Introduction
Indoleamine 2,3-dioxygenase (IDO) is expressed in
antigen-presenting cells (APCs) and has an immunoregulatory role in
various models of autoimmunity and allotransplantation (1–6).
In addition to APCs, IDO is expressed in trophoblast cells
contributing to successful semi-allogenic pregnancy (7,8),
while its expression in certain cancer cells has been incriminated
for escape of cancer from immunosurveillance (9). In hemodialysis patients, who are
characterized by impaired adaptive immunity, increased serum IDO
levels have been associated with decreased T-cell count, as well as
failure to respond to a vaccine with a T-cell dependent antigen
(10,11).
By degrading L-tryptophan along the kynurenine
pathway, IDO alters the local microenvironment in a manner that
suppresses T-cell function. More precisely, L-tryptophan depletion
activates general control non-derepressible 2 kinase (GCN2K), which
phosphorylates eukaryotic initiation factor 2α (eIF2α), altering
the translational program of T-cells (12–16). The effect of L-tryptophan
depletion on the other amino acid sensing system, the mammalian
target of rapamycin complex 1 (mTORC1), has also been investigated
although with contradictory results (12–17). Kynurenine, a product of
L-tryptophan degradation by IDO, by activating the aryl-hydrocarbon
receptor (AhR) is involved in the immunosuppressive action of this
enzyme favoring CD4+ T-cell differentiation towards
regulatory T-cells (Treg) (18,19).
Considering the above described pathways as a
starting point, the presence of IDO in APCs leads to decreased
proliferation and increased apoptosis, and promotes the
differentiation of CD4+ T-cells towards a regulatory
instead of effector (Teff) phenotype (20,21). While many intermediate events
remain to be elucidated, recent research indicates that IDO may
exert these effects by affecting the metabolism of CD4+
T-cells. Indeed, IDO suppresses aerobic glycolysis and
glutaminolysis in human alloreactive CD4+ T-cells by
affecting the expression of glucose transporter 1 and various
glycolytic and glutaminolytic enzymes (14–16). It also downregulates key enzymes
involved in fatty acid synthesis (17). The above-mentioned metabolic
pathways are prerequisites for rapid T-cell proliferation following
T-cell receptor stimulation, as well as for differentiation towards
effector cell lineages instead of Treg. Following T-cell
activation, T-cells reprogram their metabolic pathways from
pyruvate via the Krebs cycle to the glycolytic and glutaminolytic
pathways in order to fulfill the bioenergetic and biosynthetic
demands of proliferation (22–24). In parallel, fatty acid synthesis
is upregulated during the activation of CD4+ T-cells
enhancing their proliferation and promoting their differentiation
into T helper 17 cells (Th17) instead of Tregs (25).
Another metabolic pathway that plays a significant
role in the CD4+ T-cell response and differentiation is
fatty acid β-oxidation. More precisely, Tregs are dependent on
fatty acid oxidation for its differentiation, whereas, Teff
populations on aerobic glycolysis (26). The effect of IDO on fatty acid
oxidation in CD4+ T-cells remains to be investigated,
and constitutes the aim of the present study.
For the purposes of the present study, two-way mixed
lymphocyte reaction (MLR) was used as a model of alloreactivity
(27), along with the specific
IDO inhibitor, 1-DL- methyl-tryptophan (1MT) (4,7).
In order to evaluate fatty acid oxidation, cells were cultured in a
medium containing oleate. The effect of IDO on carnitine
palmitoyltransferase I (CPT1), the tightly regulated enzyme that
controls the entry of fatty acids into the mitochondria for
oxidation (28,29), was assessed as well. Treatment of
CD4+ T-cells with the CPT1 inhibitor, etomoxir, has been
shown to abrogate differentiation into Tregs (26). The effects of IDO on the
end-points of CD4+ T-cell function, proliferation,
apoptosis and differentiation were also evaluated.
Materials and methods
Subjects
Blood samples were collected from 5 healthy
volunteers (3 males and 2 females, 37±7 years of age). Informed
consent was obtained from each individual enrolled in the study and
the Ethics Committee of the University Hospital of Larissa
(Larissa, Greece) approved the study protocol.
Cell culture conditions
Peripheral blood mononuclear cells (PBMCs) were
isolated from whole blood by the Ficoll-Hypaque density gradient
centrifugation (Histopaque 1077; Sigma-Aldrich, St. Louis, MO, USA)
and counted using an optical microscope (Axiovert 40 C; Carl Zeiss
AG, Oberkochen, Germany) on a Neubauer plaque. Cell viability was
assessed by trypan blue assay (Sigma-Aldrich). Cell cultures were
performed in RPMI-1640 medium with L-glutamine, 10 mM
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) and
supplemented with 10% fetal bovine serum and antibiotic-antimycotic
solution (both from Sigma-Aldrich). The cultures were performed at
37°C in a humidified atmosphere containing 5% CO2.
Ten MLRs were performed in the presence or absence
of the IDO inhibitor 1MT (Sigma-Aldrich) at a concentration of 100
μM. The above concentration was chosen according to previous
experiments that revealed efficacy without toxicity (13–16). Unless otherwise stated, in all the
experiments, oleate (Sigma-Aldrich) at a final concentration of 1
mM was added from the beginning of the MLRs.
To determine cell proliferation, 10 MLRs were
performed in 96-well plates for 7 days. Peripheral blood
mononuclear cells from each member of the MLR couple were
5×104, measuring to 1×105 PBMCs in total in
each well. Cultures of 1×105/well resting PMBCs from
each member of the MLR couple were used as controls.
To assess various components in the supernatant, as
well as the expression of certain proteins and CPT1 activity in
CD4+ T-cells, 10 MLRs were performed in 12-well plates
for 7 days. The number of PBMCs for each member of the MLR couple
was 5×105, reaching a total of 1×106 PBMCs in
each well. At the end of the 7-day period supernatants were
collected and stored at −80°C, whereas CD4+ T-cells were
isolated from the MLRs by negative selection using the
CD4+ T-cell isolation kit, human (Miltenyi Biotec GmbH,
Bergisch Gladbach, Germany).
Cell proliferation in two-way mixed
lymphocyte reactions
Cell proliferation enzyme-linked immunosorbent assay
(ELISA) (Roche Diagnostics, Indianapolis, IN, USA), based on
bromodeoxyuridine (BrdU) labeling and immunoenzymatic detection,
was used to examine cell proliferation. The proliferation index was
calculated as the ratio of the optical density (OD) derived from
each MLR to the mean of the ODs derived from the control resting
PBMC cultures of the two subjects that constituted the specific
MLR. These experiments were performed in triplicate and the results
refer to the mean of the three measurements.
L-tryptophan and oleate consumption in
MLRs
L-tryptophan consumption was assessed by measuring
its concentration in the supernatants of MLRs by means of ELISA
(BlueGene Biotech, Shanghai, China). The sensitivity of the above
ELISA kit is 1 ng/ml.
Similarly, oleate consumption was assessed by
measuring its concentration in the supernatants of MLRs
colorimetrically using the Free Fatty Acid Quantification kit
(Abcam, Cambridge, UK). The detection limit of the above kit was 2
μM.
Expression of certain proteins in
CD4+ T-cells isolated from the MLRs
The expression of certain proteins in
CD4+ T-cells was assessed by western blot analysis.
Isolated CD4+ T-cells were counted via optical
microscopy on a Neubauer plaque and cell viability was determined
by trypan blue assay (Sigma-Aldrich). Equal numbers of T-cells from
each MLR were lysed using the T-PER tissue protein extraction
reagent (Thermo Fisher Scientific Inc., Rockford, IL, USA)
supplemented with protease and phosphatase inhibitors
(Sigma-Aldrich and Roche Diagnostics). Protein was quantified using
the Bradford assay (Sigma-Aldrich) and 10 μg from each
sample were used for western blot analysis. The blots were
incubated with the primary antibodies for 16 h, followed by the
secondary antibody (anti-rabbit IgG, HRP-linked antibody; Cell
Signaling Technology, Danvers, MA, USA) incubation for 30 min. In
case of reprobing PVDF blots, the previous primary and secondary
antibodies were removed using the Restore Western Blot Stripping
Buffer (Thermo Fisher Scientific Inc.) according to the
manufacturer's protocol. The PVDF blot was then reused and western
blot analysis resumed as previously described, using a different
primary antibody. Analysis of the western blots was performed using
the ImageJ software (National Institute of Health, Bethesda, MD,
USA).
The primary antibodies used in western blot analysis
were specific for eIF2α phosphorylated at serine 51 (p-eIF2α; Cat.
no. 9721) (Cell Signaling Technology), cytochrome P450, family 1,
subfamily A, polypeptide 1 (CYP1A1; Cat. no. sc-20772) (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), p-70S6 kinase phosphorylated
at threonine 389 (p-p70S6K; Cat. no. 9234) (Cell Signaling
Technology), CPT1A (Cat. no. 12252S; Cell Signaling Technology),
CPT1B (Cat. no. ab134988), CPT1C (Cat. no. ab87498) (both from
Abcam), acetyl-CoA carboxylase 2 (ACC2; Cat. no. 8578) (Cell
Signaling Technology), ACC2 phosphorylated at serine 221 (p-ACC2;
Cat. no. ab109540) (Abcam), activated cleaved at aspartate 175
caspase-3 (Cat. no. 9664), forkhead box P3 (FoxP3; Cat. no. 5298)
(both from Cell Signaling Technology), retinoic acid receptor
related orphan receptor γt (RORγt; Cat. no. orb6888) (Biorbyt,
Cambridge, UK) and β-actin (Cat. no. 4967; Cell Signaling
Technology).
Carnitine palmitoyltransferase I
activity
To determine CPT1 enzyme activity, a non-radioactive
method was performed in whole cell lysates according to the method
of Bieber and Fiol (30).
CD4+ T-cell lysates were prepared as described for
western blot analysis and the method was based on measurement of
the initial release of CoA-SH from palmitoyl CoA
specrtrophotometrically using the reagent
5,5′-dithio-bis-(2-nitrobenzoic acid) (DTNB). Briefly, 50 μl
buffer solution (containing 116 mM Tris, 2.5 mM EDTA, 2 mM DTNB,
0.2% Triton X-100, pH 8.0) and 50 μg protein extract were
added to the reaction mixture. After 5 min preincubation at 28°C,
50 μl of 1 mM palmitoyl-CoA was added and the reaction was
initiated with a final addition of 5 μl of 1.2 mM
L-carnitine solution, followed by an immediate photometric
measurement at 412 nm. These reagents were purchased from
Sigma-Aldrich.
Statistical analysis
The normality of the evaluated variables was
assessed and confirmed by the one-sample Kolmogorov-Smirnov test.
For comparison of means the paired-sample t-test or unpaired-sample
t-test were used. Results were presented as the means ± standard
deviation (SD). A value of P<0.05 was considered to indicate a
statistically significant difference.
The results obtained from the western blot analysis
and enzyme activity assay are expressed as optical densities (OD),
thus p-values were calculated by comparing the means of OD.
Statistical analysis after normalization for the control OD values
was avoided to prevent violation of the prerequisite for normal
distribution of the compared variables when applying parametric
statistical tests. However, for the reader's convenience, in the
text and figures the results are noted and depicted after
normalization of means for the control group.
Results
IDO increases L-tryptophan degradation in
MLRs, enhances eIF2α phosphorylation and CYP1A1 expression in
MLR-derived CD4+ T-cells, but does not affect p70S6K
phosphorylation in MLR-derived CD4+ T-cells
In MLRs, IDO increased L-tryptophan degradation
since its inhibitor 1MT increased L-tryptophan concentration in the
supernatants from 2.47±0.44 to 6.19±0.47 μg/ml (p<0.001,
paired t-test) (Fig. 1A).
By degrading L-tryptophan, IDO enhanced the p-eIF2α
level in MLR-derived CD4+ T-cells since the treatment of
MLRs with 1MT altered the p-eIF2α level by a factor of 0.52±0.18
(p<0.001, paired t-test) (Fig. 1B
and C). Similarly, in CD4+ T-cells derived from
1MT-treated MLRs, CYP1A1 expression was altered by a factor of
0.51±0.29 (p<0.001, paired t-test) indicating that IDO increases
CYP1A1 expression (Fig. 1B and
D). By contrast, 1MT treatment of the MLRs did not affect the
content of p-p70S6K in CD4+ T-cells, since its level was
altered only by a factor of 1.07±0.16 (p=0.316, paired t-test)
(Fig. 1B and E).
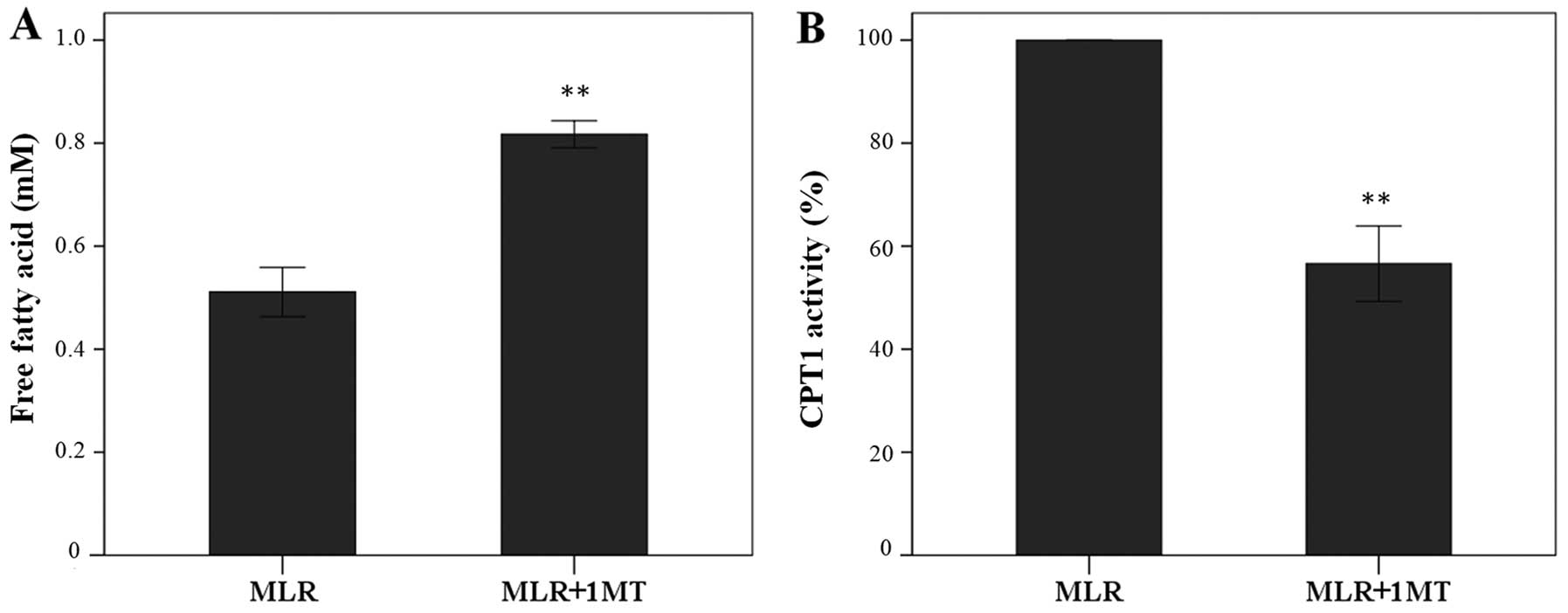
IDO increases fatty acid oxidation in
MLRs and CPT1 enzymatic activity in MLR-derived CD4+
T-cells
In MLRs, IDO increased fatty acid oxidation since
its inhibitor 1MT increased the oleate concentration in the
supernatants from 0.51±0.05 mM to 0.82±0.03 mM (p<0.001, paired
t-test) (Fig. 2A).
In CD4+ T-cells derived from 1MT-treated
MLRs CPT1 activity was at the 56.61±7.32% of the activity found in
cells derived from the control MLRs (p<0.001, paired t-test),
indicating that IDO enhanced CPT1 enzymatic activity in
CD4+ T-cells (Fig.
2B).
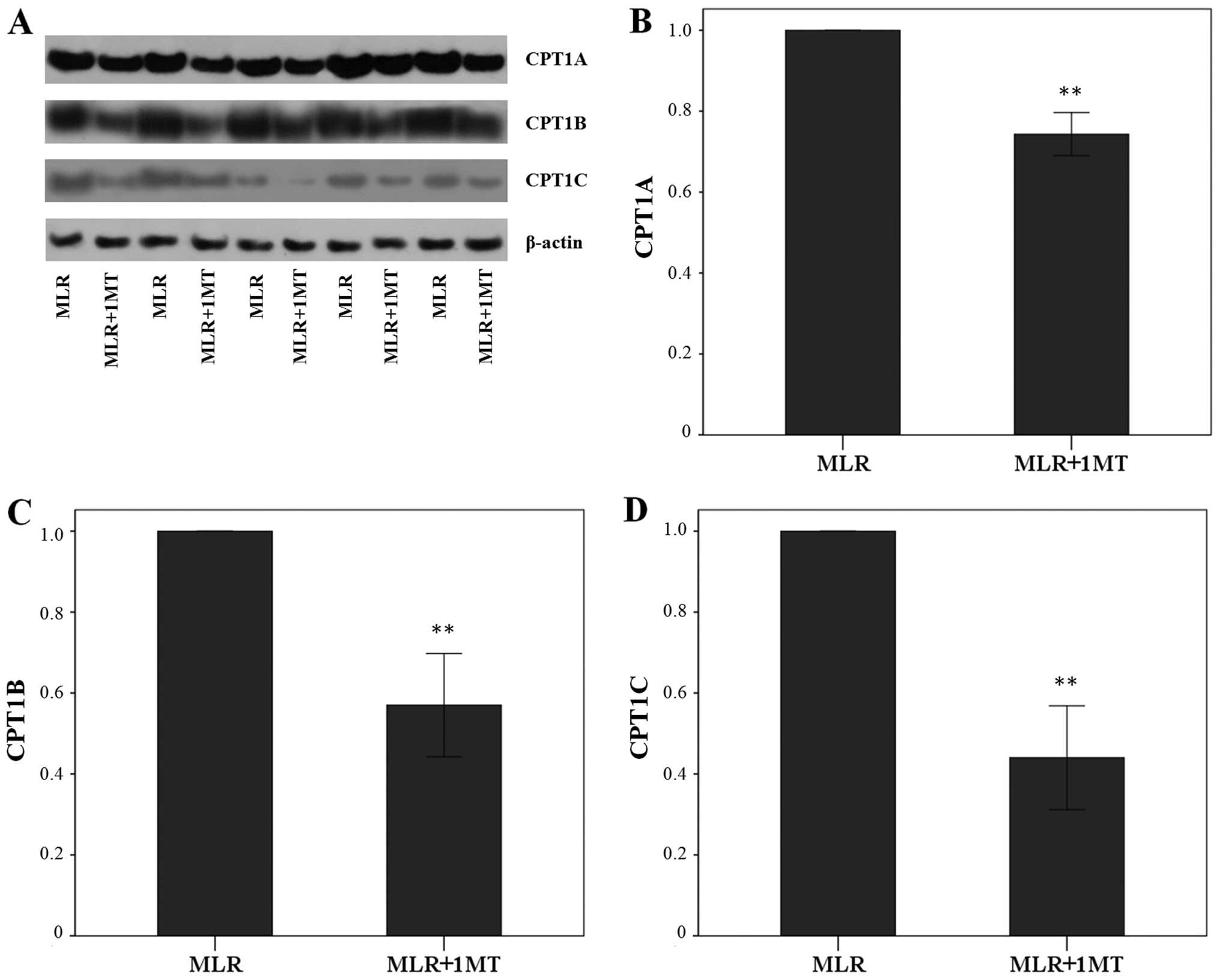
IDO increases CPT1A, CPT1B and CPT1C
expression in MLR-derived CD4+ T-cells
Unblocked IDO activity in MLRs increased CPT1A
expression in MLR-derived CD4+ T-cells since the
treatment of MLRs with the IDO inhibitor, 1MT, altered the CPT1A
level by a factor of 0.74±0.05 (p<0.001, paired t-test)
(Fig. 3A and B). This was even
more profound in the case of CPT1B, which was altered due to 1MT by
a factor of 0.57±0.13 (p<0.001, paired t-test) (Fig. 3A and C), and of CPT1C, which was
altered by a factor of 0.44±0.13 (p<0.001, paired t-test)
(Fig. 3A and D).
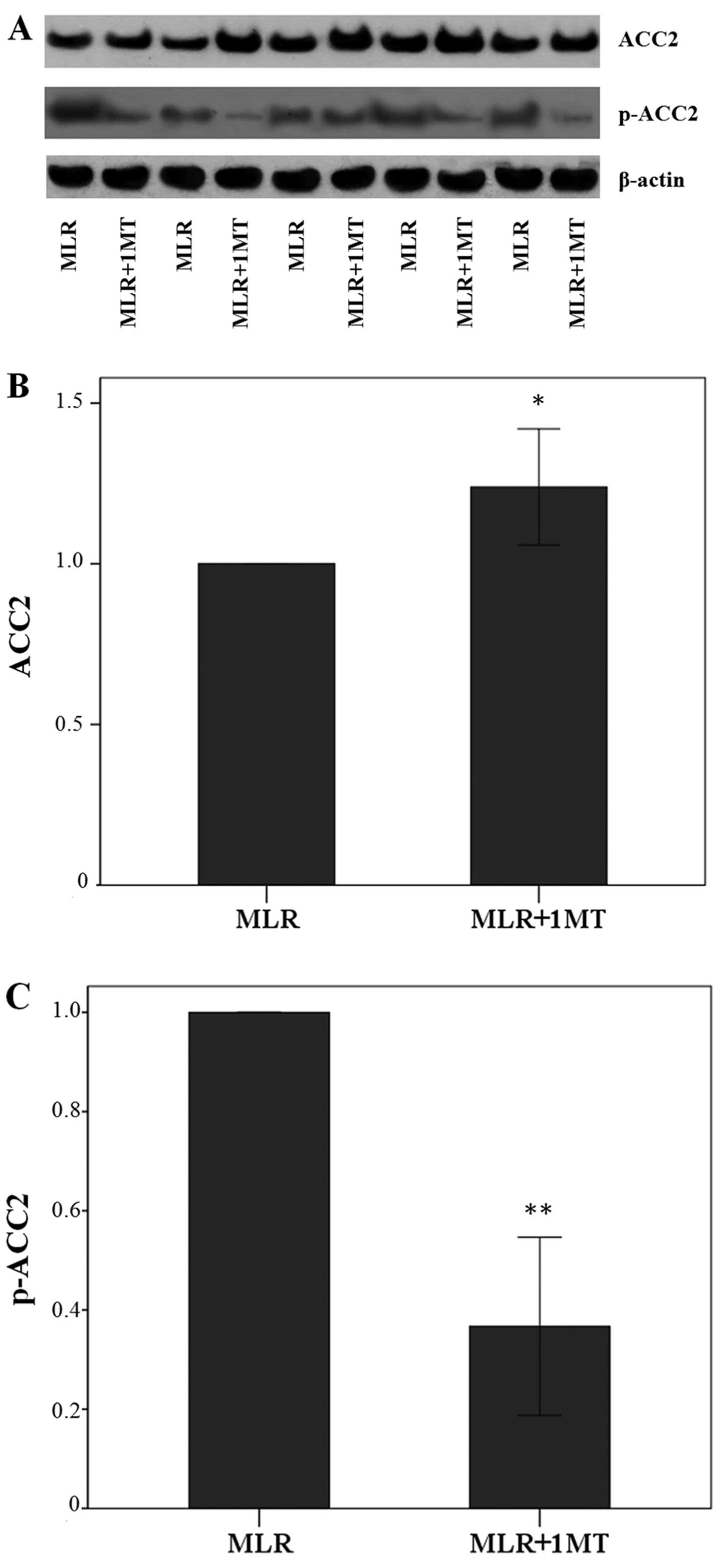
IDO decreases ACC2 expression, whereas it
increases the level of phosphorylated ACC2 in MLR-derived
CD4+ T-cells
IDO activity in the MLRs decreased the total ACC2
expression in MLR-derived CD4+ T-cells since the
treatment of MLRs with the IDO inhibitor, 1MT, led to alterations
in the levels of ACC2 by a factor of 1.24±0.18 (p=0.001, paired
t-test) (Fig. 4A and B).
The effect of IDO on the level of p-ACC2 was more
profound since in the CD4+ T-cells derived from the
1MT-treated MLRs, the level of p-ACC2 was altered by a factor of
0.37±0.18 (p<0.001, paired t-test) (Fig. 4A and C). Thus, IDO, by degrading
L-tryptophan in the MLRs, increased the content of the inactivated
phosphorylated form of ACC2 in CD4+ T-cells.
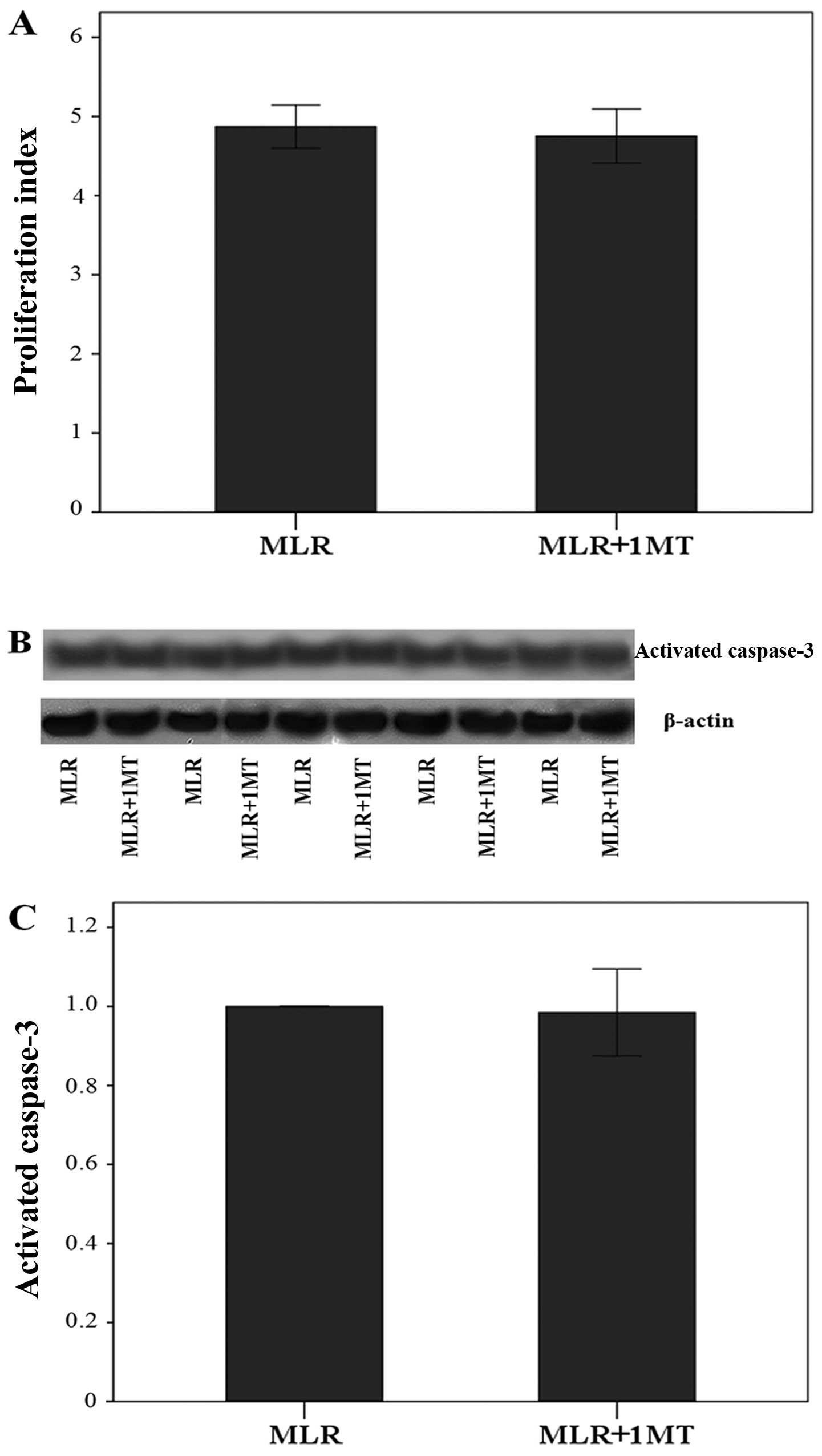
IDO does not affect cell proliferation in
MLRs nor activated caspase-3 in MLR-derived CD4+
T-cells
Using culture media containing oleate, IDO did not
affect cell proliferation in MLRs, since the addition of 1MT did
not affect the proliferation index significantly. More precisely,
the proliferation index was 4.87±0.27 in the untreated MLRs and
4.75±0.34 in the 1MT-treated MLRs (p=0.313, paired t-test)
(Fig. 5A).
Similarly, in the presence of oleate, IDO did not
affect the content of activated caspase-3 in CD4+
T-cells, which is the terminal caspase of the apoptotic pathways.
Compared to the activated caspase-3 level in CD4+
T-cells derived from the control MLRs, its level did show a
negligible variation only by a factor of 0.98±0.11 in
CD4+ T-cells derived from 1MT-treated MLRs (p=0.523,
paired t-test) (Fig. 5B and
C).
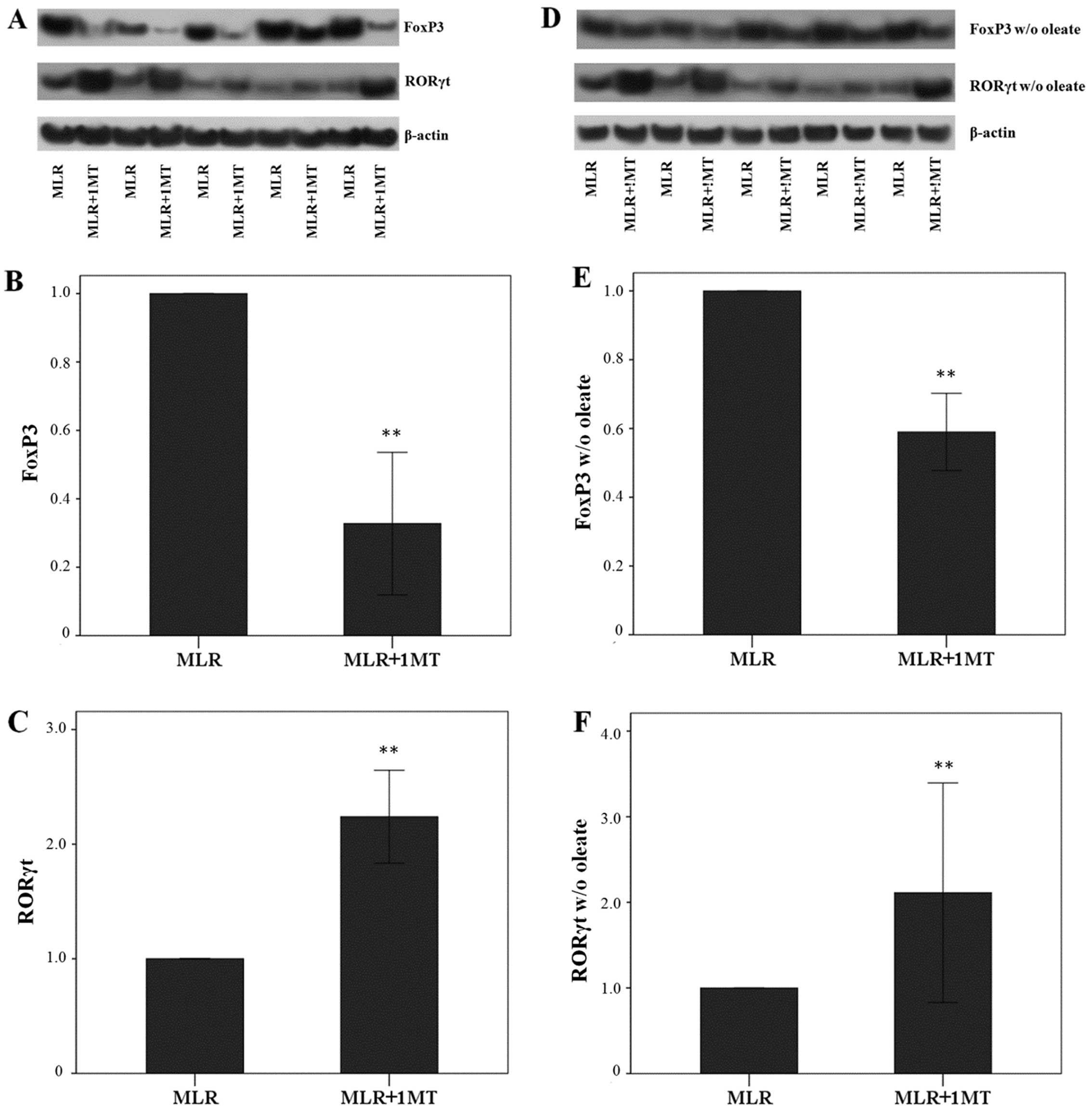
IDO, particularly in the presence of
oleate, induces FoxP3 expression, but suppresses RORγt expression
in MLR-derived CD4+ T-cells
Unblocked IDO activity in MLRs increased FoxP3
expression in MLR-derived CD4+ T-cells as the treatment
of MLRs with the IDO inhibitor, 1MT, altered the FoxP3 level by a
factor of 0.33±0.21 (p<0.001, paired t-test) (Fig. 6A and B).
The opposite was observed with the expression of
RORγt. Treatment of the MLRs with the IDO inhibitor, 1MT, induced a
significant increase in RORγt levels by a factor of 2.24±0.41
(p<0.001, paired t-test) (Fig. 6A
and C). Thus, by degrading L-tryptophan, IDO decreased RORγt
expression in CD4+ T-cells.
In the absence of oleate, treatment of the MLRs with
1MT also resulted in a decrease in FoxP3 expression in
CD4+ T-cells by a factor of 0.59±0.11 (p<0.001)
(Fig. 6D and E). However, this
decrease was significantly less than that observed in MLRs in the
presence of oleate (p=0.003, unpaired t-test) suggesting that
oleate is beneficial for FoxP3 expression.
In the absence of oleate from the MLRs, 1MT
treatment also resulted in an increase in RORγt expression in
CD4+ T-cells by a factor of 2.11±1.28 (p<0.001)
(Fig. 6D and F). This increase
did not differ from the increase observed in MLRs performed in the
presence of oleate (p=0.772, unpaired t-test).
Discussion
Indoleamine 2,3-dioxygenase is expressed in APCs and
by degrading L-tryptophan in the microenvironment where the immune
response occurs, it suppresses CD4+ T-cell function by
inhibiting cell proliferation, inducing apoptosis and promoting
differentiation into Tregs (20,21).
In order to define which of the described pathways
are involved in the effect of IDO on CD4+ T cells
(12–14,17–19), a model of alloreactivity, the MLR,
was used. In this model, IDO induced L-tryptophan degradation.
Decreased L-tryptophan activated the GCN2K pathway since the
phosphorylation of its substrate eIF2α was increased when IDO
activity was not blocked by 1MT. This observation is in accordance
with previous studies (12–14). Additionally, the present study
recapitulates the results of other studies that failed to detect an
effect on the other amino-acid sensing system, the mTORC1, since
the level of phosphorylation of its substrate, p70S6K, remained
unaffected by 1MT (12–14). This is in accordance with findings
showing that mTORC1 is sensitive to the depletion of certain amino
acids; and more precisely of leucine, isoleucine, valine and
possibly arginine, but not of tryptophan (31). Furthermore, L-tryptophan depletion
and its degradation by IDO results in the production of kynurenine,
which may affect CD4+ T-cell function (18,19). In the MLR-derived CD4+
T cells, the expression of CYP1A1, a transcriptional target of AhR,
was increased in the absence of 1MT, indicating that the IDO
kynurenine AhR pathway is associated with our experimental
model.
Recent studies have confirmed that IDO may exert its
effect on CD4+ T cells by affecting their metabolism
(13–16). Specifically, L-tryptophan
degradation by IDO has been shown to decrease aerobic glycolysis,
glutaminolysis and fatty acid synthesis (13–16), all required for rapid
CD4+ T-cell proliferation and differentiation towards
Teff lineages (22–25). The results of the present study
confirmed that L-tryptophan degradation by IDO increased fatty acid
consumption in MLRs. In parallel, the activity of CPT1 in
MLR-derived CD4+ T cells increased. Fatty acid
β-oxidation occurs in the mitochondrial matrix. However, acyl-CoAs
cannot pass the inner mitochondrial membrane, unless they are
converted to acylcarnitine in the cytoplasmic surface of the inner
mitochondrial membrane. This reaction is catalyzed by CPT1, which,
by controlling the entry of fatty acid into the mitochondrial
matrix regulates the rate of fatty acid oxidation (28,29).
The reason for the increased CPT1 activity in
CD4+ T cells derived from MLR without the IDO inhibitor,
1MT, may be due to the increased levels of the three CPT1
isoenzymes identified in the current study. A possible explanation
may depend on the confirmed effect of IDO-induced L-tryptophan
degradation in transcription factors such as p53 and cMyc that
control cell metabolism in CD4+ T cells (14,16). However, the exact mechanism for
this IDO-related increase in CPT1A, CPT1B and CPT1C expression
remains to be elucidated.
In addition to CPT1 expression, the activity of this
enzyme is tightly regulated and more precisely, is allosterically
inhibited by malonyl-CoA. Malonyl-CoA is produced by ACC2, an
enzyme associated with the outer mitochondrial membrane (28,29). When IDO activity was not inhibited
in MLRs, ACC2 expression in the MLR-derived CD4+ T cells
decreased. In addition, possibly due to AMP-activated protein
kinase (AMPK) (32), the
phosphorylated inactivated form of ACC2 increased markedly. This
IDO-induced alteration in ACC2 is expected to lead to decreased
ACC2 activity, decreased malonyl-CoA production and increased CPT1
activity and fatty acid β-oxidation.
We also evaluated the effect of IDO-induced
L-tryptophan degradation on two terminal points of CD4+
T-cell immune response, proliferation and apoptosis. Contrary to
what has been shown in a similar experimental model (13–16), IDO did not affect cell
proliferation in MLRs, or CD4+ T-cell apoptosis as
assessed by activated caspase-3, the terminal caspase at which all
the apoptotic pathways converge (33). However, the presence of oleate in
the culture medium in the present study yielded different results.
Oleate, as a fatty acid, along with the IDO-induced increase in
fatty acid oxidation may protect CD4+ T-cells from
energy deprivation, since IDO is known to decrease glucose influx
in the cell, aerobic glycolysis and glutaminolysis (13–16). The presence of a fatty acid in the
culture medium may protect cells from energy deprivation, thus also
preventing the inhibition of cell proliferation and the induction
of apoptosis. These results also raise the question of whether it
is more appropriate to perform immunological experiments using more
'normal' culture medium, which contains fatty acids.
The effect of IDO-induced L-tryptophan degradation
on the expression of the Treg signature transcription factor FoxP3,
and of the Th17 signature transcription factor, RORγt, was
evaluated (34). The two
CD4+ T-cell lineages are formed reciprocally as regards
fatty acid metabolism. Fatty acid synthesis favors differentiation
into the Th17 lineage, whereas fatty acid oxidation favors
differentiation into Tregs (25,26). According to what is generally
considered (35–37), IDO increased FoxP3, but decreased
RORγt expression in MLR-derived CD4+ T-cells. In order
to define the effect of the presence of fatty acid in the culture
medium, we repeated the experiments without oleate. No difference
was detected s regards RORγt; however, the presence of oleate IDO
induced a greater increase in FoxP3 expression. The reason remains
to be defined, since various aspects regarding the mechanisms that
connect fatty acid metabolism with CD4+ T-cell function,
such as post-translational protein modification by lipids or the
availability of acetyl-CoA for epigenetic modifications, are under
investigation (38). Thus, this
raises the question of whether a culture medium containing fatty
acids more closely mimics the in vivo conditions and may
thus be more suitable for lymphocyte culture studies.
In conclusion, the present study demonstrated that
IDO, by degrading L-tryptophan, enhanced CPT1 activity and fatty
acid oxidation, and exerted fatty acid-dependent effects in human
alloreactive CD4+ T cells.
References
|
1
|
Seo SK, Choi JH, Kim YH, Kang WJ, Park HY,
Suh JH, Choi BK, Vinay DS and Kwon BS: 4-1BB-mediated immunotherapy
of rheumatoid arthritis. Nat Med. 10:1088–1094. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Gurtner GJ, Newberry RD, Schloemann SR,
McDonald KG and Stenson WF: Inhibition of indoleamine
2,3-dioxygenase augments trinitrobenzene sulfonic acid colitis in
mice. Gastroenterology. 125:1762–1773. 2003. View Article : Google Scholar
|
|
3
|
Kwidzinski E, Bunse J, Aktas O, Richter D,
Mutlu L, Zipp F, Nitsch R and Bechmann I: Indolamine
2,3-dioxygenase is expressed in the CNS and down-regulates
autoimmune inflammation. FASEB J. 19:1347–1349. 2005.PubMed/NCBI
|
|
4
|
Alexander AM, Crawford M, Bertera S,
Rudert WA, Takikawa O, Robbins PD and Trucco M: Indoleamine
2,3-dioxygenase expression in transplanted NOD Islets prolongs
graft survival after adoptive transfer of diabetogenic splenocytes.
Diabetes. 51:356–365. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Beutelspacher SC, Pillai R, Watson MP, Tan
PH, Tsang J, McClure MO, George AJ and Larkin DF: Function of
indoleamine 2,3-dioxygenase in corneal allograft rejection and
prolongation of allograft survival by over-expression. Eur J
Immunol. 36:690–700. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li Y, Tredget EE, Ghaffari A, Lin X,
Kilani RT and Ghahary A: Local expression of indoleamine
2,3-dioxygenase protects engraftment of xenogeneic skin substitute.
J Invest Dermatol. 126:128–136. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Munn DH, Zhou M, Attwood JT, Bondarev I,
Conway SJ, Marshall B, Brown C and Mellor AL: Prevention of
allogeneic fetal rejection by tryptophan catabolism. Science.
281:1191–1193. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mellor AL, Sivakumar J, Chandler P, Smith
K, Molina H, Mao D and Munn DH: Prevention of T cell-driven
complement activation and inflammation by tryptophan catabolism
during pregnancy. Nat Immunol. 2:64–68. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Munn DH and Mellor AL: Indoleamine
2,3-dioxygenase and tumor-induced tolerance. J Clin Invest.
117:1147–1154. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Eleftheriadis T, Yiannaki E, Antoniadi G,
Liakopoulos V, Pissas G, Galaktidou G and Stefanidis I: Plasma
indoleamine 2,3-dioxygenase and arginase type I may contribute to
decreased blood T-cell count in hemodialysis patients. Ren Fail.
34:1118–1122. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eleftheriadis T, Liakopoulos V, Antoniadi
G, Stefanidis I and Galaktidou G: Indoleamine 2,3-dioxygenase is
increased in hemodialysis patients and affects immune response to
hepatitis B vaccination. Vaccine. 29:2242–2247. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Munn DH, Sharma MD, Baban B, Harding HP,
Zhang Y, Ron D and Mellor AL: GCN2 kinase in T cells mediates
proliferative arrest and anergy induction in response to
indoleamine 2,3-dioxygenase. Immunity. 22:633–642. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Eleftheriadis T, Pissas G, Antoniadi G,
Liakopoulos V and Stefanidis I: Indoleamine 2,3-dioxygenase
depletes tryptophan, activates general control non-derepressible 2
kinase and down-regulates key enzymes involved in fatty acid
synthesis in primary human CD4+ T cells. Immunology.
146:292–300. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Eleftheriadis T, Pissas G, Antoniadi G,
Spanoulis A, Liakopoulos V and Stefanidis I: Indoleamine
2,3-dioxygenase increases p53 levels in alloreactive human T cells,
and both indoleamine 2,3-dioxygenase and p53 suppress glucose
uptake, glycolysis and proliferation. Int Immunol. 26:673–684.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Eleftheriadis T, Pissas G, Yiannaki E,
Markala D, Arampatzis S, Antoniadi G, Liakopoulos V and Stefanidis
I: Inhibition of indoleamine 2,3-dioxygenase in mixed lymphocyte
reaction affects glucose influx and enzymes involved in aerobic
glycolysis and glutaminolysis in alloreactive T-cells. Hum Immunol.
74:1501–1509. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Eleftheriadis T, Pissas G, Antoniadi G,
Tsogka K, Sounidaki M, Liakopoulos V and Stefanidis I: Indoleamine
2,3 dioxygenase downregulates T cell receptor complex ζ chain and c
Myc, and reduces proliferation, lactate dehydrogenase levels and
mitochondrial glutaminase in human T cells. Mol Med Rep.
13:925–932. 2016.
|
|
17
|
Cobbold SP, Adams E, Farquhar CA, Nolan
KF, Howie D, Lui KO, Fairchild PJ, Mellor AL, Ron D and Waldmann H:
Infectious tolerance via the consumption of essential amino acids
and mTOR signaling. Proc Natl Acad Sci USA. 106:12055–12060. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mezrich JD, Fechner JH, Zhang X, Johnson
BP, Burlingham WJ and Bradfield CA: An interaction between
kynurenine and the aryl hydrocarbon receptor can generate
regulatory T cells. J Immunol. 185:3190–3198. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Opitz CA, Litzenburger UM, Sahm F, Ott M,
Tritschler I, Trump S, Schumacher T, Jestaedt L, Schrenk D, Weller
M, et al: An endogenous tumour-promoting ligand of the human aryl
hydrocarbon receptor. Nature. 478:197–203. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
King NJ and Thomas SR: Molecules in focus:
Indoleamine 2,3-dioxygenase. Int J Biochem Cell Biol. 39:2167–2172.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Curti A, Trabanelli S, Salvestrini V,
Baccarani M and Lemoli RM: The role of indoleamine 2,3-dioxygenase
in the induction of immune tolerance: Focus on hematology. Blood.
113:2394–2401. 2009. View Article : Google Scholar
|
|
22
|
Maciver NJ, Jacobs SR, Wieman HL, Wofford
JA, Coloff JL and Rathmell JC: Glucose metabolism in lymphocytes is
a regulated process with significant effects on immune cell
function and survival. J Leukoc Biol. 84:949–957. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Fox CJ, Hammerman PS and Thompson CB: Fuel
feeds function: Energy metabolism and the T-cell response. Nat Rev
Immunol. 5:844–852. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang R, Dillon CP, Shi LZ, Milasta S,
Carter R, Finkelstein D, McCormick LL, Fitzgerald P, Chi H, Munger
J, et al: The transcription factor Myc controls metabolic
reprogramming upon T lymphocyte activation. Immunity. 35:871–882.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Berod L, Friedrich C, Nandan A, Freitag J,
Hagemann S, Harmrolfs K, Sandouk A, Hesse C, Castro CN, Bähre H, et
al: De novo fatty acid synthesis controls the fate between
regulatory T and T helper 17 cells. Nat Med. 20:1327–1333. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Michalek RD, Gerriets VA, Jacobs SR,
Macintyre AN, MacIver NJ, Mason EF, Sullivan SA, Nichols AG and
Rathmell JC: Cutting edge: Distinct glycolytic and lipid oxidative
metabolic programs are essential for effector and regulatory
CD4+ T cell subsets. J Immunol. 186:3299–3303. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Sato T, Deiwick A, Raddatz G, Koyama K and
Schlitt HJ: Interactions of allogeneic human mononuclear cells in
the two-way mixed leucocyte culture (MLC): Influence of cell
numbers, subpopulations and cyclosporin. Clin Exp Immunol.
115:301–308. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Lopaschuk GD, Ussher JR, Folmes CD, Jaswal
JS and Stanley WC: Myocardial fatty acid metabolism in health and
disease. Physiol Rev. 90:207–258. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Schreurs M, Kuipers F and van der Leij FR:
Regulatory enzymes of mitochondrial beta-oxidation as targets for
treatment of the metabolic syndrome. Obes Rev. 11:380–388. 2010.
View Article : Google Scholar
|
|
30
|
Bieber LL and Fiol C: Purification and
assay of carnitine acyltransferases. Methods Enzymol. 123:276–284.
1986. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gallinetti J, Harputlugil E and Mitchell
JR: Amino acid sensing in dietary-restriction-mediated longevity:
Roles of signal-transducing kinases GCN2 and TOR. Biochem J.
449:1–10. 2013. View Article : Google Scholar :
|
|
32
|
Mihaylova MM and Shaw RJ: The AMPK
signalling pathway coordinates cell growth, autophagy and
metabolism. Nat Cell Biol. 13:1016–1023. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Fadeel B and Orrenius S: Apoptosis: A
basic biological phenomenon with wide-ranging implications in human
disease. J Intern Med. 258:479–517. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Raphael I, Nalawade S, Eagar TN and
Forsthuber TG: T cell subsets and their signature cytokines in
autoimmune and inflammatory diseases. Cytokine. 74:5–17. 2015.
View Article : Google Scholar :
|
|
35
|
Fallarino F, Grohmann U, You S, McGrath
BC, Cavener DR, Vacca C, Orabona C, Bianchi R, Belladonna ML, Volpi
C, et al: The combined effects of tryptophan starvation and
tryptophan catabolites down-regulate T cell receptor zeta-chain and
induce a regulatory phenotype in naive T cells. J Immunol.
176:6752–6761. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sharma MD, Baban B, Chandler P, Hou DY,
Singh N, Yagita H, Azuma M, Blazar BR, Mellor AL and Munn DH:
Plasmacytoid dendritic cells from mouse tumor-draining lymph nodes
directly activate mature Tregs via indoleamine 2,3-dioxygenase. J
Clin Invest. 117:2570–2582. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Sharma MD, Hou DY, Liu Y, Koni PA, Metz R,
Chandler P, Mellor AL, He Y and Munn DH: Indoleamine
2,3-dioxygenase controls conversion of Foxp3+ Tregs to
TH17-like cells in tumor-draining lymph nodes. Blood.
113:6102–6111. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lochner M, Berod L and Sparwasser T: Fatty
acid metabolism in the regulation of T cell function. Trends
Immunol. 36:81–91. 2015. View Article : Google Scholar : PubMed/NCBI
|