR with a Perkin-Elmer ABI Prism 7500 sequence
detection system and Power SYBR-Green PCR Master Mix (Applied
Biosystems, Life Technologies, Grand Island, NY, USA). The
following primers were used: ACE forward, 5′-ATC AAG CGG ATC ATA
AAG AAG-3′ and reverse, 5′-CAC GCT GTA GGT GGT TTC C-3′; ACE2
forward, 5′-TCT GAA TGA CAA CAG CCT AG-3′ and reverse, 5′-CAC TCC
CAT CAC AAC TCC-3′; MAS forward, 5′-TAT TCC TCA TCT TCG CTA T-3′
and reverse, 5′-GCC CTG GTC AGA ACA ACT-3′; AT1R forward, 5′-TCA
CCT GCA TCA TCA TCT GG-3′ and reverse, 5′-AGC TGG TAA GAA TGA TTA
GG-3′; and GAPDH forward, 5′-GAC CCA GAA TAC CAA GTG CAG ATG TA-3′
and reverse, 5′-CTG TTT CAG GAT TTA AGG TTG GAG ATT-3′. Gene
expression was normalized to GAPDH. Data analysis was performed
using the 2−ΔΔCt method (24).
Immunohistochemistry
Immunohistochemical staining was performed as
previously described (25).
Briefly, the sections were placed in 10 ml glass centrifuge tubes
and dewaxed using two changes of xylene (3 ml) for 10 min at room
temperature, and then rehydrated in a sequence of 100, 95, 70 and
50% graded ethanol (3 ml). After blocking with 5% BSA for 4 h, the
sections were incubated overnight at 4°C with anti-ACE2 (1:100;
Abcam, Cambridge, UK), anti-ACE (1:200; Santa Cruz Biotechnology,
Dallas, TX, USA), anti-MAS (1:300; Alomone Labs Ltd., Jerusalem,
Israel) or anti-AT1R (1:200; Abcam) antibodies. The sections were
washed with phosphate-buffered saline (PBS) three times (5 min ×3)
and incubated with biotinylated secondary antibodies (1:200),
followed by incubation with an avidin-biotin peroxidase complex
(both from ZSGB-Biotechnology, Beijing, China). The sections
incubated with normal rabbit serum (1:10) served as negative
controls. Peroxidase conjugate localization was determined using
3,3′-diaminobenzidine tetrahydrochloride (DAB; Sigma, Milwaukee,
WI, USA) as the chromogen, and the sections were counterstained
with hematoxylin (Beijing Solarbio Science & Technology Co.,
Ltd., Beijing, China). Images were captured using an IX80
microscope (Olympus, Tokyo, Japan) and analyzed using Image Pro
Plus v3.0 software (Media Cybernetics, Carlsbad, CA, USA). The
positive staining was defined as localization of brown
staining.
Western blot analysis
Western blot analysis was performed as previously
described (26,27). The heart tissues were swiftly
harvested, washed in PBS three times and then homogenized in lysis
buffer (Tris-HCl pH 7.5, 20 mmol/l; EDTA, 2 mmol/l; NP-40, 1%; and
Triton X-100, 1%) with protease inhibitors [phenylmethylsulfonyl
fluoride (PMSF), 2 mmol/l; leupeptin, 50 µg/ml; aprotinin,
25 µg/ml; pepstatin A, 10 µg/ml; and dithiothreitol,
2 mmol/l]. Total protein concentrations were determined using a
bicinchoninic acid assay (BCA) assay (Beyotime, Haimen, China).
Approximately 30 µg of the homogenate samples were run on a
10% sodium dodecyl sulfate (SDS)-polyacrylamide gel, and the
proteins were electrotransferred to nitrocellulose membranes
(Bio-Rad Laboratories, Hercules, CA, USA). The membranes were
blocked using 5% evaporated milk in PBS and then incubated for 4 h
at room temperature with primary antibodies [anti-ACE2 (1:100;
ab15348; Abcam), anti-ACE (1:200; sc-20791; Santa Cruz
Biotechnology), anti-AT1R (1:100; ab18801; Abcam) and anti-MAS
(1:300; AAR-013, Alomone Labs Ltd.) and β-actin (ab8226, Abcam)].
Following incubation, the blots were washed with PBS, and then
incubated for 2 h with HRP-conjugated secondary antibodies
(1:1,000). HRP signals were detected using an ECL kit (Amersham
Biosciences, Little Chalfont, UK). Quantitative analysis of the
data was performed by measuring the intensity of protein band
signals using Bio-Rad Image Lab Software (Bio-Rad
Laboratories).
Transmission electron microscopy
The heart tissues were carefully isolated and fixed
with 2.5% glutaraldehyde in 0.1 M phosphate buffer (pH 7.2–7.4) for
2 h at 4°C. After washing in phosphate buffer, the tissues were
post-fixed in 1% osmium tetroxide (Sigma-Aldrich, Shanghai, China)
in 0.1 M phosphate buffer for 2 h. The tissues were dehydrated in a
graded ethanol series, infiltrated with propylene oxide, embedded
in epoxy resin and sliced into ultrathin sections (1 µm).
After double-staining with uranyl acetate (Spectrum Chemical
Manufacturing Corp., Shanghai, China) and lead citrate
(Sigma-Aldrich, Shanghai, China), electron microphotographs were
captured using a transmission electron microscope (H-7650; Hitachi,
Tokyo, Japan).
Statistical analysis
The results are expressed as the means ± SD.
Differences at various time-points within groups were compared with
a repeated measures analysis of variance (ANOVA) followed by a
Tukey post-hoc test. Continuous variables were adjusted to a normal
distribution and equal variances were compared using the
Kolmogorov-Smirnov test and homogeneity of variance test. Survival
rate comparisons were performed using Chi-square analysis.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Enalapril does not improve survival rates
in a swine model of CA and resuscitation
There were no significant differences in baseline
parameters (including body weight, HR, average arterial pressure,
average pulmonary arterial pressure and CO) among the sham, saline-
and enalapril-treated groups (Table
I). Moreover, there were no significant differences in the
number of defibrillation shocks, shock energy and outcome between
enalapril-treated pigs and saline-treated pigs (P>0.05)
(Table II).
 | Table IPhysiological baseline measurements
across experimental groups. |
Table I
Physiological baseline measurements
across experimental groups.
| Baseline
parameters | Sham
group
(n=8) | Saline
group
(n=12) | Enalapril
group
(n=12) |
|---|
| Body weight
(kg) | 29.13±2.16 | 30.63±0.92 | 30.38±0.92 |
| Heart rate
(beats/min) | 99.00±7.44 | 101.38±8.30 | 100.50±10.04 |
| Average arterial
pressure (mmHg) | 103.12±5.19 | 101.88±5.22 | 87.00±5.81 |
| Average pulmonary
arterial pressure (mmHg) | 23.42±4.32 | 24.13±5.24 | 24.34±4.56 |
| Cardiac output
(l/min) | 2.86±0.22 | 2.99±0.20 | 2.99±0.19 |
| CPP (mmHg) | 43.5±2.4 | 41.7± 5.1 | 42.6± 6.2 |
| Table IIOutcome measures after
post-resuscitation in sham, saline- and enalapril-treated groups in
a swine model of myocardial I/R injury. |
Table II
Outcome measures after
post-resuscitation in sham, saline- and enalapril-treated groups in
a swine model of myocardial I/R injury.
| Outcome
measures | Sham | Saline | Enalapril |
|---|
| Number of
shocks | 0 | 3.7±1.5a | 2.8±0.9a |
| Energy of shock
(J) | 0 | 292.5±39.8a | 276.7±54.6a |
| Time to ROSC
(min) | 0 | 5±1.8a | 5.5±1.7a |
| Number of pigs
surviving at 6 h | 8 (8/8) | 6 (6/12)a | 9 (9/12)a |
Effects of enalapril on hemodynamic
indices in a swine model of CA and resuscitation
We measured hemodynamic indices in the sham, saline-
and enalapril-treated groups. Resuscitation significantly boosted
HR (Fig. 1A) and MAP (Fig. 1C), but reduced CO (Fig. 1B) and CPP (Fig. 1D). Enalapril treatment did not
change the HR, CO or CPP (Fig. 1A, B
and D). However, enalapril treatment exhibited a marked blood
pressure-lowing effect, partly inhibiting the enhancement of MAP at
4 and 6 h post-ROSC (Fig.
1C).
 | Figure 1Cardiac functions of pigs
post-resuscitation in the sham, saline- and enalapril-treated
groups. (A) Heart rate (HR, beats/sec), (B) cardiac output (CO,
l/min), (C) mean arterial pressure (MAP, mmHg) and (D) coronary
perfusion pressure (CPP, mmHg) were monitored at different time
points (baseline, after infusion, 0, 0.5, 1, 2, 4 and 6 h)
following restoration of spontaneous circulation (post-ROSC, h).
*P<0.05 and **P<0.01 vs. sham group;
#P<0.05 vs. saline group. n=8–12/group initially. |
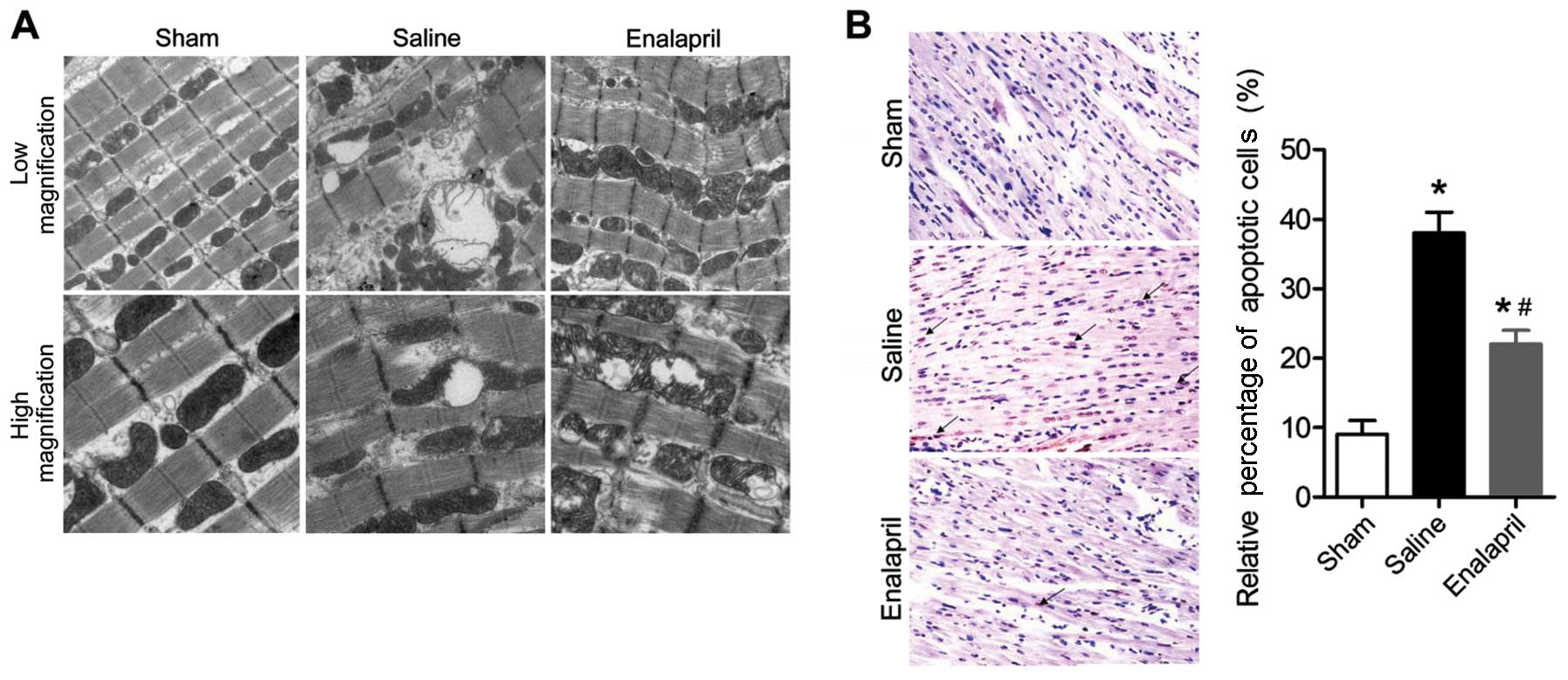
Enalapril ameliorates I/R-induced
myocardial injury in a swine model of CA and resuscitation
The effects of enalapril treatment on
ultrastructural changes in the myocytes are shown in Fig. 2A. Transmission electron
micrographs showed that acute CA and resuscitation resulted in
immediate and serious injury to the myocardium of animals in the
saline group. The myocytes in the saline group exhibited gigantic
vacuoles, disintegration of myofibrils, severe fraying of
intercellular organelles and considerable Z-band disruption.
Moreover, deranged cristae and significant swelling were observed
in mitochondria. By contrast, these injuries were less severe in
the myocardium of animals in the enalapril-treated group. In
addition, the results of the TUNEL assay showed that myocardial
apoptosis in the saline group was markedly induced by
CA-resuscitation, an effect that was reversed by enalapril
treatment (Fig. 2B). The plasma
levels of cTNI, an enzymatic marker for acute myocardial infarction
and cardiac cell death, were also markedly enhanced in the saline
group at 2 and 6 h after ROSC (Table III). Enalapril treatment
significantly inhibited the increase in plasma cTNI levels at 6 h
after ROSC (Table III). Taken
together, the findings of the present study indicate that enalapril
ameliorates I/R-induced myocardial injury in a swine model of CA
and resuscitation.
| Table IIIPlasma cTNI levels in sham, saline-
and enalapril-treated groups in a swine model of myocardial I/R
injury. |
Table III
Plasma cTNI levels in sham, saline-
and enalapril-treated groups in a swine model of myocardial I/R
injury.
| Time after
ROSC | Sham | Saline | Enalapril |
|---|
| 0 h after ROSC | 0.01±0.005 | 0.01±0.003 | 0.01±0.007 |
| 2 h after ROSC | 0.01±0.006 | 1.99±0.74a | 2.12±0.98a |
| 6 h after ROSC | 0.01±0.004 | 4.25±1.20a | 3.17±1.05a,b |
Enalapril decreases plasma Ang II levels
but not Ang-(1–7) levels in a swine model of CA and
resuscitation
There was no significant difference in the baseline
plasma levels of Ang II (Fig. 3A)
and Ang-(1–7) (Fig.
3B) among the three groups. The plasma Ang II levels in the
saline group were markedly increased at 2, 4 and 6 h post-ROSC
(Fig. 3A). Enalapril treatment
significantly reduced the plasma Ang II levels at 4 and 6 h
post-ROSC (P<0.05) compared with levels in the saline group
(Fig. 3A). The plasma
Ang-(1–7) levels in the saline group were also
increased at 4 and 6 h post-ROSC (Fig. 3B). Enalapril treatment did not
reduce the plasma Ang-(1–7) levels, although there was a trend
toward a reduction of Ang-(1–7)
levels in the enalapril-treated pigs at 4 and 6 h post-ROSC
(Fig. 3B).
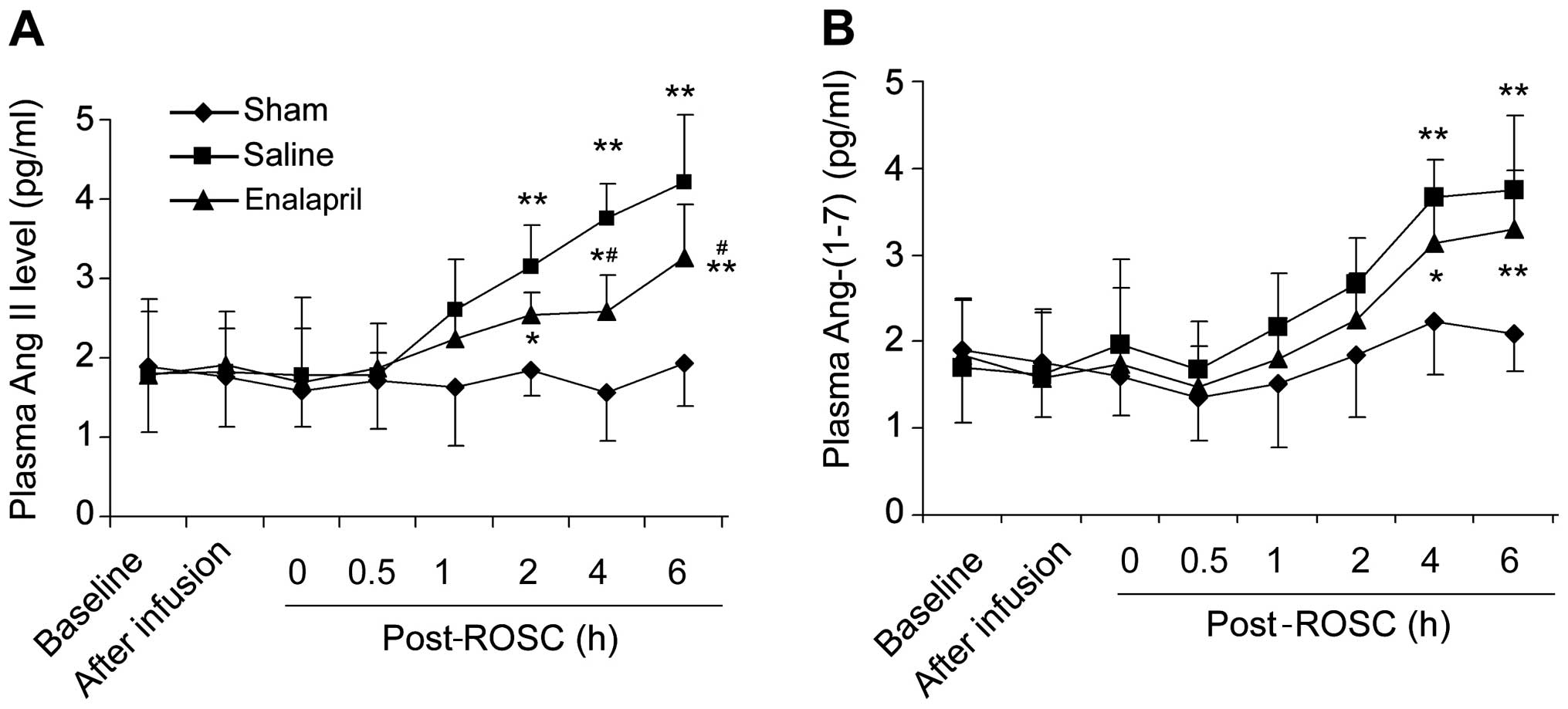 | Figure 3Plasma levels of the
renin-angiotensin system components, angiotensin (Ang) II and
Ang-(1–7), in a swine model of cardiac arrest
(CA) and resuscitation. ELISA determination of (A) Ang II and (B)
Ang-(1–7) levels at different time points
(baseline, after infusion, 0, 0.5, 1, 2, 4 and 6 h) in sham,
saline- and enalapril-treated groups following the return of
spontaneous circulation (post-ROSC, h) were determined by ELISA.
*P<0.05 and **P<0.01 vs. sham group;
#P<0.05 vs. saline group. n=6-9/group. |
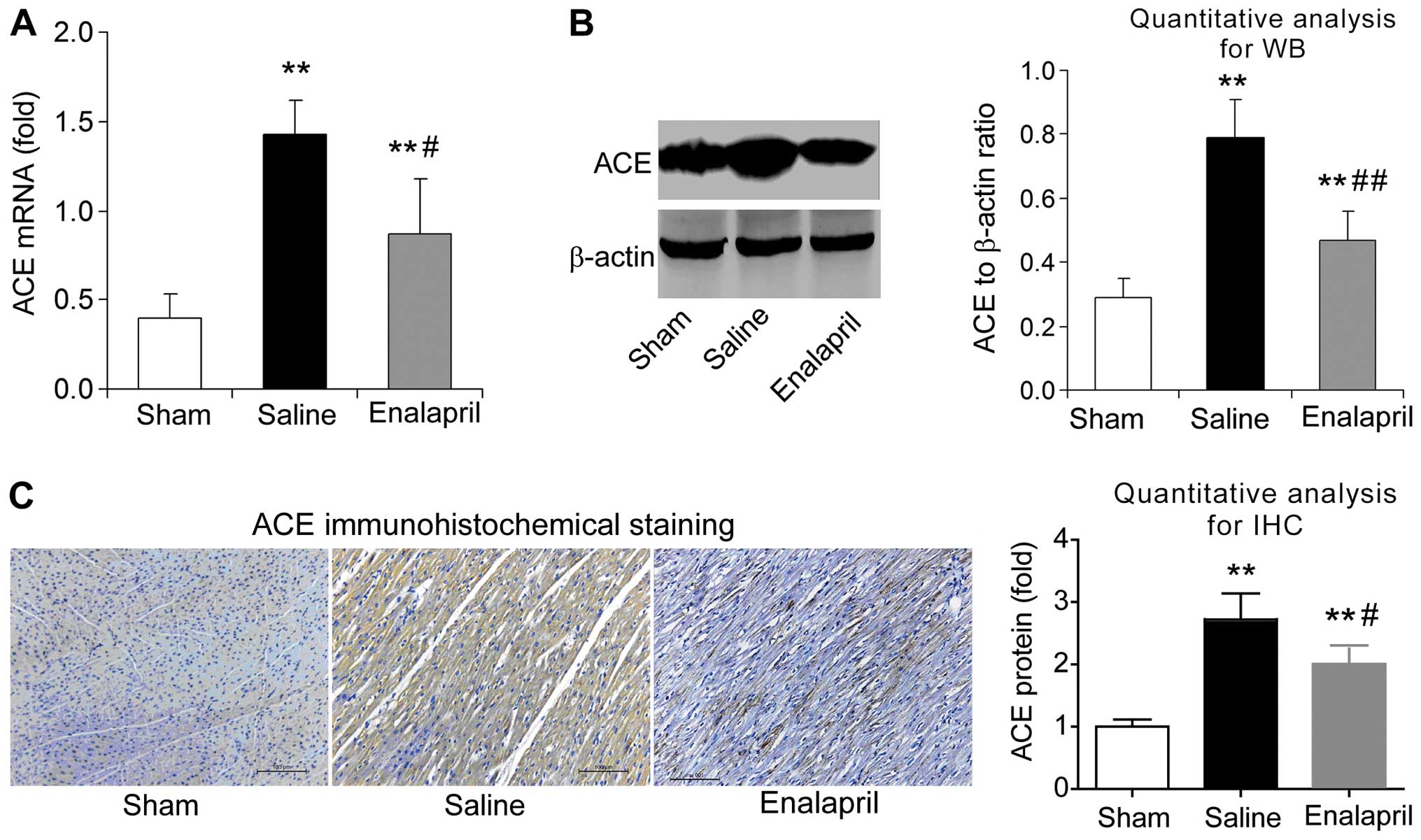
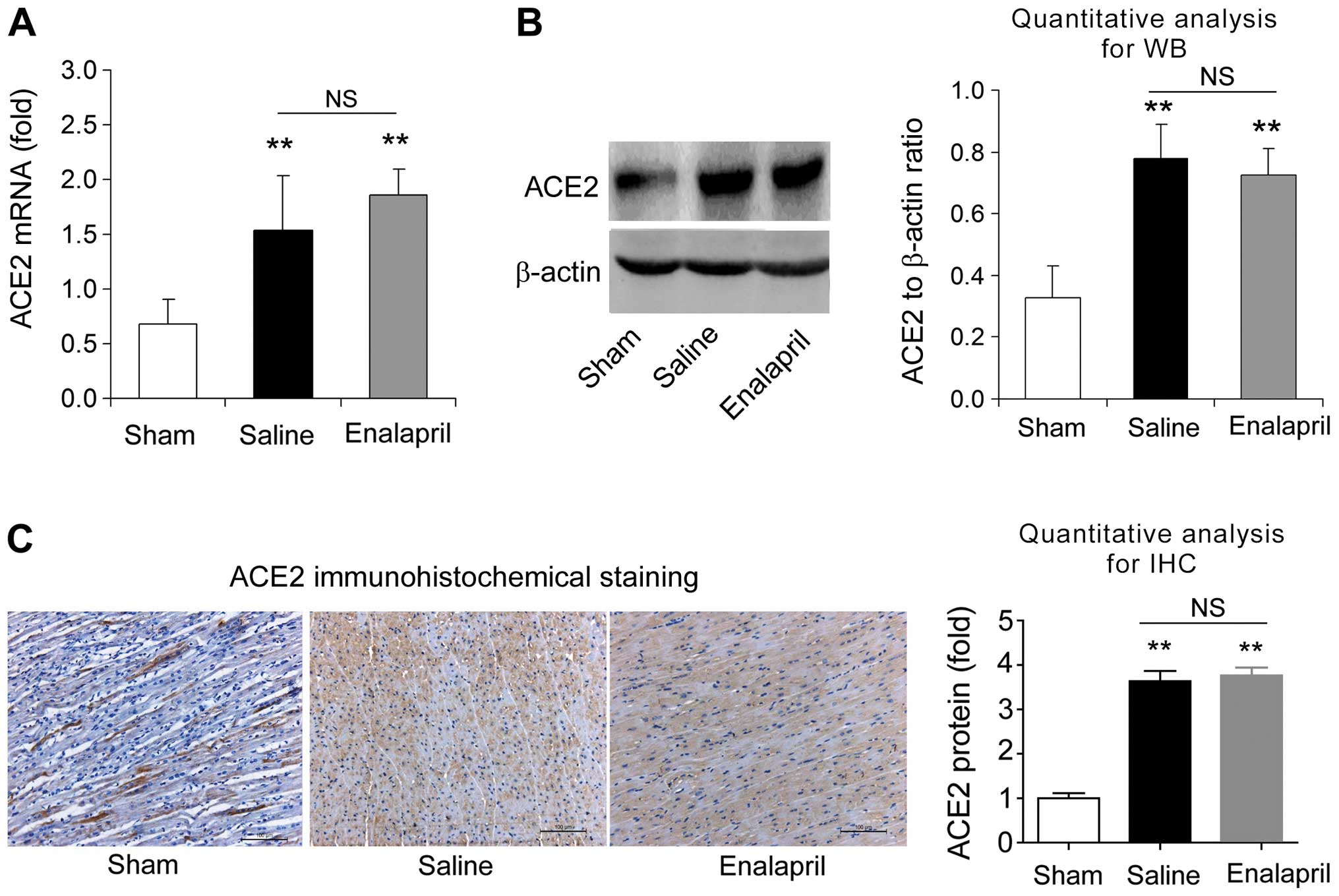
Enalapril reduces myocardial ACE
expression whereas it has no effect on ACE2 expression in a swine
model of CA and resuscitation
The effects of enalapril on myocardial ACE and ACE2
expression were assessed by RT-qPCR, western blot analysis and
immunohistochemistry. We found that the mRNA levels of ACE
increased by ~3-fold after resuscitation in the saline group
compared with that in the sham group (P<0.05) (Fig. 4A). Enalapril treatment inhibited
this upregulation of myocardial ACE after resuscitation (P<0.05)
(Fig. 4A). The results of western
blot analysis revealed that the myocardial ACE protein level in the
saline group was ~2.5-fold of that in the sham group, and enalapril
suppressed this upregulation (P<0.01) (Fig. 4B). Immunohistochemical analysis
confirmed that ACE protein levels were increased after
resuscitation and that this effect was partly inhibited by
enalapril treatment (P<0.05) (Fig.
4C). RT-qPCR, western blot analysis and immunohistochemistry
revealed that the myocardial mRNA and protein levels of ACE2 were
significantly enhanced after resuscitation (P<0.05) (Fig. 5A–C). In contrast to the changes
observed in ACE expression, enalapril treatment failed to modulate
the myocardial mRNA and protein expression of ACE2 (P>0.05)
(Fig. 5A–C). These data indicate
that enalapril reduces myocardial ACE expression whereas it has no
effect on ACE2 expression after resuscitation in swine.
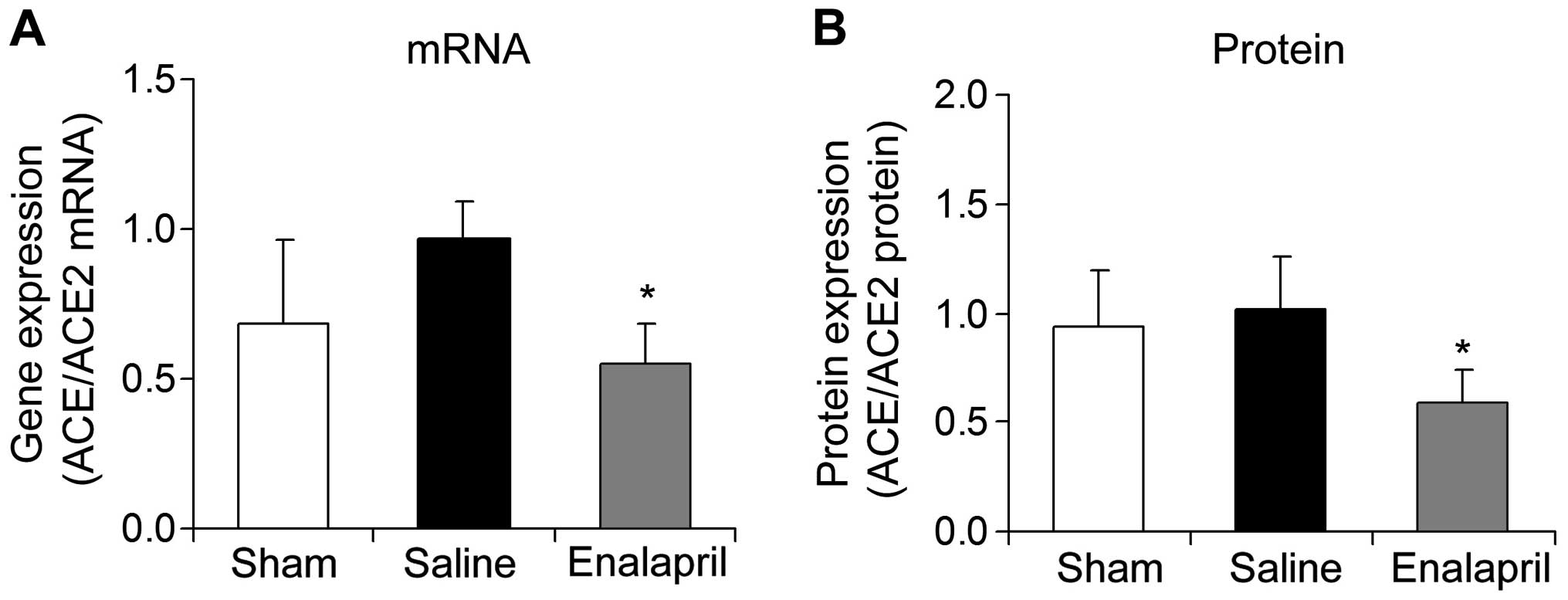
Enalapril decreases the myocardial
ACE/ACE2 mRNA ratio in a swine model of CA and resuscitation
The mRNA and protein ratios of myocardial ACE to
ACE2 were determined in each group following resuscitation. The
ratios of myocardial ACE/ACE2 mRNA (Fig. 6A) and protein (Fig. 6B) were significantly decreased by
enalapril treatment (P<0.05).
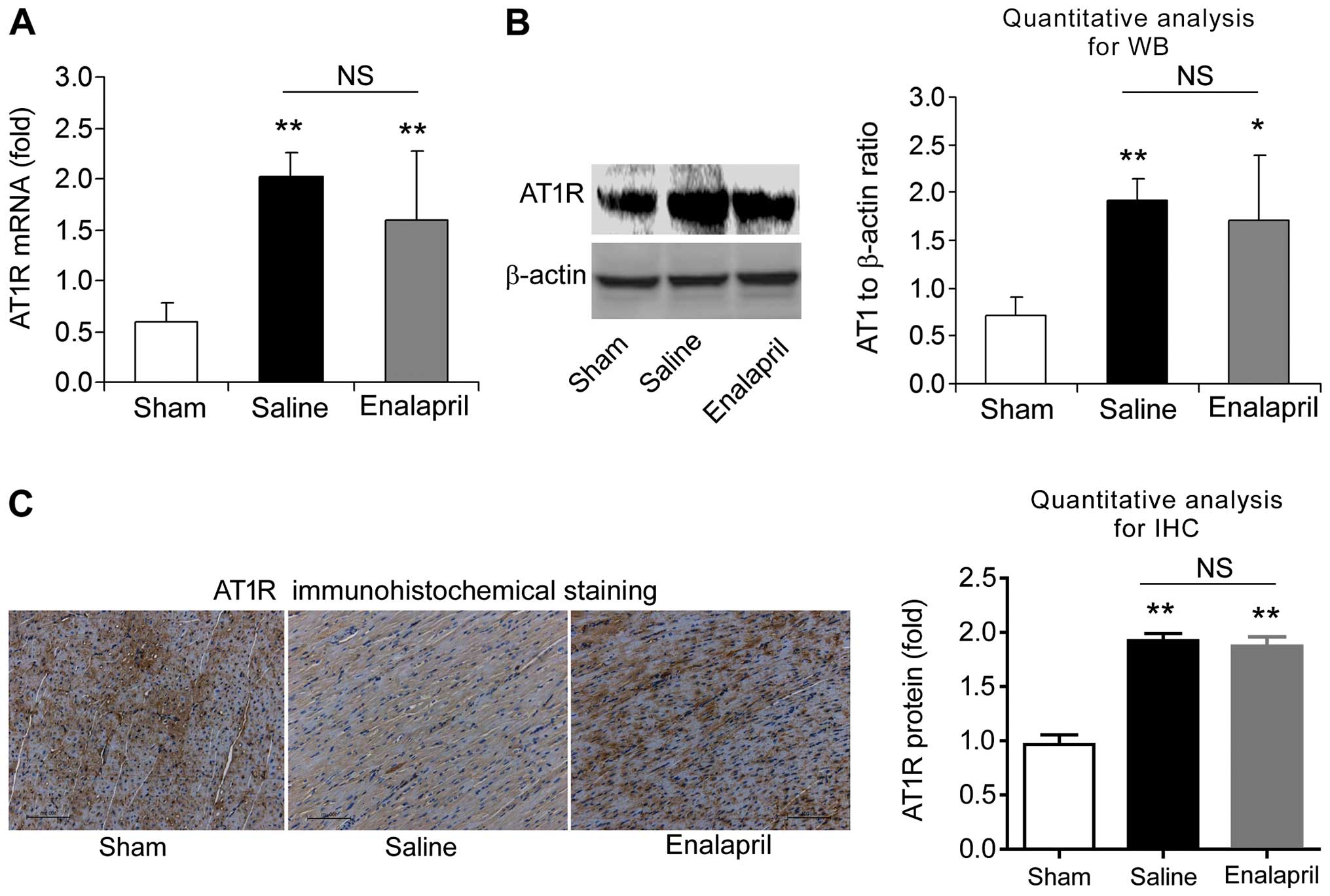
Enalapril does not alter AT1R and MAS
expression in a swine model of CA and resuscitation
We also measured the mRNA and protein levels of AT1R
and MAS in each experimental group using RT-qPCR, western blot
analysis and immunohistochemistry. The mRNA level of AT1R was
increased ~4-fold after resuscitation in the saline group compared
with that in the sham group (P<0.01) (Fig. 7A). Western blot analysis also
showed an increase in myocardial AT1R protein levels in the saline
group (P<0.01) (Fig. 7B).
Immunohistochemical analysis confirmed the upregulation of AT1R
protein (Fig. 7C). Enalapril
treatment had no effect on the upregulation of AT1R (Fig. 7A–C). Similarly, RT-qPCR, western
blot analysis and immunohistochemistry revealed that the mRNA and
protein levels of myocardial MAS were significantly enhanced after
resuscitation (P<0.01) (Fig.
8). However, enalapril treatment did not modulate the mRNA or
protein expression of myocardial MAS (Fig. 8). Taken together, these findings
indicate that AT1R and MAS are upregulated after CA and
resuscitation, whereas enalapril does not affect their
upregulation.
Discussion
In the present study, we demonstrated a
cardioprotective role for enalapril through modulation of the RAAS
in a swine model of CA and resuscitation. Although enalapril did
not improve the survival rate in this model, it greatly ameliorated
I/R-induced ultrastructural changes in myocytes, plasma enzyme
levels and myocardial apoptosis. Additionally, the enhancement of
plasma Ang II levels at 4 and 6 h post-ROSC was significantly
inhibited by enalapril pre-treatment. By contrast, enalapril
pre-treatment did not affect plasma Ang-(1–7)
levels. RT-qPCR, western blot analysis and immunohistochemistry
assays demonstrated that enalapril pre-treatment successfully
suppressed the upregulation of ACE but not ACE2 upon CA and
resuscitation. Particularly, we noted that the mRNA ratio of
ACE/ACE2 was also decreased by enalapril pre-treatment. Finally,
enalapril did not alter the upregulation of AT1R and MAS upon CA
and resuscitation. These results demonstrate, for the first time to
the best of our knowledge, that pre-treatment with an ACEI protects
against myocardial I/R injury post CA and resuscitation, and that
inhibition of the ACE/Ang II/AT1R axis may contribute to the
cardioprotective effects of enalapril in this context.
The pathophysiology of PCAS is unique from
post-arrest brain injury, post-arrest myocardial dysfunction,
systemic I/R response and persistent precipitating pathology
(28). Ischemic cerebral injury
alone contributes significantly to the high morbidity and mortality
in the resuscitated cardiac arrest patient. The myocardial
dysfunction associated with PCAS is characterized largely by global
hypokinesis and also, ultimately affects the success rate of
resuscitation (28). The
following molecular mechanisms may contribute to the
pathophysiology of myocardial dysfunction in PCAS. Firstly, I/R
injury results in a large amount of oxygen free radicals which
damage cell membranes and induce the necrosis and apoptosis of
myocytes (29). Secondly,
intracellular Na+ accumulation caused by CA leads to
cytosolic Ca2+ overload through the reverse-mode
operation of the sarcolemmal Na+-Ca2+
exchanger. Limiting sarcolemmal Na+ entry during
resuscitation from ventricular fibrillation has been shown to
prevent the accumulation of excess mitochondrial Ca2+
and attenuate myocardial injury (30). Additionally, it has become
apparent that the endothelium participates in a host of responses
elicited by I/R injury in PCAS. Both biochemical and mechanical
stimulation of the endothelium elicits the production of several
mediators, including endothelium-derived nitric oxide,
prostaglandins, antithrombotics and anticoagulants (31).
Enalapril is a long-acting, sulphydryl-free ACEI
with maximal humoral and hypotensive effects at 4–8 h, which remain
detectable at 24 h, after a single dose (32). It has been widely prescribed as an
anti-hypertensive drug for many years. Previously, the beneficial
effect of enalapril on myocardial ischemic injury has been
confirmed in animal models of coronary artery ligation (7,33).
These studies showed that ACEIs limited myocardial injury and
necrosis (7,33). Moreover, a larger scale clinical
trial demonstrated that there were significant reductions in the
number of enalapril-treated patients developing myocardial
infarction or unstable angina (34). Enalapril has also been shown to
decrease blood markers of inflammation in patients hospitalized
with ischemic symptoms (34).
However, there are significant differences between the myocardial
ischemic injury induced by coronary artery ligation and
CA-resuscitation. Coronary artery ligation always induces local
myocardial ischemic injury whereas CA-resuscitation may induce
global hypoperfusion that results in acute damage to other organs,
such as cerebral I/R injury and kidney I/R injury (9,10).
Disruption of the blood-brain barrier and the formation of brain
edema after CA-resuscitation are the major factors leading to
permanent brain damage (35).
However, information regarding the application of ACEIs in patients
with CA and resuscitation is limited. Recently, Hoyer et al
provided evidence that ACE inhibition led to significantly improved
myocardial contractility after prolonged CA with Bretschneider's
solution (36). In line with the
findings of this study, our data demonstrated that enalapril also
successfully decreased myocardial injury in a swine model of
CA-resuscitation. The attenuation of ultrastructural injury, plasma
cTNI levels and myocardial apoptosis were observed in the heart
tissue of the enalapril-treated swine. We suggest that the
cardioprotective effects of enalapril are associated with
anti-apoptotic activity. Previously, the anti-apoptotic effect of
ACEIs or ARBs were illustrated in left anterior descending (LAD)
artery ligation-induced local myocardial ischemia (37,38). To the best of our knowledge, our
data provides evidence for this effect of ACEIs in a model of
CA-resuscitation for the first time.
Some researchers and clinicians have suggested that
ACEIs may induce hypoperfusion and therefore be harmful to the
myocardium during CA and resuscitation (39,40). For example, a case report by
Bjerregaard and Jaffe found that a patient presenting for cerebral
aneurysm clipping continued ACEI use on the morning of surgery, and
subsequently experienced significant post-induction hypotension
that culminated in CA (39).
However, Zhang et al reported that enalapril did not affect
coronary perfusion pressure during the resuscitation period after
VF (41). Our data showed that
although MAP was slightly decreased, enalapril had no detrimental
effect on the survival rate post CA-resuscitation. These results
suggest that blockade of the RAAS would not increase the mortality
of CA. Clinical investigation has also demonstrated that there was
no difference in the incidence of CA between patients treated with
ACEIs and without ACEIs (2).
Thus, this study supports the notion that the application of ACEIs
is a promising therapy in post-resuscitation patients.
The present study further explored the potential
effects of enalapril pre-treatment on the RAAS system. The RAAS is
composed of a series of hormones and corresponding enzymes.
Angiotensinogen is the precursor molecule of the RAAS and its
cleavage product is Ang I, which is converted to Ang II by ACE. Ang
II is the major effector of the RAAS through binding to 2 receptor
subtypes, AT1R and AT2R. Most detrimental actions of Ang II on the
heart are mediated by AT1 (42,43). Ang-(1–7)
may be catalyzed by ACE2 from Ang II and activates the MAS receptor
to exert vasodilatatory, anti-angiogenic, anti-thrombotic and
anti-proliferative effects (6).
Thus, the balance between the ACE/Ang II/AT1R axis and the
ACE2/Ang-(1–7)/MAS axis elegantly modulates
cardiovascular biology. An imbalance between these two axes leads
to dysfunction of the RAAS, which may underlie the pathophysiology
of cardiovascular disorders such as ischemic heart disease,
vascular remodeling, hypertension, cardiac failure,
atherosclerosis, neointima formation and chronic kidney disease.
During these processes, an increased circulating level of Ang II
may be a critical adverse factor. In line with previous findings
(44), we observed elevations in
plasma Ang II and Ang-(1–7) in the saline group; however,
enalapril successfully lowered plasma Ang II levels. As a result,
we hypothesize that enalapril may protect organs from Ang
II-induced cellular damage by suppressing the ACE/Ang II/AT1R axis.
By contrast, enalapril did not affect Ang-(1–7)
levels, suggesting that ACEIs do not alter plasma Ang-(1–7)
levels.
Enalapril reduced the mRNA and protein expression of
ACE, as well as the ACE/ACE2 mRNA ratio. The ratio of ACE/ACE2 in
myocardial tissue is a marker of RAAS activation (45). Particularly, ACE and ACE2 often go
in opposite directions in renal pathological states, and the
ACE/ACE2 ratio is a convenient way to reflect this altered pattern
(45). It has been demonstrated
that the ACE/ACE2 ratio correlated positively with mean blood
pressure, fasting blood glucose, serum creatinine, proteinuria and
hemoglobin A1c, and inversely correlated with the estimated
glomerular filtration rate (34).
The ACE/ACE2 ratio may serve as a novel biomarker or independent
risk factor relevant for the diagnosis and the prognosis of
cardiorenal syndrome (46). An
increased ACE/ACE2 ratio in diabetic patients with overt
nephropathy suggests that RAAS activation has occurred, which may
contribute to renal injury as a result of Ang II accumulation
(45,47). Liu et al showed that a
change in the ACE/ACE2 ratio correlates with the severity of
cerulein-induced acute pancreatitis in mice (48). Moreover, the ACE/ACE2 ratio is a
potential biomarker for pulmonary disease (49). Consequently, the enalapril-induced
attenuation of the local ACE/ACE2 ratio may ultimately contribute
to the protective effects of enalapril on myocytes.
There are several limitations of this study.
Firstly, we used healthy pigs in this model of CA and
resuscitation; however, most humans suffering from CA are at risk
of hyperglycemia, hyperlipidemia and arrhythmia. These malfunctions
may affect the outcome of enalapril treatment in humans. Secondly,
we selected the dose of enalapril according to the findings of
previous studies and did not test other dosages and time windows
for enalapril pre-treatment in the CA and resuscitation model. In
fact, the dosage and time window for enalapril directly determines
the therapeutic outcome. Thirdly, enalapril was administered prior
to CA to illustrate the protective effects of enalapril against
global ischemic injury rather than against limited myocardial
tissue ischemia. Whether enalapril would still exert
cardioprotective effects post-CA was not evaluated.
In conclusion, our data demonstrate that enalapril
protects against myocardial I/R injury in a swine model of CA and
resuscitation by suppressing the ACE/Ang II/AT1R axis. These
results enhance our understanding of the cardioprotective effects
of enalapril targeted against CA-induced global I/R injury.
Acknowledgments
This study was supported by the National Natural
Science Foundation of China (no. 81374004).
References
|
1
|
Lerakis S, Hayek S, Arepalli CD, Thourani
V and Babaliaros V: Cardiac magnetic resonance for paravalvular
leaks in post-transcatheter aortic valve replacement. Circulation.
129:e430–e431. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Go AS, Mozaffarian D, Roger VL, Benjamin
EJ, Berry JD, Blaha MJ, Dai S, Ford ES, Fox CS, Franco S, et al
American Heart Association Statistics Committee and Stroke
Statistics Subcommittee: Heart disease and stroke statistics - 2014
update: a report from the American Heart Association. Circulation.
129:e28–e292. 2014. View Article : Google Scholar
|
|
3
|
Dumas F, White L, Stubbs BA, Cariou A and
Rea TD: Long-term prognosis following resuscitation from out of
hospital cardiac arrest: role of percutaneous coronary intervention
and therapeutic hypothermia. J Am Coll Cardiol. 60:21–27. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hypothermia after Cardiac Arrest Study
Group: Mild therapeutic hypothermia to improve the neurologic
outcome after cardiac arrest. N Engl J Med. 346:549–556. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Stub D, Bernard S, Duffy SJ and Kaye DM:
Post cardiac arrest syndrome: a review of therapeutic strategies.
Circulation. 123:1428–1435. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Paul M, Poyan Mehr A and Kreutz R:
Physiology of local renin-angiotensin systems. Physiol Rev.
86:747–803. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li K and Chen X: Protective effects of
captopril and enalapril on myocardial ischemia and reperfusion
damage of rat. J Mol Cell Cardiol. 19:909–915. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sharma JN: The kallikrein-kinin system:
from mediator of inflammation to modulator of cardioprotection.
Inflammopharmacology. 12:591–596. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kusch A, Hoff U, Bubalo G, Zhu Y, Fechner
M, Schmidt-Ullrich R, Marko L, Müller DN, Schmidt-Ott KM, Gürgen D,
et al: Novel signalling mechanisms and targets in renal ischaemia
and reperfusion injury. Acta Physiol (Oxf). 208:25–40. 2013.
View Article : Google Scholar
|
|
10
|
Geng Y, Li E, Mu Q, Zhang Y, Wei X, Li H,
Cheng L and Zhang B: Hydrogen sulfide inhalation decreases early
blood-brain barrier permeability and brain edema induced by cardiac
arrest and resuscitation. J Cereb Blood Flow Metab. 35:494–500.
2015. View Article : Google Scholar
|
|
11
|
Guo ZJ, Li CS, Yin WP, Hou XM, Gu W and
Zhang D: Comparison of shock-first strategy and cardiopulmonary
resuscitation-first strategy in a porcine model of prolonged
cardiac arrest. Resuscitation. 84:233–238. 2013. View Article : Google Scholar
|
|
12
|
Idris AH, Becker LB, Ornato JP, Hedges JR,
Bircher NG, Chandra NC, Cummins RO, Dick W, Ebmeyer U, Halperin HR,
et al Writing Group: Utstein-style guidelines for uniform reporting
of laboratory CPR research A statement for healthcare professionals
from a task force of the American Heart Association the American
College of Emergency Physicians, the American College of
Cardiology, the European Resuscitation Council, the Heart and
Stroke Foundation of Canada, the Institute of Critical Care
Medicine, the Safar Center for Resuscitation Research, and the
Society for Academic Emergency Medicine. Circulation. 94:2324–2336.
1996. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang S, Li C, Ji X, Yang L, Su Z and Wu J:
Effect of continuous compressions and 30:2 cardiopulmonary
resuscitation on global ventilation/perfusion values during
resuscitation in a porcine model. Crit Care Med. 38:2024–2030.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Pereira AJ, Jeger V, Fahrner R,
Djafarzadeh S, Lensch M, Takala J and Jakob SM: Interference of
angiotensin II and enalapril with hepatic blood flow regulation. Am
J Physiol Gastrointest Liver Physiol. 307:G655–G663. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Gu W, Li CS, Yin WP, Hou XM, Zhang J,
Zhang D and Guo Z: Expression imbalance of transcription factors
GATA-3 and T-bet in post-resuscitation myocardial immune
dysfunction in a porcine model of cardiac arrest. Resuscitation.
84:848–853. 2013. View Article : Google Scholar
|
|
16
|
Zhang Q and Li C: Combination of
epinephrine with esmolol attenuates post-resuscitation myocardial
dysfunction in journalporcine model of cardiac arrest. PLoS One.
8:e826772013. View Article : Google Scholar
|
|
17
|
Ji XF, Li CS, Wang S, Yang L and Cong LH:
Comparison of the efficacy of nifekalant and amiodarone in a
porcine model of cardiac arrest. Resuscitation. 81:1031–1036. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Valenzuela TD, Roe DJ, Cretin S, Spaite DW
and Larsen MP: Estimating effectiveness of cardiac arrest
interventions: a logistic regression survival model. Circulation.
96:3308–3313. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang P, Xu TY, Wei K, Guan YF, Wang X, Xu
H, Su DF, Pei G and Miao CY: ARRB1/β-arrestin-1 mediates
neuroprotection through coordination of BECN1-dependent autophagy
in cerebral ischemia. Autophagy. 10:1535–1548. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wang YG, Li Y, Wang CY, Ai JW, Dong XY,
Huang HY, Feng ZY, Pan YM, Lin Y, Wang BX and Yao LL:
L-3-n-Butylphthalide protects rats' cardiomyocytes from
ischaemia/reperfusion-induced apoptosis by affecting the
mitochondrial apoptosis pathway. Acta Physiol (Oxf). 210:524–533.
2014. View Article : Google Scholar
|
|
21
|
Wang P, Du H, Zhou CC, Song J, Liu X, Cao
X, Mehta JL, Shi Y, Su DF and Miao CY: Intracellular
NAMPT-NAD+-SIRT1 cascade improves post-ischaemic
vascular repair by modulating Notch signalling in endothelial
progenitors. Cardiovasc Res. 104:477–488. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Ragone MI, Torres NS and Consolini AE:
Energetic study of cardioplegic hearts under ischaemia/reperfusion
and [Ca(2+)] changes in cardiomyocytes of guinea-pig:
mitochondrial role. Acta Physiol (Oxf). 207:369–384. 2013.
View Article : Google Scholar
|
|
23
|
Wang P, Xu TY, Guan YF, Zhao Y, Li ZY, Lan
XH, Wang X, Yang PY, Kang ZM, Vanhoutte PM and Miao CY: Vascular
smooth muscle cell apoptosis is an early trigger for hypothyroid
atherosclerosis. Cardiovasc Res. 102:448–459. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang P, Xu TY, Guan YF, Tian WW, Viollet
B, Rui YC, Zhai QW, Su DF and Miao CY: Nicotinamide
phosphoribosyltransferase protects against ischemic stroke through
SIRT1-dependent adenosine monophosphate-activated kinase pathway.
Ann Neurol. 69:360–374. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang P, Guan YF, Du H, Zhai QW, Su DF and
Miao CY: Induction of autophagy contributes to the neuroprotection
of nicotinamide phosphoribosyltransferase in cerebral ischemia.
Autophagy. 8:77–87. 2012. View Article : Google Scholar
|
|
26
|
Wang P, Xu TY, Guan YF, Su DF, Fan GR and
Miao CY: Perivascular adipose tissue-derived visfatin is a vascular
smooth muscle cell growth factor: role of nicotinamide
mononucleotide. Cardiovasc Res. 81:370–380. 2009. View Article : Google Scholar
|
|
27
|
Mapanga RF, Joseph D, Symington B, Garson
KL, Kimar C, Kelly-Laubscher R and Essop MF: Detrimental effects of
acute hyperglycaemia on the rat heart. Acta Physiol (Oxf).
210:546–564. 2014. View Article : Google Scholar
|
|
28
|
Reynolds JC and Lawner BJ: Management of
the post-cardiac arrest syndrome. J Emerg Med. 42:440–449. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Gu W, Li CS, Yin WP, Guo ZJ, Hou XM and
Zhang D: Apoptosis is involved in the mechanism of
postresuscitation myocardial dysfunction in a porcine model of
cardiac arrest. Am J Emerg Med. 30:2039–2045. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wang S, Radhakrishnan J, Ayoub IM,
Kolarova JD, Taglieri DM and Gazmuri RJ: Limiting sarcolemmal
Na+ entry during resuscitation from ventricular
fibrillation prevents excess mitochondrial Ca2+
accumulation and attenuates myocardial injury. J Appl Physiol
(1985). 103:55–65. 2007. View Article : Google Scholar
|
|
31
|
Adams JA: Endothelium and cardiopulmonary
resuscitation. Crit Care Med. 34(Suppl): S458–S465. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lant AF, McNabb RW and Noormohamed FH:
Kinetic and metabolic aspects of enalapril action. J Hypertens
Suppl. 2:S37–S42. 1984.PubMed/NCBI
|
|
33
|
Doğan R, Farsak B, Tuncer M and Demirpençe
E: Attenuation of ischemia-reperfusion injury by enalapril maleat.
Gen Pharmacol. 31:203–208. 1998. View Article : Google Scholar
|
|
34
|
Montalescot G, Drexler H, Gallo R, Pearson
T, Thoenes M and Bhatt DL: Effect of irbesartan and enalapril in
non-ST elevation acute coronary syndrome: Results of the
randomized, double-blind ARCHIPELAGO study. Eur Heart J.
30:2733–2741. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Miclescu A, Sharma HS, Martijn C and
Wiklund L: Methylene blue protects the cortical blood-brain barrier
against ischemia/reperfusion-induced disruptions. Crit Care Med.
38:2199–2206. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Hoyer A, Kempfert J, Pritzwald-Stegmann P,
Mohr FW and Dhein S: Acute hemodynamic effects of
angiotensin-converting enzyme inhibition after prolonged cardiac
arrest with Bretschneider's solution. Naunyn Schmiedebergs Arch
Pharmacol. 387:1221–1229. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Wu B, Lin R, Dai R, Chen C, Wu H and Hong
M: Valsartan attenuates oxidative stress and NF-κB activation and
reduces myocardial apoptosis after ischemia and reperfusion. Eur J
Pharmacol. 705:140–147. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kobara M, Tatsumi T, Kambayashi D, Mano A,
Yamanaka S, Shiraishi J, Keira N, Matoba S, Asayama J, Fushiki S
and Nakagawa M: Effects of ACE inhibition on myocardial apoptosis
in an ischemia-reperfusion rat heart model. J Cardiovasc Pharmacol.
41:880–889. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Bjerregaard J and Jaffe RA: Intraoperative
cardiac arrest: was it the ACE inhibitor? J Clin Anesth. 26:62–64.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chua HR, Glassford N and Bellomo R: Acute
kidney injury after cardiac arrest. Resuscitation. 83:721–727.
2012. View Article : Google Scholar
|
|
41
|
Zhang Y, Boddicker KA, Rhee BJ, Davies LR
and Kerber RE: Effect of nitric oxide synthase modulation on
resuscitation success in a swine ventricular fibrillation cardiac
arrest model. Resuscitation. 67:127–134. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Marques Neto SR, da H Silva A, dos Santos
MC, Ferraz EF and Nascimento JH: The blockade of angiotensin AT1
and aldosterone receptors protects rats from synthetic
androgen-induced cardiac autonomic dysfunction. Acta Physiol (Oxf).
208:166–171. 2013. View Article : Google Scholar
|
|
43
|
Biwer LA, Broderick TL, Xu H, Carroll C
and Hale TM: Protection against L-NAME-induced reduction in cardiac
output persists even after cessation of angiotensin-converting
enzyme inhibitor treatment. Acta Physiol (Oxf). 207:156–165. 2013.
View Article : Google Scholar
|
|
44
|
Paradis NA, Rose MI and Garg U: The effect
of global ischemia and reperfusion on the plasma levels of
vasoactive peptides. The neuroendocrine response to cardiac arrest
and resuscitation Resuscitation. 26:261–269. 1993.
|
|
45
|
Soler MJ, Wysocki J and Batlle D: ACE2
alterations in kidney disease. Nephrol Dial Transplant.
28:2687–2697. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Varagic J, Ahmad S, Nagata S and Ferrario
CM: ACE2: Angiotensin II/angiotensin-(1–7) balance in cardiac and
renal injury. Curr Hypertens Rep. 16:4202014. View Article : Google Scholar
|
|
47
|
Bernardi S, Toffoli B, Zennaro C, Tikellis
C, Monticone S, Losurdo P, Bellini G, Thomas MC, Fallo F, Veglio F,
et al: High-salt diet increases glomerular ACE/ACE2 ratio leading
to oxidative stress and kidney damage. Nephrol Dial Transplant.
27:1793–1800. 2012. View Article : Google Scholar
|
|
48
|
Liu R, Qi H, Wang J, Wang Y, Cui L, Wen Y
and Yin C: Angiotensin-converting enzyme (ACE and ACE2) imbalance
correlates with the severity of cerulein-induced acute pancreatitis
in mice. Exp Physiol. 99:651–663. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Hsieh WY, Kuan TC, Cheng KS, Liao YC, Chen
MY, Lin PH, Hsu YC, Huang CY, Hsu WH, Yu SY and Lin CS: ACE/ACE2
ratio and MMP-9 activity as potential biomarkers in tuberculous
pleural effusions. Int J Biol Sci. 8:1197–1205. 2012. View Article : Google Scholar : PubMed/NCBI
|