1. Introduction
Malformations of cortical development (MCDs) play a
major role in the aetiology of epilepsy. One of the MCD subtypes,
focal cortical dysplasia (FCD), is particularly important as a
frequent cause of epilepsy resistant to drugs, as approximately one
half (46.5%) of patients with epilepsy has some form of this
pathology (1). The disorder is
the result of neuronal migration, proliferation and differentiation
disruption during brain development, leading to regional cortical
lamination, neuronal maturation and differentiation abnormalities.
The first description of FCD in resected specimens from 10 patients
treated surgically for refractory epilepsy was made by Taylor et
al in 1971 (2). Taylor
described FCD as a malformative disorganisation and dyslamination
of the neocortex with the presence of giant, dysmorphic neurones
and bizarre 'balloon cells', similar to giant cells in tuberous
sclerosis (TS). Since then, the classification of FCD has changed,
distinguishing architectural dysplasias with or without the
presence of dysmorphic neurons or balloon cells (Taylor cells).
None of these, however, have met the requirements of clinical
practice. Additionally, some neurodevelopmental studies have shed
light on the possibility of postnatal neurogenesis failure due to
various pathogenic states of the brain (dysmature cerebral
developmental hypothesis) as the background of epileptogenesis
(3). Against this background, an
ad hoc Task Force of the International League Against Epilepsy
(ILAE), the Diagnostic Methods Commission, have proposed a new
clinicopathological classification system to help clinical practice
(4). The ILAE consensus
classification included both isolated and associated FCD variants
(Table I) and its recent
evaluation showed good inter- and intra-observer agreement
(5).
 | Table ICharacteristics of FCD types in the
ILAE classification system (25). |
Table I
Characteristics of FCD types in the
ILAE classification system (25).
| ILAE consensus
classification system for FCDs |
| FCD I:
Corticaldyslamination | FCD IA: FCD with
abnormal radial cortical lamination
• Abundant microcolumnar organization
FCD IB: FCD with abnormal tangential cortical lamination
• Failure to establish a six-layered tangential composition of the
isocortex
• Less clear demarcation of white/grey matter transition
• Cellular abnormalities (immature neurons, hypertrophic pyramidal
neurons outside layer 5, disoriented dendrites)
FCD IC: FCD with abnormal radial and tangential cortical
lamination |
| FCD II: Cortical
dyslamination with dysmorphic neurons | FCD IIA: FCD with
dysmorphic neurons, without balloon cells
• Dysmorphic neurons
• Cortical dyslamination (only layer 1 is identifiable)
• Usually blurred grey/white matter transition
FCD IIB: FCD with dysmorphic neurons and balloon cells
• Balloon cells
• Dysmorphic neurons
• Intermediate cells (cells of glial and neuronal
features)
• Cortical dyslamination
• Blurred grey/white matter transition
• Usually reduced myelin content in the underlying white
matter |
| FCD III: Cortical
laminationabnormalitiesrelated to additionalpathology (in
theadjoining area) | FCD IIIA: Cortical
lamination abnormalities in the temporal lobe associated with
hippocampal sclerosis
• Patient with hippocampal sclerosis (HS; Ammon's horn sclerosis)
plus:
• Alterations of cytoarchitectural composition or temporal lobe
sclerosis
FCD IIIB: Cortical lamination abnormalities (dyslamination,
hypertrophic neurons, cortical hypoplasia) adjacent to a glial or
glioneuronal tumor
FCD IIIC: Cortical lamination abnormalities adjacent to vascular
malformation
FCD IIID: Cortical lamination abnormalities adjacent to any other
lesion acquired during early life (e.g., traumatic brain injury,
glial scarring after ischaemia or haemorrhage, inflammatory or
infectious diseases, which were not included in types
IIIA-C)
FCD III NOS (not otherwise specified): Focal cortical dysplasia
associated with clinically suspected principal lesion, but lesion
not available for histopathological examination |
2. Aetiology and pathophysiology
The aetiology and pathogenesis of FCD is still
uncertain; however, many histopathological and molecular findings
point to abnormal neuronal and glial proliferation and migration
processes (see below). Several hypotheses for FCD II have been
proposed. The extrinsic damage hypothesis postulates that the
damage to the developing brain is made by extrinsic factors, such
as ischemia, hypoxia or toxins. Alternative somatic mutation
hypothesis points to a mutant cortical progenitor cell to produce a
clonal population of abnormal cells, forming FCD lesions (6). The latter hypothesis seems
convincing, as FCD lesions have a characteristic funnel-shaped
morphological distribution, typical of clonally-related neurons, as
well as the co-expression of glial and neuronal antigens in
different combinations, in balloon cells and dysmorphic neurons in
FCD specimens (7). Some studies
have also suggested that FCD lesions originate from the abnormal
retention of pre-plate cells in the subplate and marginal zones, as
the consequence of the partial failure of events during late
corticogenesis (3,8).
The recent study by Rossini et al (9) on the layer-specific gene expression
in FCD II, revealed the presence of hidden cortical laminations
among normal-looking neurons, but not dysmorphic neurons or balloon
cells, which may suggest that only selected precursor cells could
be subjected to damage during cortical development.
Recently, a number of proteins involved in the
regulation of cortical development has been observed to be
deregulated and the cellular distribution alterations of these
proteins have been found in FCD specimens. Among these proteins
are: bone morphogenic protein-4, whose expression has been found to
be reduced and altered in FCD IIB and cortical tubers from TS
(10); the increased expression
of doublecortin-like protein, found within the dysplastic cortex of
FCD IIB and cortical tubers from TS (11); a study on the expression and
distribution of interleukin-2 (IL-2), IL-2 receptors (IL-2R) and
the downstream factors of the IL-2 signalling pathway revealed
higher IL-2 and IL-2R levels in the FCD samples vs. the control and
in FCD II vs. FCD I, as well as increased levels of Janus kinases 1
and 3 in FCD (12).
The new theory of FCD pathogenesis, based on HPV16
infection and HPV16 oncoprotein E6 expression in the developing
foetal brain, has been postulated by Chen et al (13). They demonstrated for the first
time the presence of HPV16 E6 oncoprotein in FCD IIB specimens and
in the human brain in general. The 100% association of HPV16
infection with FCD IIB in 50 samples resected from patients with
intractable epilepsy was demonstrated; however, the authors did not
find the presence of HPV16 in other types of FCD, as such an
association was found only in balloon cells. In addition, the
functional correlation between HPV16 E6 oncoprotein expression and
focal cortical malformation development associated with enhanced
mammalian target of rapamycin (mTOR) complex 1 (mTORC1) signalling
in an animal model was demonstrated. A similar study by Liu et
al, detecting HPV16 and three other viral species:
cytomegalovirus, herpes simplex virus, and human herpes virus in
FCD IIA, seems to support these data (14). Furthermore, it is already known
that all the above-mentioned viruses are able to activate the mTOR
pathway (15), which is also
implicated in FCD pathogenesis. In spite of these findings, whether
we can classify FCD as a viral pathology, remains uncertain,
particularly, as some data contradicting this hypothesis have been
published (16). It is generally
known that extensive cortical malformations can be caused by
prenatal infections (e.g., TORCH spectrum syndromes); however,
whether more localised malformation, such as FCD may result from
intrauterine infections, warrants further investigation.
3. Molecular and genetic studies
The most convincing data point to mTOR cascade
abnormalities, as the cause of FCD. Some authors have suggested TS
complex 1 (TSC1) gene involvement in FCD pathogenesis. Amino
acid polymorphisms of hamartin (TSC1 gene product),
involving exons 5 and 17, and silent base substitution in exons 14
and 22, are more frequent in FCD in comparison to controls. In
addition, the loss of TSC1 heterozygosity has been reported.
Hamartin, forming a complex with tuberin encoded by the TS complex
2 (TSC2) gene, is a negative regulator of mTOR.
TSC1/TSC2 mutations, or sequence alterations, lead to the
loss of hamartin/tuberin protein complex activity which, in
consequence, enables mTOR cascade activation, cell growth and
proliferation, in response to diverse external or intracellular
signals (17,18) (Fig.
1). The activation of upstream signalling pathways, e.g.,
protein kinase B or AKT (PKB) and exracellular signal-regulated
protein kinase (ERK) pathways, results in mTOR upregulation; there
are some reports on AKT pathway activation in balloon cells and,
recently, one study on ERK hyperactivation in FCD IIB has been
published (19–21). It has also been recently suggested
that FCD may be caused by de novo somatic mutations of mTOR
occurring during brain development (22).
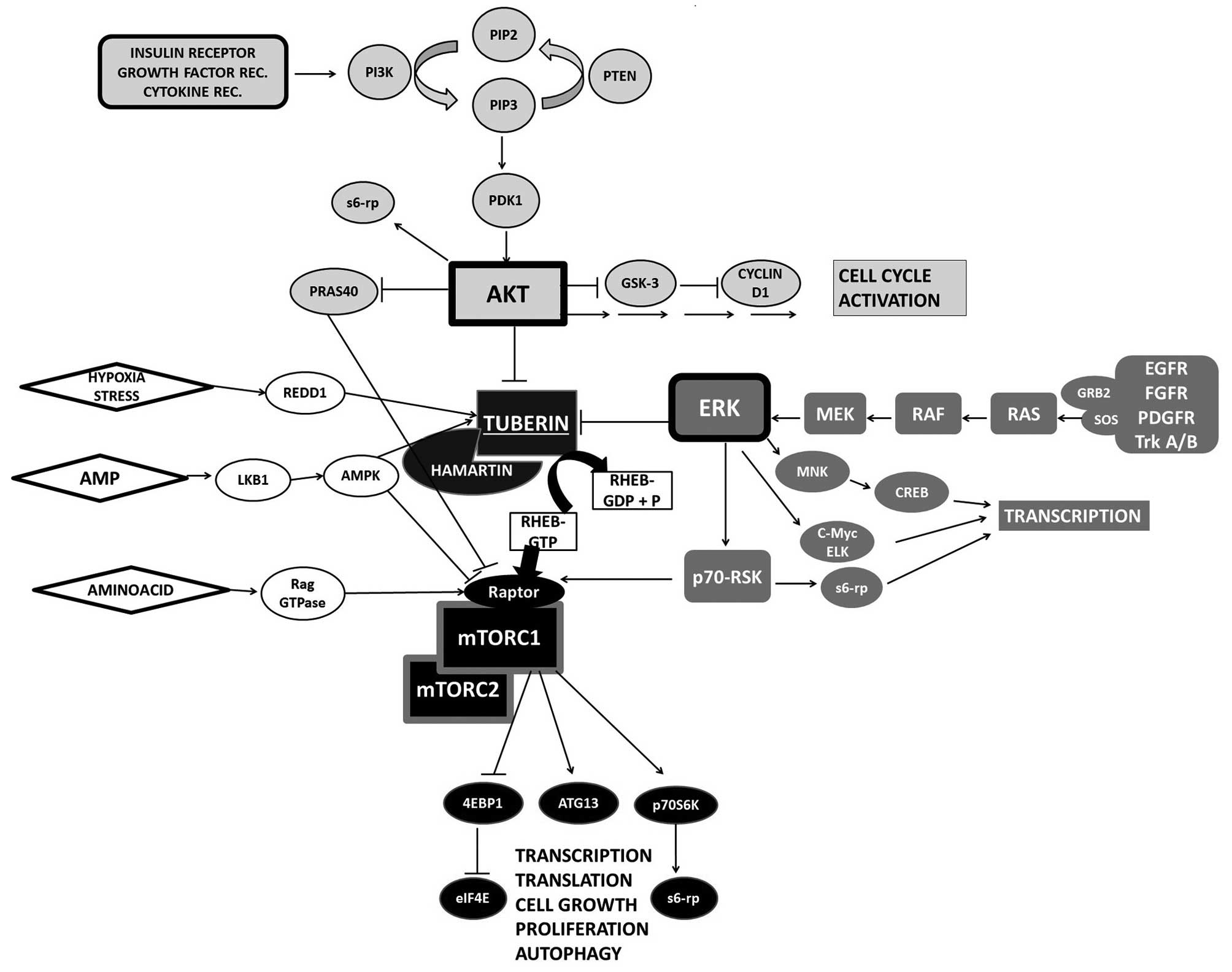 | Figure 1Upstream and downstream mammalian
target of rapamycin (mTOR) regulation. Growth factor control plays
a crucial role in mTOR regulation. Two main kinase cascades, AKT
and exracellular signal-regulated protein kinase (ERK), mediate
signal leading to phosphorylation and inhibition of tuberous
sclerosis complex 2 (TSC2) which, subsequently, upregulates the
mTOR activator, Rheb. mTORC1 activates S6K and inhibits 4EBP1,
accelerating translation. PI3K, phosphatidylinositol 3-kinases;
PIP3, phosphatidylinositol (3,4,5)-triphosphate; PDK1, 3-phosphoinositide
dependent protein kinase 1; GSK-3, glycogen synthase kinase 3; Mek,
mitogen-activated protein kinase kinase; EGFR, epidermal growth
factor receptor; FGFR, fibroblast growth factor receptor; PDGFR,
platelet-derived growth factor receptor; RSK, ribosomal s6 kinase;
s6-rp, S6 ribosomal protein; Rheb, Ras homolog enriched in brain;
Raptor, regulatory-associated protein of mTOR; AMPK, 5′AMP,
activated protein kinase; 4EBP1, eukaryotic translation initiation
factor 4E-binding protein 1; eIF4E, eukaryotic translation
initiation factor 4E; p70S6K, ribosomal protein S6 kinase β-1;
ATG13, autophagy-related protein 13. |
It is unlikely that FCD could be connected with only
one gene mutation. The alterations of Notch and Wnt pathway
proteins, which are involved in neurogenesis, neuroglial cell fate
determination, neuronal migration and neural tube development, have
been shown to be altered as well. The study by Cotter et al
found elevated levels of disheveled and adenomatous polyposis coli
(APC) proteins, the absence of Notch-1 protein and alterations in
β-catenin levels, with decreased levels of nuclear β-catenin in
balloon cells (23) (Fig. 2).
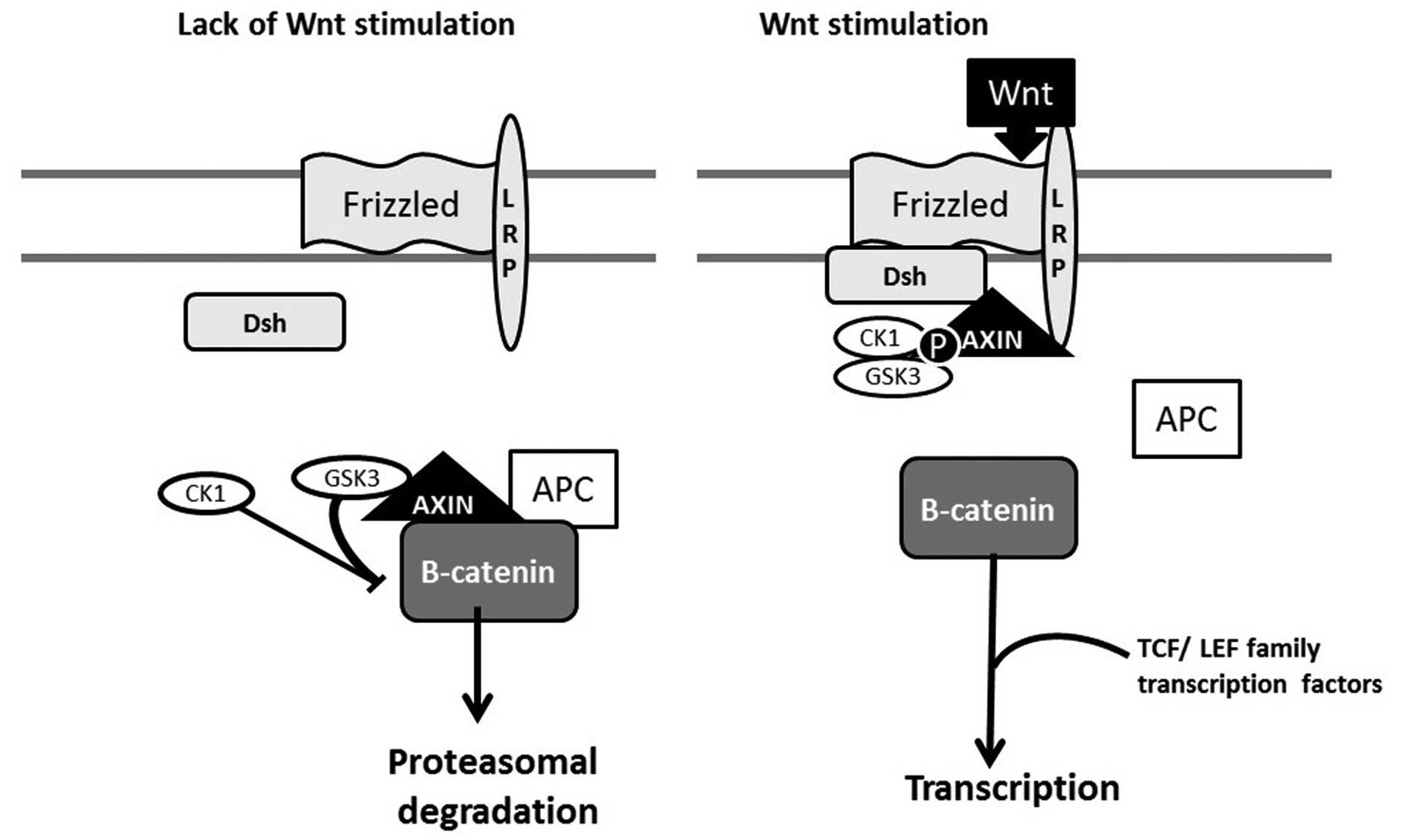 | Figure 2Wnt/β-catenin sygnaling pathway:
'off' and 'on' states. Wnt presence activates the receptor complex,
leading to 'destruction complex' (APC, axin and GSK3)
disassembling, axin binding to the receptor complex and β-catenin
translocation to the nucleus, where it performs a variety of
functions. LRP, lipoprotein receptor-related protein; Dsh,
dishevelled; GSK3, glycogen synthase kinase 3; APC, adenomatous
polyposis coli protein; CK1, casein kinase 1; P, phosphate. |
As only polymorphysms or allelic TSC variants
have been shown in FCD, and no disease-causing mutations of
TSC genes, as well as no signalling pathway alterations
specific for FCD, it is currently unjustifiable to use
histopathological staining in FCD diagnoses. Such studies, however,
could extend our knowledge on genetic disturbances in FCD, bringing
new diagnostic options in the future.
4. Cytopathology and epileptogenesis
Balloon cells in FCD IIB have glial features and
seem to arise from radial glial stem cells (3). For this reason, there is a
supposition of the aberrant differentiation of these cells or
possible pluripotency, with the abnormal destination of the cell
(10). In addition, cell markers
indicating immaturity and developmental abnormality, such as glial
fibrillary acidic protein (GFAP), nestin, vimentin, CD133, CD34 are
all expressed in balloon cells (7,24).
Moreover, dysmorphic neurons exhibit a strong expression of
phosphorylated and non-phosphorylated neurofilament proteins,
maturity markers, including NeuN, as well as cell markers pointing
to cell immaturity or abnormalities in development or survival
(7,8,25).
The above-mentioned data support the idea that the neuronal as
opposed to the glial differentiation of these cells is
disrupted.
The specific morphology of balloon cells may be a
result of uptriggered cellular pathways. mTOR, the central
regulator of the cellular state, is a kinase responsible for the
production of even 5–10% of all proteins in the cell. mTOR
upregulation has been consistently found in balloon cells, which
may contribute to their volume. A similar phenomenon has also been
described in seemingly identical giant cells found in TS, where
mTOR upregulation results usually from ERK hyperactivity (26).
Balloon cells are unable to generate epileptic
discharges. They lack dendritic spines and axons, and have a very
high input resistance. The newest findings suggest even their
protective anti-seizure role by glutamate neutralisation, as
glutamine synthetase immunopositivity in these cells, but not in
dysmorphic neurons, was shown (27). On the other hand, cytomegalic
neurons exhibit very low input resistance and atypical
hyperexcitable properties of the intrinsic membrane, which allows
the cells to generate repeated spikes after reaching firing
threshold (28,29).
Several mechanisms leading to neuronal
hyperexcitability and epilepsy in FCD have been postulated. There
is evidence of both decreased inhibition and increased stimulation
states, including the overexpression of NMDA and AMPA receptor
subunits in FCD neurons, the loss of GABAergic neurons,
differential composition of GABA receptor subunits, altered GABAA
responses in paediatric cortical dysplasia (similar to immature
cortex), as well as the dysfunction of the synaptic inhibition of
pyramidal neurons due to disordered migration and the
maldistribution of interneurons (3,30,31). In addition, the deregulation of
cation-chloride cotransporter expression:
Na+-K+-2Cl− cotransporter (NKCC1)
and K+-Cl− cotransporter (KCC2), has been
observed in FCD tissue (32),
while other studies point to post-translational voltage potassium
channel (Kv4.2) modifications (33). The authors suggested that the
abnormal expression of these cotransporters in neuronal and glial
cells may contribute to increased network excitability, as their
distribution is comparable to the immature cortex. It is, however,
not certain as to whether this finding actively contributes to
epileptogenesis in FCD and other malformations of cortical
development. Finally, the activation of various inflammatory
pathways connected with cells of microglial/macrophage lineage has
been observed in FCD as well (34).
5. Clinical presentation
In recent years, as the entity recognition
increased, rare, as it was thought, FCD became one of the most
frequent pathologies in paediatric patients undergoing surgical
treatment (35). It is the most
prevalent lesion found in paediatric patients undergoing epilepsy
surgery and is one of the three most common lesions in adult
patients undergoing epilepsy surgery (35,36). Its prevalence in patients with
epilepsy patients has not been ascertained. In a summary prepared
by Spreafico and Blümcke, based on the European Epilepsy Brain Bank
and German Reference Center for Epilepsy surgery, among 748
patients with intractable epilepsy or histopathologically confirmed
MCDs, FCDs constitute >75%, with FCD IIB being the most frequent
(>39%) (37).
FCDs may affect any part of the brain cortex,
varying in size and location. Clinically, the most frequent symptom
of FCD is drug-refractory epilepsy, beginning in most cases, in
early childhood. Adult-onset epilepsy in most studies is limited to
several cases (38,39). Seizures are generally of high
frequency, up to dozens a day (40) and can be localised or generalise
secondarily. A recent study comparing the characteristics of
patients with FCD and epilepsy onset in the first year showed that
the presence epileptic spasms is connected with the frontal lobe
localisation of the lesion (41).
The same study also found that patients with focal seizures and
epileptic spasms have an earlier epilepsy onset compared to those
with only partial seizures. The early onset of seizures is also
presumed to be associated with the posterior localisation of the
lesion, compared to other sitings (42). A recent FCD II subgroup analysis
pointed out that FCD IIB patients have an earlier epilepsy onset in
comparison with FCD IIA patients. Subtype IIB was observed
predominantly in the frontal lobe and subtype IIA was more often
observed in the temporal lobe (40). In cases where cortical lamination
abnormalities co-exist with the principal lesion, such as glial or
glioneuronal tumours (FCD IIIB), the frequency of seizures reaches
100%, particularly when the tumour is slow-growing (43). In addition, male predominance and
a higher seizure frequency in tumours associated with FCDs compared
to solitary tumours has been noted (44).
The clinical consequences of FCD are also congnitive
impairment and neurological deficits, as well as a decrease in
intelligence quotient (IQ) in the majority of patients (42). It is thought that the grade of
this deficit and the clinical manifestations of the disorder
correlate with lesion localisation, size as well as subtype. In the
study by Widdess-Walsh et al, cognitive impairment was
observed more often in FCD II (42). Moreover, the study by Krsek et
al showed that 96% of FCD I patients had mental retardation,
with 55% of patients retarded severely (IQ <35), while in FCD
II, the proportion was 67 and 27%, respectively (45). Patients with FCD I also exhibit
higher maladaptive scales, whilst behavioural disorders due to
brain dysfunction are not observed in FCD II patients at all.
Psychiatric features have been observed to be connected with the
early onset and the posterior localisation of the lesion. A minor
delay in development and normal development are associated with
patients with more circumscribed lesions. Early onset is more
frequently associated with mild to severe mental retardation and
more often leads to adverse outcomes in comparison with adult-onset
seizures (46).
6. Imaging
Magnetic resonance imaging studies allow the
separation of FCD IIB from other cortical dysplasias. The
indicative features are local cortical thickening, blurring of the
white-grey matter interface and the focally increased signal of the
subcortical white matter on T2-weighted imaging, often tapering
toward the underlying ventricle ('transmantle sign'-nearly unique
for FCD IIB) (47).
As the neuroimaging techniques became more advanced
and precise, the identification of the lesion is more prevalent and
classification is easier. Still, however, there are cases with
pathologically proven FCD without any abnormalities in neuroimaging
studies.
The recent study by Colombo et al, reviewing
retrospectively MR images of 118 FCD II patients, showed that the
MRI abnormalities were present in 79% of cases, whilst the correct
diagnosis of FCD II, made on the basis of abnormal MRI scans, was
made in 89% of patients. A strong correlation between the
transmantle sign and FCD IIB was emphasised (48). Recently, Mühlebner et al
pointed to some quantitative parameters, which may be useful for
the higher sensitivity of MRI evaluation in patients with
drug-resistant focal epilepsy (49). Other recent studies have
demonstrated the predominance of morphometric MRI analysis as more
sensitive for FCD II detection than conventional visual analysis
alone (50). Lately, the Arterial
Spin Labeling MRI technique, evaluating the cerebral perfusion
abnormalities during interictal periods, turned out to be helpful
in the evaluation of epileptic zones in FCD patients (51). Furthermore, novel techniques of
ShearWave elastography and magnetoencephalography have been
demonstrated to detect MRI-negative FCD lesions, facilitating
presurgical evaluation (52).
Finally, in uncertain situations, functional neuroimaging
techniques, including ictal SPECT or FDG-PET, are used.
7. Treatment
FCD treatment depends on individual patient
presentation and remains mainly symptomatic, including cognitive
and neurological deficit therapy, as well as epilepsy management.
As epilepsy in FCD is commonly pharmacoresistant, and the basis of
the resistance seem to be multifactorial, anti-epileptic treatment
is particularly challenging, and should reflect different
therapeutic strategies.
One of these strategies involves well-known
mediators of drug resistance: multidrug resistance gene-1
P-glycoprotein (MDR1) and multidrug resistance-associated protein 1
(MRP1). These participate in the formation of blood-cerebrospinal
fluid (CSF) and blood-brain barriers, and in FCD lesions they have
been found to be overexpressed in many brain tissue components of
the epileptogenic zone (53).
They have been found to be most frequently expressed in glial
fibrillary acidic protein-positive balloon cells (glial type) and
microtubule-associated protein 2-positive balloon cells (neuronal
type) (54). In addition, major
vault protein, a protein associated with drug resistance, was found
to be upregulated in dysmorphic neurons in FCD (55). Therefore, in order to design the
treatment for drug-resistant seizures, specific knowledge on the
above-mentioned proteins seems to be of uttermost significance.
Secondly, mTOR pathway inhibitors may be a novel
targeted therapeutic option for epilepsy in FCD. Due to mTOR
signalling dysregulation, FCD is sometimes termed 'mTORopathy'. As
the mTOR pathway is involved in protein synthesis, its activity
abnormalities may promote neurotransmitter receptor or ion channel
alterations and, consequently, neuronal hiperexcitability, leading
to epileptogenesis (at least in animal models) (56,57). mTOR inhibitors, such as rapamycin
or everolimus, are used in the treatment of another 'mTORopathy',
TS. Their potential anti-epileptogenic and disease-modifying
effects have been described and their learning deficit-reversing
activity has been demonstrated in a mouse model (58,59). Finally, other recent studies,
pointing to the link between FCD and HPV16, may also, potentially,
identify some novel therapeutic strategies for the prevention of
FCD (13). Molecular and genetic
studies, however, are still required and should bring new concepts
of efficient therapies.
At the moment, the treatment of choice for
epileptogenic, drug-resistant lesions, is surgical resection. Its
long-term efficacy and safety in certain ILAE classification
subgroups is evaluated in order to determine the associations
between pathological subtypes and clinical relevance and prognosis.
Particularly, type III FCD and its post-surgical outcome needs to
be evaluated according to the new classification, as it was not
specified previously.
Surgical resection of the epileptic lesion may not
only liberate patients from seizures, but also has a positive
effect on cognition and intellectual outcomes, improving full-scale
IQ (60). Early surgical
intervention in children with complete resection of the
epileptogenic lesion seems to be of particular importance. If
accomplished before two years of seizure duration, it not only
leads to better seizure control, but it also improves cognitive
function development and quality of life (61).
Failure to achieve a seizure-free state is often due
to incomplete cortical resection. However, a recent study by Wagner
et al showed that subcortical hyperintense zone resection is
not essential for a positive post-operative outcome, which may
reduce the risk of surgery (62),
whilst according to the study by Mühlebner et al, complete
resection of the lesion is the only predictor of surgical outcome,
irrespective of the ILAE subtype (63). Other predictors of surgical
outcome are also pre-operative seizure frequency, pre-operative
ability of epileptogenic focus localisation in imaging techniques,
multilobar lesion and dual pathology presence (64).
8. Clinicopathological associations and
outcome
As a series of factors affects epilepsy surgery
outcomes, it is hard to compare particular studies to one another.
The probability of being free from seizures ranges in different
studies, and on average, seizure recurrence affects approximately
one half of patients treated surgically (65–68). Seizure outcome depends, however,
on the time after surgery, and Kapplan-Meier curves (in the
above-mentioned and other similar studies) analysis points at the
highest epilepsy recurrence frequency within the first 3 years
after surgery. A recent study by Ryzi et al (69) showed however, that approximately
30% of patients with seizure recurrence within one year after
surgery remained seizure-free in a long term observation (at least
5 years) and the next 18% has auras only. Such data are, though,
only general predictions for the patient, and recently, Jehi et
al elaborated some nomograms for individualised predictions of
post-surgical seizure outcome (66).
The seizure-free post-operative outcome rate in FCD
varies in different reports and the proportion ranged from
approximately 50 to 83% in a recent study by Tassi et al
(70). The meta-analysis by
Rowland et al (71)
presented freedom from seizures in 58% of patients out of a group
of over 2,000 patients with FCD-related epilepsy. The result is
not, however, stable in time. According to Mrelashvili et al
(65) about one third of patients
has early seizure recurrence (<3 months), with a median
recurrence time of 38 months.
As far as FCD subtypes associated with principal
lesions are concerned, their clinical presentation seems to have
similar characteristics to epilepsy associated with isolated
principal lesions, as well as at least similar (and often better)
outcomes, in comparison to isolated FCD I cases (44,72–76). According to the most recent study
by Fauser et al (77),
there are no statistical differences in the outcome in FCD I, II
and IIIA patients. The rate of (Engel's) class I post-operative
results (i.e., patient free of disabling seizures) in this largest
FCD patient cohort reported to date, was 65% after one year, and
remained stable over time. The authors also noted higher numbers of
febrile seizures and auras in FCD IIIA, compared to FCD I and II,
which is also supported by the literature (72,76).
In another recent study by Giulioni et al
(75), patients with hippocampal
sclerosis associated with FCD I (i.e., FCD IIIA) showed a similar
seizure outcome to patients with isolated hippocampal sclerosis (84
vs. 82%, respectively) and had better seizure outcome than patients
with isolated FCD I (63%). FCD IIIA was also the most common
pathology. Aforegoing and other similar findings [e.g., Thom et
al (80) or Tassi et
al (78,79)] are in contrast with the
controversial results obtained recently by Johnson et al
(81). In the study by Johnson
et al, FCD IIIA had the poorest seizure outcome, compared to
major pathologies: isolated hippocampal sclerosis (HS) and isolated
tumours, and similar to temporal FCD I. The presented data
contradict the ILAE classification. However, there was similar
electro-clinical presentation between FCD IIIA and HS, taking into
account e.g., the frequency of seizures, the age of epilepsy onset
or the presence of auras. In the letter to the editor, Giulioni
et al commented on this study critically (73).
Other associated FCD subtypes also seem to have a
good prognosis. Santos et al (75) pointed to a very good seizure
outcome of FCD IIIB (Engel I rate of 87%) following appropriate
surgical resection. In their study, a better outcome was connected
with the extended resection of the tumour and the whole area of
epileptogenicity. Another study by Cossu et al (44) compared the outcome between
associated and isolated forms of FCD, finding that the solitary FCD
I outcome is worse than FCD IIIB or FCD II associated with tumours
(47 vs. 83 vs. 76%, respectively). The clinical characteristics and
outcome in tumour-associated FCDs in that study was more similar to
the cases of solitary tumour than FCD I, which is consistent with
results obtained by the above-mentioned authors (74,78–80).
There is not much information on the outcome in FCD
IIIC and FCD IIID subtypes. One of the latest studies suggests that
FCD IIIC differs from the other FCD III subtypes (76). In that study, the mean age of
epilepsy in FCD III C was higher than in other FCD III types and
was similar to that of vascular malformations in the temporal lobe.
The duration of epilepsy in FCD III B and III C was shorter than
that in other associated types. The characteristics of epilepsy in
these subtypes were similar to the seizures associated with
isolated principal lesions.
Little is known about the clinicopathological
associations of FCD IIID. In one study (76) on a group of FCD IIID patients, six
had traumatic brain injury, three patients had glial scarring after
perinatal ischaemic injury, and two patients underwent infections.
The mean age of epilepsy onset in this group was 14 years and the
seizure frequency was not higher than in other subtypes. There is,
unfortunately, no information on seizure outcome in these
patients.
As far as FCD I and II is concerned, there is much
literature on prognosis in these subtypes, as it had been present
in previous classifications. The study by Simpson and Prayson,
taking into consideration the ILAE classification, including the
new FCD IC subtype, did not reveal any differences in Engel I rate
after surgery, which was 48%, in all three FCD I subtypes (82). The outcome in FCD II cases is
better, it fluctuates between 50–88% (40,71). There is a higher seizure outcome
in FCD IIB, compared to FCD IIA, and there is no correlation
between outcome and age at onset or the duration of epilepsy
(71).
To sum up these findings, FCD III outcome is rather
similar to that in isolated principal lesions connected with
particular subtypes and seems distinct from FCD I and II. In
particular, the difference between a relatively good seizure
outcome in FCD IIIA and a worse outcome in isolated FCD I suggests
that in FCD III, the principal lesion is a very important factor of
the outcome. A summary of the largest recent studies on the
post-surgical seizure outcome in epilepsy due to FCD and other
causes is presented in Table
II.
| Table IIPost-surgical seizure outcome in
epilepsy due to FCD and other causes, in recent studies (40,44,64,67,68,70,71,75,87–90). |
Table II
Post-surgical seizure outcome in
epilepsy due to FCD and other causes, in recent studies (40,44,64,67,68,70,71,75,87–90).
|
Authors/(Refs.) | Post-surgical
seizure outcome in FCD (regarding FCD subtype)
|
|---|
Surgical
outcome
Engel I class: % of PTS | Follow-up data |
|---|
| Fauser et al
(77) | Total: 65
FCD I: 61
FCD II: 67
FCD IIIA: 65 | 5-year follow-up;
outcome stable over time |
| Simpson and Prayson
(82) | FCD I: 48 | Median follow-up:
63 months (32–93 months);
Total follow-up of all the patients: 58 months |
| Yao et al
(40) | FCD IIA:
50
FCD IIB: 75 | Mean follow-up: 2.5
years |
| Santos et al
(75) | FCD IIIB: 87 | 5-year
follow-up |
| Cossu et al
(44) | FCD I: 47
FCD II: 84
FCD II + tumour 76
FCD IIIB: 83 | No data on mean
follow-up available |
| Giulioni et
al (74) | FCD I: 63
HS: 82
FCD IIIA: 84 | Mean follow-up: 6
years |
| Tassi et al
(70) | Total: 83
FCDIIA: 74
FCD IIB: 88 | Mean follow-up: 81
months; very rare seizure recurrences |
| Tassi et al
(78) | Isolated FCD I:
46
FCD I + HS: 82
FCD I + tumour: 82 | At least 2-year
follow-up |
|
|
Authors/(Refs.) | General
post-surgical seizure outcome in epilepsy
|
| Surgical outcome:
% | Follow-up and other
information |
|
| Jehi et al
(66) | Baseline risk of
seizure freedom: 0.52 at 2 years, 0.4 at 5 years;
Baseline risk of Engel I score: 0.69 at 2 years, 0.62 at 5
years. | 27% of studied
patients had MCD-related epilepsy;
Median follow up: 3.3 years |
| Mrelashvili et
al (65) | Engel I:
53
Engel I/II: 21
Engel III/IV: 26
Early recurrence: 32
Median recurrence: 38 months | FCD-related
epilepsy;
Median time to last follow up: 13.5 months; |
| Simasathien et
al (68) | Engel I:
57
Overall seizure-free probability:
After 1 year: 66
After 2 years: 52
After 5 years: 33 | 59% of studied
patients had MCD-related frontal lobe epilepsy;
mean follow up: 4.3 years; |
| Bulacio et
al (67) | Ovarall
seizure-free probability:
After 1 year: 61
After 5 years: 42
After 10 years: 33 | Various causes of
epilepsy were taken into consideration
Comparable results in the group with cortical dysplasia only |
9. FCD IIB: local form of tuberous
sclerosis?
Taylor et al observed a histological
similarity between FCD and TS, and postulated it to be an attenuate
or atypical form of TS (2).
Nowadays, after over 40 years, this issue is still under
investigation, and it is sometimes assumed that FCD IIB may be a
local form of TS (83,84).
Histomorphologically, balloon cells found in FCD and
giant cells in TS are similar (85). Common TS brain lesions,
subependymal giant cell tumour (SGCT), subependymal nodules and
cortical tubers, as well as FCD IIB lesions express both neuronal
and glial characteristics (7,86).
This suggests that the above lesions originate from progenitor,
undifferentiated cells, which either remain undifferentiated or
tend to pursue one of three possible directions: differentiation as
neuronal cells, glial (astrocytic) cells or cells of mixed
glio-neuronal features (24,86). Immunohistochemical studies on
balloon and giant cells have shown the expression of the same
markers (e.g., CD34, nestin, wimentin, GFAP and doublecortin)
(86,87). In addition, a strong MDR-1 and
MRP-1 immunoreactivity was shown to be present in giant and other
tuber cells of TS epileptogenic lesions, which (similarly to FCD),
can possibly explain the drug resistance of epilepsy (88).
Apart from immunohistochemical markers, there are
other molecular and morphological similarities between brain
tumours in TS and FCD IIB. It is already known that mutations or
genetic alterations of TSC1 and TSC2 suppressor genes
are involved in the pathogenesis of both TS and FCDIIB (17,89). Both TS and FCD are mTORopathies,
as the development of balloon and giant cells probably results from
mTOR activation. Recent studies point also to apoptosis signalling
pathways and the activation of neurodegeneration in both FCD II and
TS (90), as well as at the
defect in autophagy, both in balloon cells in FCD and TS cells,
connected with abnormal mTOR activation and reversible by mTOR
inhibition (91,92). Indeed, mTOR is responsible for the
control over protein translation, and mTOR activation leads to
hypertrophy of the cell (93).
Thus, morphological similarity between balloon and giant cells may
be a sign of mTOR activation.
The recent study by Kotulska et al also
revealed the clinical association between TSC presentation and FCD
presence. Patients with FCD have more severe epilepsy with higher
drug-resistance as well as more severe mental retardation (94).
The above similarities suggest that both TS and FCD
IIB may be pathogenically identical malformations, with FCD IIB
being focal form of TS in CNS. However, in order to make a
consistent statement, further molecular and genetic studies are
required.
10. Conclusions
In the present review, we discuss different forms of
FCD, specifying the similarities and differences between them. In
light of recent findings, we also describe the molecular mechanisms
leading to the development of the lesion e.g., the most convincing
data point to mTOR cascade abnormalities, as the cause of FCD. As
far as clinicopathological associations and outcome are concerned,
we review current data, while focusing on the outcome data for FCD
III, which has not been summarised to date, at least to the best of
our knowledge. Finally, we describe why FCD IIB is sometimes known
as the local form of TS, and what are the similarities between the
two entities.
Acknowledgments
This study was supported by the grant no. 231/14
from the Childrens' Health Institute, Warsaw, Poland.
References
|
1
|
Bingaman WE: Surgery for focal cortical
dysplasia. Neurology. 62(Suppl 3): S30–S34. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Taylor DC, Falconer MA, Bruton CJ and
Corsellis JA: Focal dysplasia of the cerebral cortex in epilepsy. J
Neurol Neurosurg Psychiatry. 34:369–387. 1971. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Cepeda C, André VM, Levine MS, Salamon N,
Miyata H, Vinters HV and Mathern GW: Epileptogenesis in pediatric
cortical dysplasia: The dysmature cerebral developmental
hypothesis. Epilepsy Behav. 9:219–235. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Blümcke I, Thom M, Aronica E, Armstrong
DD, Vinters HV, Palmini A, Jacques TS, Avanzini G, Barkovich AJ,
Battaglia G, et al: The clinicopathologic spectrum of focal
cortical dysplasias: A consensus classification proposed by an ad
hoc Task Force of the ILAE Diagnostic Methods Commission.
Epilepsia. 52:158–174. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Coras R, de Boer OJ, Armstrong D, Becker
A, Jacques TS, Miyata H, Thom M, Vinters HV, Spreafico R, Oz B, et
al: Good interobserver and intraobserver agreement in the
evaluation of the new ILAE classification of focal cortical
dysplasias. Epilepsia. 53:1341–1348. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Crino PB, Miyata H and Vinters HV:
Neurodevelopmental disorders as a cause of seizures:
Neuropathologic, genetic, and mechanistic considerations. Brain
Pathol. 12:212–233. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Englund C, Folkerth RD, Born D, Lacy JM
and Hevner RF: Aberrant neuronal-glial differentiation in
Taylor-type focal cortical dysplasia (type IIA/B). Acta
Neuropathol. 109:519–533. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Andres M, Andre VM, Nguyen S, Salamon N,
Cepeda C, Levine MS, Leite JP, Neder L, Vinters HV and Mathern GW:
Human cortical dysplasia and epilepsy: An ontogenetic hypothesis
based on volumetric MRI and NeuN neuronal density and size
measurements. Cereb Cortex. 15:194–210. 2005. View Article : Google Scholar
|
|
9
|
Rossini L, Medici V, Tassi L, Cardinale F,
Tringali G, Bramerio M, Villani F, Spreafico R and Garbelli R:
Layer-specific gene expression in epileptogenic type II focal
cortical dysplasia: Normal-looking neurons reveal the presence of a
hidden laminar organization. Acta Neuropathol Commun. 2:452014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo W, Zhang CQ, Shu HF, Yang MH, Yin Q
and Yang H: Expression of bone morphogenetic protein-4 in the
cortical lesions of focal cortical dysplasia IIb and the tuberous
sclerosis complex. J Mol Neurosci. 50:7–13. 2013. View Article : Google Scholar
|
|
11
|
Boer K, Lucassen PJ, Spliet WG,
Vreugdenhil E, van Rijen PC, Troost D, Jansen FE and Aronica E:
Doublecortin-like (DCL) expression in focal cortical dysplasia and
cortical tubers. Epilepsia. 50:2629–2637. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Guo W, Zheng DH, Sun FJ, Yang JY, Zang ZL,
Liu SY, Yin Q, Zhang CQ and Yang H: Expression and cellular
distribution of the interleukin 2 signaling system in cortical
lesions from patients with focal cortical dysplasia. J Neuropathol
Exp Neurol. 73:206–222. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen J, Tsai V, Parker WE, Aronica E,
Baybis M and Crino PB: Detection of human papillomavirus in human
focal cortical dysplasia type IIB. Ann Neurol. 72:881–892. 2012.
View Article : Google Scholar
|
|
14
|
Liu S, Lu L, Cheng X, Xu G and Yang H:
Viral infection and focal cortical dysplasia. Ann Neurol.
75:614–616. 2014. View Article : Google Scholar
|
|
15
|
Buchkovich NJ, Yu Y, Zampieri CA and
Alwine JC: The TORrid affairs of viruses: Effects of mammalian DNA
viruses on the PI3K-Akt-mTOR signalling pathway. Nat Rev Microbiol.
6:266–275. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shapiro KA, McGuone D, Deshpande V, Sadow
PM, Stemmer-Rachamimov A and Staley KJ: Failure to detect human
papillomavirus in focal cortical dysplasia type IIb. Ann Neurol.
78:63–67. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Becker AJ, Urbach H, Scheffler B, Baden T,
Normann S, Lahl R, Pannek HW, Tuxhorn I, Elger CE, Schramm J, et
al: Focal cortical dysplasia of Taylor's balloon cell type:
Mutational analysis of the TSC1 gene indicates a pathogenic
relationship to tuberous sclerosis. Ann Neurol. 52:29–37. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jóźwiak S, Kwiatkowski D, Kotulska K,
Larysz-Brysz M, Lewin-Kowalik J, Grajkowska W and Roszkowski M:
Tuberin and hamartin expression is reduced in the majority of
subependymal giant cell astrocytomas in tuberous sclerosis complex
consistent with a two-hit model of pathogenesis. J Child Neurol.
19:102–106. 2004.
|
|
19
|
Schick V, Majores M, Engels G, Hartmann W,
Elger CE, Schramm J, Schoch S and Becker AJ: Differential
Pi3K-pathway activation in cortical tubers and focal cortical
dysplasias with balloon cells. Brain Pathol. 17:165–173. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Miyata H, Chiang AC and Vinters HV:
Insulin signaling pathways in cortical dysplasia and TSC-tubers:
Tissue microarray analysis. Ann Neurol. 56:510–519. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Patil VV, Guzman M, Carter AN, Rathore G,
Yoshor D, Curry D, Wilfong A, Agadi S, Swann JW, Adesina AM, et al:
Activation of extracellular regulated kinase and mechanistic target
of rapamycin pathway in focal cortical dysplasia. Neuropathology.
36:146–156. 2016. View Article : Google Scholar
|
|
22
|
Lim JS, Kim WI, Kang HC, Kim SH, Park AH,
Park EK, Cho YW, Kim S, Kim HM, Kim JA, et al: Brain somatic
mutations in MTOR cause focal cortical dysplasia type II leading to
intractable epilepsy. Nat Med. 21:395–400. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cotter D, Honavar M, Lovestone S, Raymond
L, Kerwin R, Anderton B and Everall I: Disturbance of Notch-1 and
Wnt signalling proteins in neuroglial balloon cells and abnormal
large neurons in focal cortical dysplasia in human cortex. Acta
Neuropathol. 98:465–472. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lamparello P, Baybis M, Pollard J, Hol EM,
Eisenstat DD, Aronica E and Crino PB: Developmental lineage of cell
types in cortical dysplasia with balloon cells. Brain.
130:2267–2276. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Crino PB, Trojanowski JQ and Eberwine J:
Internexin, MAP1B, and nestin in cortical dysplasia as markers of
developmental maturity. Acta Neuropathol. 93:619–627. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Jozwiak J, Jozwiak S and Wlodarski P:
Possible mechanisms of disease development in tuberous sclerosis.
Lancet Oncol. 9:73–79. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Buccoliero AM, Barba C, Giordano F, Baroni
G, Genitori L, Guerrini R and Taddei GL: Expression of glutamine
synthetase in balloon cells: A basis of their antiepileptic role?
Clin Neuropathol. 34:83–88. 2015. View Article : Google Scholar
|
|
28
|
Cepeda C, Hurst RS, Flores-Hernández J,
Hernández-Echeagaray E, Klapstein GJ, Boylan MK, Calvert CR, Jocoy
EL, Nguyen OK, André VM, et al: Morphological and
electro-physiological characterization of abnormal cell types in
pediatric cortical dysplasia. J Neurosci Res. 72:472–486. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cepeda C, André VM, Vinters HV, Levine MS
and Mathern GW: Are cytomegalic neurons and balloon cells
generators of epileptic activity in pediatric cortical dysplasia?
Epilepsia. 46(Suppl 5): 82–88. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Cepeda C, André VM, Wu N, Yamazaki I,
Uzgil B, Vinters HV, Levine MS and Mathern GW: Immature neurons and
GABA networks may contribute to epileptogenesis in pediatric
cortical dysplasia. Epilepsia. 48(Suppl 5): 79–85. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Calcagnotto ME, Paredes MF, Tihan T,
Barbaro NM and Baraban SC: Dysfunction of synaptic inhibition in
epilepsy associated with focal cortical dysplasia. J Neurosci.
25:9649–9657. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Aronica E, Boer K, Redeker S, Spliet WG,
van Rijen PC, Troost D and Gorter JA: Differential expression
patterns of chloride transporters,
Na+-K+-2Cl− - cotransporter and
K+-Cl− - cotransporter, in
epilepsy-associated malformations of cortical development.
Neuroscience. 145:185–196. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Aronica E, Boer K, Doorn KJ, Zurolo E,
Spliet WG, van Rijen PC, Baayen JC, Gorter JA and Jeromin A:
Expression and localization of voltage dependent potassium channel
Kv4.2 in epilepsy associated focal lesions. Neurobiol Dis.
36:81–95. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Iyer A, Zurolo E, Spliet WG, van Rijen PC,
Baayen JC, Gorter JA and Aronica E: Evaluation of the innate and
adaptive immunity in type I and type II focal cortical dysplasias.
Epilepsia. 51:1763–1773. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Harvey AS, Cross JH, Shinnar S and Mathern
GW; ILAE Pediatric Epilepsy Surgery Survey Taskforce: Defining the
spectrum of international practice in pediatric epilepsy surgery
patients. Epilepsia. 49:146–155. 2008. View Article : Google Scholar
|
|
36
|
Becker AJ, Blümcke I, Urbach H, Hans V and
Majores M: Molecular neuropathology of epilepsy-associated
glioneuronal malformations. J Neuropathol Exp Neurol. 65:99–108.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Spreafico R and Blümcke I: Focal Cortical
Dysplasias: Clinical implication of neuropathological
classification systems. Acta Neuropathol. 120:359–367. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Siegel AM, Cascino GD, Elger CE, Devinsky
O, Laff R, Najjar S, Sperling MR, LoRusso G, Cossu M, Urbach H, et
al: Adult-onset epilepsy in focal cortical dysplasia of Taylor
type. Neurology. 64:1771–1774. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Fauser S, Schulze-Bonhage A, Honegger J,
Carmona H, Huppertz HJ, Pantazis G, Rona S, Bast T, Strobl K,
Steinhoff BJ, et al: Focal cortical dysplasias: Surgical outcome in
67 patients in relation to histological subtypes and dual
pathology. Brain. 127:2406–2418. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Yao K, Mei X, Liu X, Duan Z, Liu C, Bian
Y, Ma Z and Qi X: Clinical characteristics, pathological features
and surgical outcomes of focal cortical dysplasia (FCD) type II:
Correlation with pathological subtypes. Neurol Sci. 35:1519–1526.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Serino D, Freri E, Ragona F, D'Incerti L,
Bernardi B, Di Ciommo V, Granata T, Vigevano F and Fusco L: Focal
seizures versus epileptic spasms in children with focal cortical
dysplasia and epilepsy onset in the first year. Epilepsy Res.
109:203–209. 2015. View Article : Google Scholar
|
|
42
|
Widdess-Walsh P, Kellinghaus C, Jeha L,
Kotagal P, Prayson R, Bingaman W and Najm IM: Electro-clinical and
imaging characteristics of focal cortical dysplasia: Correlation
with pathological subtypes. Epilepsy Res. 67:25–33. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Blümcke I: Neuropathology of focal
epilepsies: A critical review. Epilepsy Behav. 15:34–39. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Cossu M, Fuschillo D, Bramerio M, Galli C,
Gozzo F, Pelliccia V, Casaceli G, Tassi L and Lo Russo G: Epilepsy
surgery of focal cortical dysplasia-associated tumors. Epilepsia.
54(Suppl 9): 115–122. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Krsek P, Pieper T, Karlmeier A,
Hildebrandt M, Kolodziejczyk D, Winkler P, Pauli E, Blümcke I and
Holthausen H: Different presurgical characteristics and seizure
outcomes in children with focal cortical dysplasia type I or II.
Epilepsia. 50:125–137. 2009. View Article : Google Scholar
|
|
46
|
Chassoux F, Devaux B, Landré E, Turak B,
Nataf F, Varlet P, Chodkiewicz JP and Daumas-Duport C:
Stereoelectroencephalography in focal cortical dysplasia: A 3D
approach to delineating the dysplastic cortex. Brain.
123:1733–1751. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Barkovich AJ, Kuzniecky RI, Bollen AW and
Grant PE: Focal transmantle dysplasia: A specific malformation of
cortical development. Neurology. 49:1148–1152. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Colombo N, Tassi L, Deleo F, Citterio A,
Bramerio M, Mai R, Sartori I, Cardinale F, Lo Russo G and Spreafico
R: Focal cortical dysplasia type IIa and IIb: MRI aspects in 118
cases proven by histopathology. Neuroradiology. 54:1065–1077. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Mühlebner A, Coras R, Kobow K, Feucht M,
Czech T, Stefan H, Weigel D, Buchfelder M, Holthausen H, Pieper T,
et al: Neuropathologic measurements in focal cortical dysplasias:
Validation of the ILAE 2011 classification system and diagnostic
implications for MRI. Acta Neuropathol. 123:259–272. 2012.
View Article : Google Scholar
|
|
50
|
Wagner J, Weber B, Urbach H, Elger CE and
Huppertz HJ: Morphometric MRI analysis improves detection of focal
cortical dysplasia type II. Brain. 134:2844–2854. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Blauwblomme T, Boddaert N, Chémaly N,
Chiron C, Pages M, Varlet P, Bourgeois M, Bahi-Buisson N, Kaminska
A, Grevent D, et al: Arterial Spin Labeling MRI: A step forward in
non-invasive delineation of focal cortical dysplasia in children.
Epilepsy Res. 108:1932–1939. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Chan HW, Pressler R, Uff C, Gunny R, St
Piers K, Cross H, Bamber J, Dorward N, Harkness W and Chakraborty
A: A novel technique of detecting MRI-negative lesion in focal
symptomatic epilepsy: Intraoperative ShearWave elastography.
Epilepsia. 55:e30–e33. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Sisodiya SM, Lin WR, Squier MV and Thom M:
Multidrug-resistance protein 1 in focal cortical dysplasia. Lancet.
357:42–43. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Aronica E, Gorter JA, Jansen GH, van
Veelen CW, van Rijen PC, Leenstra S, Ramkema M, Scheffer GL,
Scheper RJ and Troost D: Expression and cellular distribution of
multidrug transporter proteins in two major causes of medically
intractable epilepsy: Focal cortical dysplasia and glioneuronal
tumors. Neuroscience. 118:417–429. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Sisodiya SM, Martinian L, Scheffer GL, van
der Valk P, Cross JH, Scheper RJ, Harding BN and Thom M: Major
vault protein, a marker of drug resistance, is upregulated in
refractory epilepsy. Epilepsia. 44:1388–1396. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Wong M, Ess KC, Uhlmann EJ, Jansen LA, Li
W, Crino PB, Mennerick S, Yamada KA and Gutmann DH: Impaired glial
glutamate transport in a mouse tuberous sclerosis epilepsy model.
Ann Neurol. 54:251–256. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Uhlmann EJ, Wong M, Baldwin RL, Bajenaru
ML, Onda H, Kwiatkowski DJ, Yamada K and Gutmann DH:
Astrocyte-specific TSC1 conditional knockout mice exhibit abnormal
neuronal organization and seizures. Ann Neurol. 52:285–296. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ehninger D, Han S, Shilyansky C, Zhou Y,
Li W, Kwiatkowski DJ, Ramesh V and Silva AJ: Reversal of learning
deficits in a Tsc2+/− mouse model of tuberous sclerosis.
Nat Med. 14:843–848. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
59
|
Krueger DA, Care MM, Holland K, Agricola
K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T and Franz
DN: Everolimus for subependymal giant-cell astrocytomas in tuberous
sclerosis. N Engl J Med. 363:1801–1811. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Skirrow C, Cross JH, Cormack F, Harkness
W, Vargha-Khadem F and Baldeweg T: Long-term intellectual outcome
after temporal lobe surgery in childhood. Neurology. 76:1330–1337.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Chen HH, Chen C, Hung SC, Liang SY, Lin
SC, Hsu TR, Yeh TC, Yu HY, Lin CF, Hsu SP, et al: Cognitive and
epilepsy outcomes after epilepsy surgery caused by focal cortical
dysplasia in children: Early intervention maybe better. Childs Nerv
Syst. 30:1885–1895. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Wagner J, Urbach H, Niehusmann P, von Lehe
M, Elger CE and Wellmer J: Focal cortical dysplasia type IIb:
Completeness of cortical, not subcortical, resection is necessary
for seizure freedom. Epilepsia. 52:1418–1424. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Mühlebner A, Gröppel G, Dressler A,
Reiter-Fink E, Kasprian G, Prayer D, Dorfer C, Czech T, Hainfellner
JA, Coras R, et al: Epilepsy surgery in children and adolescents
with malformations of cortical development - outcome and impact of
the new ILAE classification on focal cortical dysplasia. Epilepsy
Res. 108:1652–1661. 2014. View Article : Google Scholar
|
|
64
|
Kim DW, Lee SK, Chu K, Park KI, Lee SY,
Lee CH, Chung CK, Choe G and Kim JY: Predictors of surgical outcome
and pathologic considerations in focal cortical dysplasia.
Neurology. 72:211–216. 2009. View Article : Google Scholar
|
|
65
|
Mrelashvili A, Witte RJ, Wirrell EC,
Nickels KC and Wong-Kisiel LC: Seizure Freedom in Children With
Pathology-Confirmed Focal Cortical Dysplasia. Pediatr Neurol.
53:513–518. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Jehi L, Yardi R, Chagin K, Tassi L, Russo
GL, Worrell G, Hu W, Cendes F, Morita M, Bartolomei F, et al:
Development and validation of nomograms to provide individualised
predictions of seizure outcomes after epilepsy surgery: A
retrospective analysis. Lancet Neurol. 14:283–290. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Bulacio JC, Jehi L, Wong C,
Gonzalez-Martinez J, Kotagal P, Nair D, Najm I and Bingaman W:
Long-term seizure outcome after resective surgery in patients
evaluated with intracranial electrodes. Epilepsia. 53:1722–1730.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
68
|
Simasathien T, Vadera S, Najm I, Gupta A,
Bingaman W and Jehi L: Improved outcomes with earlier surgery for
intractable frontal lobe epilepsy. Ann Neurol. 73:646–654. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
69
|
Ryzí M, Ošlejšková H, Rektor I, Novák Z,
Hemza J, Chrastina J, Svoboda M, Hermanová M and Brázdil M:
Long-term approach to patients with postsurgical seizures.
Epilepsia. 57:597–604. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
70
|
Tassi L, Garbelli R, Colombo N, Bramerio
M, Russo GL, Mai R, Deleo F, Francione S, Nobili L and Spreafico R:
Electroclinical, MRI and surgical outcomes in 100 epileptic
patients with type II FCD. Epileptic Disord. 14:257–266.
2012.PubMed/NCBI
|
|
71
|
Rowland NC, Englot DJ, Cage TA, Sughrue
ME, Barbaro NM and Chang EF: A meta-analysis of predictors of
seizure freedom in the surgical management of focal cortical
dysplasia. J Neurosurg. 116:1035–1041. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
72
|
Fauser S, Essang C, Altenmüller DM, Staack
A, Steinhoff BJ, Strobl K, Bast T, Schubert-Bast S, Doostkam S,
Zentner J and Schulze-Bonhage A: Is there evidence for clinical
differences related to the new classification of temporal lobe
cortical dysplasia? Epilepsia. 54:909–917. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
73
|
Giulioni M, Martinoni M and Marucci G:
About focal cortical dysplasia (FCD) type IIIa. Epilepsy Res.
108:1955–1957. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
74
|
Giulioni M, Marucci G, Martinoni M, Volpi
L, Riguzzi P, Marliani AF, Bisulli F, Tinuper P, Tassinari CA,
Michelucci R, et al: Seizure outcome in surgically treated
drug-resistant mesial temporal lobe epilepsy based on the recent
histopathological classifications. J Neurosurg. 119:37–47. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
75
|
Santos MV, de Oliveira RS and Machado HR:
Approach to cortical dysplasia associated with glial and
glioneuronal tumors (FCD type IIIb). Childs Nerv Syst.
30:1869–1874. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
76
|
Wu J, Li W, Chen Y, Kang L and Zhao W:
Clinical characteristics of 92 patients with temporal lobe focal
cortical dysplasia identified by pathological examination. J Clin
Neurosci. 21:2170–2174. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
77
|
Fauser S, Essang C, Altenmüller DM, Staack
AM, Steinhoff BJ, Strobl K, Bast T, Schubert-Bast S, Stephani U,
Wiegand G, et al: Long-term seizure outcome in 211 patients with
focal cortical dysplasia. Epilepsia. 56:66–76. 2015. View Article : Google Scholar
|
|
78
|
Tassi L, Garbelli R, Colombo N, Bramerio
M, Lo Russo G, Deleo F, Milesi G and Spreafico R: Type I focal
cortical dysplasia: Surgical outcome is related to histopathology.
Epileptic Disord. 12:181–191. 2010.PubMed/NCBI
|
|
79
|
Tassi L, Meroni A, Deleo F, Villani F, Mai
R, Russo GL, Colombo N, Avanzini G, Falcone C, Bramerio M, et al:
Temporal lobe epilepsy: Neuropathological and clinical correlations
in 243 surgically treated patients. Epileptic Disord. 11:281–292.
2009.PubMed/NCBI
|
|
80
|
Thom M, Eriksson S, Martinian L, Caboclo
LO, McEvoy AW, Duncan JS and Sisodiya SM: Temporal lobe sclerosis
associated with hippocampal sclerosis in temporal lobe epilepsy:
Neuropathological features. J Neuropathol Exp Neurol. 68:928–938.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
81
|
Johnson AM, Sugo E, Barreto D, Cunningham
AM, Hiew CC, Lawson JA, Somerville ER, Connolly AM and Bye AM:
Clinicopathological associations in temporal lobe epilepsy patients
utilising the current ILAE focal cortical dysplasia classification.
Epilepsy Res. 108:1345–1351. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
82
|
Simpson SL and Prayson RA: Post-surgical
outcome for epilepsy associated with type I focal cortical
dysplasia subtypes. Mod Pathol. 27:1455–1460. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
83
|
Yagishita A and Arai N: Cortical tubers
without other stigmata of tuberous sclerosis: Imaging and
pathological findings. Neuroradiology. 41:428–432. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
84
|
Jozwiak J, Kotulska K and Jozwiak S:
Similarity of balloon cells in focal cortical dysplasia to giant
cells in tuberous sclerosis. Epilepsia. 47:8052006. View Article : Google Scholar : PubMed/NCBI
|
|
85
|
Hyman MH and Whittemore VH: National
Institutes of Health consensus conference: Tuberous sclerosis
complex. Arch Neurol. 57:662–665. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
86
|
Jozwiak J, Jozwiak S and Skopinski P:
Immunohistochemical and microscopic studies on giant cells in
tuberous sclerosis. Histol Histopathol. 20:1321–1326.
2005.PubMed/NCBI
|
|
87
|
Oh HS, Lee MC, Kim HS, Lee JS, Lee JH, Kim
MK, Woo YJ, Kim JH, Kim HI and Kim SU: Pathophysiologic
characteristics of balloon cells in cortical dysplasia. Childs Nerv
Syst. 24:175–183. 2008. View Article : Google Scholar
|
|
88
|
Lazarowski A, Lubieniecki F, Camarero S,
Pomata H, Bartuluchi M, Sevlever G and Taratuto AL: Multidrug
resistance proteins in tuberous sclerosis and refractory epilepsy.
Pediatr Neurol. 30:102–106. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
89
|
Lugnier C, Majores M, Fassunke J,
Pernhorst K, Niehusmann P, Simon M, Nellist M, Schoch S and Becker
A: Hamartin variants that are frequent in focal dysplasias and
cortical tubers have reduced tuberin binding and aberrant
subcellular distribution in vitro. J Neuropathol Exp Neurol.
68:1136–1146. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
90
|
Iyer A, Prabowo A, Anink J, Spliet WG, van
Rijen PC and Aronica E: Cell injury and premature neurodegeneration
in focal malformations of cortical development. Brain Pathol.
24:1–17. 2014. View Article : Google Scholar
|
|
91
|
McMahon J, Huang X, Yang J, Komatsu M, Yue
Z, Qian J, Zhu X and Huang Y: Impaired autophagy in neurons after
disinhibition of mammalian target of rapamycin and its contribution
to epileptogenesis. J Neurosci. 32:15704–15714. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
92
|
Yasin SA, Ali AM, Tata M, Picker SR,
Anderson GW, Latimer-Bowman E, Nicholson SL, Harkness W, Cross JH,
Paine SM and Jacques TS: mTOR-dependent abnormalities in autophagy
characterize human malformations of cortical development: Evidence
from focal cortical dysplasia and tuberous sclerosis. Acta
Neuropathol. 126:207–218. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
93
|
Lee CH, Inoki K and Guan KL: mTOR pathway
as a target in tissue hypertrophy. Annu Rev Pharmacol Toxicol.
47:443–467. 2007. View Article : Google Scholar
|
|
94
|
Kotulska K, Jurkiewicz E, Domańska-Pakieła
D, Grajkowska W, Mandera M, Borkowska J and Jóźwiak S: Epilepsy in
newborns with tuberous sclerosis complex. Eur J Paediatr Neurol.
18:714–721. 2014. View Article : Google Scholar : PubMed/NCBI
|














