Introduction
Three major hydrogen sulfide
(H2S)-producing enzymes have been identified:
cystathionine-γ-lyase (CSE), cystathionine-β-synthase (CBS) and
3-mercaptopyruvate sulfurtransferase (3MST) (1–10).
H2S is known to regulate a multitude of physiological
and pathophysiological functions in the vascular, immune and
nervous system (1–10).
The role of H2S in various forms of
critical illness has been a subject of intensive investigations
over the past decade. Some studies have demonstrated the
therapeutic effect of H2S donation in various models of
circulatory shock (11–16), while others have reported that the
pharmacological inhibition of H2S production (17–21) or the genetic deficiency of
H2S-producing enzymes (22,23) results in beneficial effects.
The aim of the current study was to examine the
effect of lipopolysaccharide (LPS)-induced changes in inflammatory
mediator production, indices of multiple organ injury and survival
in wild-type (WT) mice and in mice with reduced expression of one
of the three H2S-producing enzymes, CSE, CBS or 3MST. We
compared the effect of bacterial LPS in WT, CBS heterozygous
(CBS+/−), CSE knockout (CSE−/−) or 3MST
mutant (Δ3MST) mice.
Materials and methods
Materials
Unless indicated otherwise, all chemicals were
obtained from Sigma-Aldrich (St. Louis, MO, USA).
Animals and experimental design
Male WT mice (C57/BL6), CBS heterozygous mice
[CBS+/−; Jackson Laboratory, Ben Harbor, ME, USA, as
previously described (24)], CSE
knockout mice [CSE−/−; a gift from Dr Solomon Snyder,
Johns Hopkins University, as previously described (25)] and 3MST mutant mice [Δ3MST;
generated at the Texas A&M University, as previously described
(26)] (all 2 months of age) were
housed in a light-controlled room with a 12-h light-dark cycle and
were allowed ad libitum access to food and water. Current
studies utilize CBS heterozygous mice, due to the high mortality
rate of CBS−/− mice after birth (25). All investigations adhered to the
Guide for the Care and Use of Laboratory Animals published by the
National Institutes of Health (Eighth Edition, 2011) and were
performed in accordance with the IACUC, University of Texas Medical
Branch, Galveston, TX, USA.
LPS-induced endotoxemia in mice
Mice were randomly allocated into the following
groups: i) WT mice + vehicle (n=10); ii) WT mice + LPS [10 mg/kg,
intraperitoneally (i.p.)] (n=10); iii) CBS+/− mice +
vehicle (n=10); iv) CBS+/− mice + LPS (10 mg/kg, i.p.)
(n=10); v) CSE−/− mice + vehicle (n=10); vi)
CSE−/− mice + LPS (10 mg/kg, i.p) (n=10); vii) Δ3MST
mice + vehicle (n=10); and viii) Δ3MST mice + LPS (10 mg/kg, i.p.)
(n=10). The volume of saline (V) administered was equal to the
volume of LPS administered. Six hours after the LPS injection the
mice were sacrificed by isoflurane inhalation (0.25–3%) followed by
opening of the chest and exsanguination by cardiac puncture; blood
and tissue samples were then collected for further examinations.
This time point was selected based on prior studies showing that at
this point LPS-induced cytokine responses are detectable (including
those that are released early on); at the same time, multiple organ
injury is already significant (27–30); however at this time point, no
mortality ensues yet.
Expression of CBS, CSE and 3MST in lung,
spleen, liver and kidney samples
The organs were placed in RIPA buffer and sonicated
(three times for 10 sec each). The supernatants were preserved and
the protein concentration was determined by bicinchoninic acid
(BCA) assay. Protein expression was determined by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) under
reducing conditions. The supernatant extracts (40
µg/µl) were boiled in equal volumes of loading buffer
(150 mM Tris-HCl, pH 6.8; 4% SDS; 20% glycerol; 15%
β-mercaptoethanol; and 0.01% bromophenol blue) and were
electrophoresed on 8–12% polyacrylamide gels. Following
electrophoretic separation, the proteins were transferred onto PVDF
membranes for western blotting. The membranes were blocked with
StartingBlock T20 (TBS) Blocking Buffer (Thermo Fisher Scientific,
Waltham, MA, USA) for 1 h. The following primary antibodies were
used: CBS, 1:1,000 (GTX628777; GeneTex, Inc., Irvine, CA, USA);
CSE, 1:1,000 (12217-1-AF; ProteinTech Group, Inc., Chicago, IL,
USA); 3MST, 1:1,000 (HPA001240; Sigma-Aldrich); and actin, 1:5,000
(sc-1616; Cell Signaling Technology, Inc., Danvers, MA, USA). The
primary antibodies were incubated overnight at 4°C and the
membranes were washed twice in TBST. Secondary horseradish
peroxidase-conjugated antibodies [anti-rabbit (7074S), anti-mouse
(7076S); Cell Signaling Technology, Inc.)] were then applied at a
dilution of 1:5,000 for 1 h. Over a 30-min period, the blots were
washed twice in TBST, after which they were incubated in enhanced
chemiluminescence reagents (SuperSignal detection kit; Pierce
Biotechnology, Inc., Rockford, IL, USA). The band intensity of the
original blots was quantified using GeneTools (Syngene; Synoptics,
Ltd., Cambridge, UK) and normalized to actin expression.
Assessment of renal dysfunction
At 6 h post-LPS challenge, blood samples were
collected via cardiac puncture and were analyzed by using a VetScan
analyzer (Abaxis North America, Union City, CA, USA). The ratio of
the blood concentration of urea was calculated as an indicator of
glomerular function.
Malondialdehyde (MDA) assay
Tissue MDA levels, an index of cellular
injury/oxidative stress, were quantified in lung samples using a
fluorimetric MDA-Specific Lipid Peroxidation assay kit (Enzo Life
Sciences, Farmingdale, NY, USA) according to the manufacturer's
instructions. The assay is based on the BML-AK171 method in which
two molecules of the chromogenic reagent
N-methyl-2-phenylindole react with one molecule of MDA at
45°C to yield a stable carbocyanine dye with a maximum absorption
at 586 nm.
Myeloperoxidase (MPO) assay
MPO activity was measured in lung samples using a
commercially available MPO fluorometric detection kit (Enzo Life
Sciences). The assay utilizes a non-fluorescent detection reagent,
which is oxidized in the presence of hydrogen peroxide and MPO to
produce its fluorescent analog. The fluorescence is measured at
excitation wavelength of 530–571 nm and emission wavelength of
590–600 nm.
Quantification of plasma cytokine
levels
Blood from mice in all groups was collected in
K2EDTA blood collection tubes and centrifuged at 4°C for 15 min at
2,000 × g within 30 min of collection. Plasma was isolated,
aliquoted and stored at −80°C until use. The EMD Millipore's
MILLIPLEX™ MAP Mouse Cytokine Magnetic Bead Panel 1 kit (EMD
Millipore, Billerica, MA, USA) was used for the simultaneous
quantification of the following analytes: interleukin (IL)-1β,
tumor necrosis factor (TNF)α, IL-2, IL-4, IL-5, IL-6, IL-10, IL-12,
interferon (IFN)γ, granulocyte-macrophage colony-stimulating factor
(GM-CSF) (Merck Millipore, Darmstadt, Germany). Luminex uses a
proprietary technique to internally color code microspheres with
two fluorescent dyes and to create distinctly colored bead sets of
500 polystyrene microspheres (5.6 µm) or 80 magnetic
microspheres (6.45 µm), each of which is coated with a
specific capture antibody. After an analyte from a test sample is
captured by the bead, a biotinylated detection antibody is
introduced. The reaction mixture is then incubated with
streptavidin-phycoerythrin conjugate, the reporter molecule, to
complete the reaction on the surface of each microsphere. The
Luminex instrument acquires and analyzes data using the Luminex
xMAP fluorescent detection method and the Luminex xPONENT™
acquisition software (Thermo Fisher Scientific).
Survival analyses
Survival was assessed in the WT, CBS heterozygous
(CBS+/−), CSE knockout (CSE−/−) or 3MST
mutant (Δ3MST) mice (n=15 mice in each group) after i.p. injection
of LPS (20 mg/kg, i.p). Mortality of the animals was recorded over
a 48-h period.
Statistical analysis
All values described in the text and figures are
expressed as the means ± standard error of the mean (SEM) for 'n'
observations. The Student's t-test, one- and two-way ANOVA with
Tukey's post hoc test were used to detect differences between
groups. The Chi-square test was used to compare survival rates.
Prism version 5 for Windows (GraphPad Software, Inc., La Jolla, CA,
USA) was used. A value of P<0.05 was considered to indicate a
statistically significantly difference.
Results
Changes in the expression of
H2S-producing enzymes in response to LPS
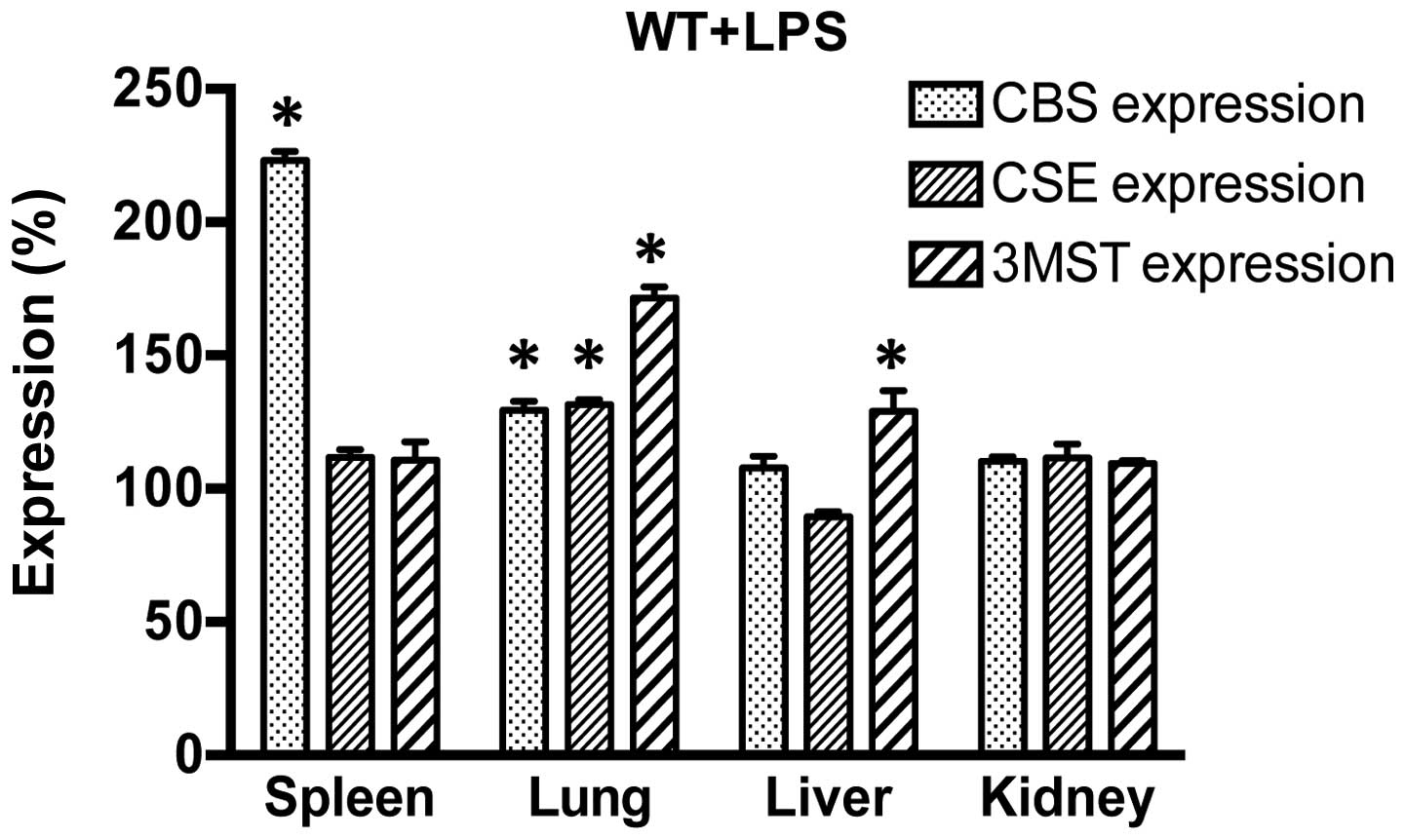
First, the effect of LPS on the expression of the
three H2S-producing enzymes (CBS, CSE and 3MST) was
examined in various tissue samples (spleen, lung, liver and kidney)
in the control (vehicle-treated) WT, CBS+/−,
CSE−/− and Δ3MST mice. We found the following basal
expression of the enzymes (Fig.
1): in CSE−/− mice, CSE protein was absent in all
tissues studied; in CBS+/− mice, CBS levels were
markedly suppressed in some tissues (liver, kidney), while they
remained unaltered in others (spleen, lung), indicating that in
some tissues a single copy of the CBS gene is sufficient to yield
physiological amounts of CBS transcripts. In addition, and as
previously observed (26), the
current strain of Δ3MST mice exhibited reduced 3MST expression in
their spleens and lungs, but not the livers and kidneys. We then
examined the effect of LPS challenge on the expression of CBS, CSE
and 3MST in WT mice. LPS induced an increase in CBS expression in
the spleen and lung; CSE expression increased in the lung and 3MST
expression increased in the lung and liver (Fig. 2). These expression patterns were,
generally, similar in the WT mice and the genetically modified
strains of mice, even though in the CSE−/− mice, the
LPS-induced upregulation of CBS occurred in the liver and kidney
and in the Δ3MST mice, it only occurred in the kidney (as opposed
to the WT mice, where it occurred in the spleen and the lung).
Moreover, in response to LPS, the upregulation of CSE in the
CBS+/− mice occurred in the spleen (whereas in the WT
mice the largest degree of CSE upregulation occurred in the lung)
(Table I).
 | Table IExpression profiles of
cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE) and
3-mercaptopyruvate sulfurtransferase (3MST) at 6 h after the
lipopolysaccharide (LPS) (10 mg/kg) injection in wild-type (WT),
CBS+/−, CSE−/− and Δ3MST mice. |
Table I
Expression profiles of
cystathionine-β-synthase (CBS), cystathionine-γ-lyase (CSE) and
3-mercaptopyruvate sulfurtransferase (3MST) at 6 h after the
lipopolysaccharide (LPS) (10 mg/kg) injection in wild-type (WT),
CBS+/−, CSE−/− and Δ3MST mice.
| WT
(%) |
CBS+/−
(%) |
CSE−/−
(%) | Δ3MST
(%) |
|---|
| CBS expression |
| Spleen | 227±29a | 168±15a | 111±6 | 107±15 |
| Lung | 134±12a | 115±6 | 109±17 | 120±10 |
| Liver | 112±12 | 19±6 | 149±11a | 107±13 |
| Kidney | 112±14 | 19±5 | 161±15a | 123±9a |
| CSE expression |
| Spleen | 116±14 | 125±15a | 0 | 91±9 |
| Lung | 134±19a | 92±16 | 0 | 120±6a |
| Liver | 92±5 | 101±6 | 0 | 103±22 |
| Kidney | 119±1 | 106±1.6 | 0 | 103±12 |
| 3MST
expression |
| Spleen | 116±8 | 125±10a | 125±11a | 1±1 |
| Lung | 177±25a | 159±20a | 241±14a | 9±1 |
| Liver | 139±9a | 129±12a | 111±11 | 107±13 |
| Kidney | 110±13 | 114±8 | 9±1 | 115±13 |
Effect of CBS+/−,
CSE−/− and Δ3MST on LPS-induced blood urea nitrogen
(BUN) plasma levels
LPS administration to all four groups of mice
studied (WT, CBS+/−, CSE−/− and Δ3MST)
induced an increase in plasma BUN levels (Fig. 3). The degree of this increase was
comparable in the WT, CSE−/− and Δ3MST mice; however,
the CBS+/− mice exhibited a reduced degree of
LPS-induced increased plasma BUN levels compared to the WT mice
(Fig. 3).
Effect of CBS+/−,
CSE−/− and Δ3MST on LPS-induced MPO and MDA tissue
levels
LPS administration induced an increase in lung MPO
and MDA levels in all four groups of mice studied (Fig. 4). The degree of the increase in
pulmonary MDA post-LPS levels tended to be less in the
CSE−/− and CBS+/− mice compared to the WT
mice (Fig. 4).
Effect of CBS+/−,
CSE−/− and Δ3MST on LPS-induced plasma cytokine
levels
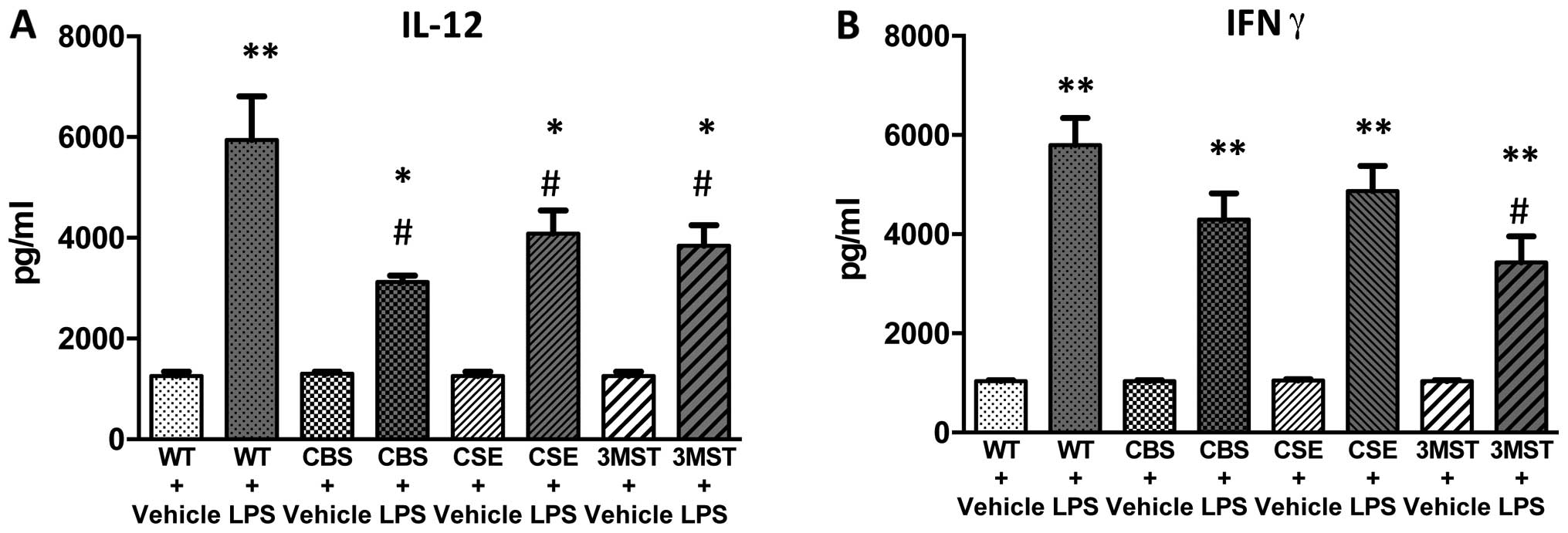
LPS administration induced an increase in the plasma
levels of multiple cytokines in all four groups of mice studied
(Figs. 5Figure 6Figure 7Figure 8–9). The degree of the increase in TNFα
tended to be less in all three groups of mice deficient in various
H2S-producing enzymes (Fig. 5), while plasma IL-5 and GM-CSF
levels tended to be higher after LPS challenge in the Δ3MST mice
(Fig. 7). The degree of the
increases in IL-10 and IL-12 levels tended to be less in all three
groups of mice deficient in various H2S-producing
enzymes (Figs. 8 and 9); plasma IFNγ levels after LPS were
lower in CBS+/−mice compared to WT mice (Fig. 9).
Effect of CBS+/−,
CSE−/− and Δ3MST on LPS-induced survival
Survival curves after LPS challenge tended to be
shifted to the right in the CSE−/− and CBS+/−
mice compared to the WT mice; however, the effect failed to reach
statistical significance, while the survival curves of the WT and
Δ3MST mice were superimposable (Fig.
10).
Discussion
The main conclusions of the current study are the
following: i) LPS induces a tissue-dependent upregulation of
H2S-producing enzymes in mice (upregulation of CBS in
the spleen, upregulation of 3MST in the liver and upregulation of
CBS, CSE and 3MST in the lung), with similar (but not identical)
patterns observed in genetically modified mice lacking either of
the three CBS-producing enzymes; ii) LPS induces the various
expected hallmarks of organ injury (elevated BUN levels, elevated
tissue levels of MPO and MDA), increased levels of circulating
cytokines and mortality over time; iii) the LPS-induced alterations
are only slightly or partially affected by the partial or complete
absence of any individual H2S-producing enzymes; with
the most pronounced changes being a) a partial attenuation of
LPS-induced BUN levels in the CBS+/− mice; b) a partial
attenuation of MDA levels in the lungs of CBS+/− and
CSE−/− mice; and c) a partial attenuat ion of TNFα,
IL-10 and IL-12 levels in all three genetically modified animal
groups. Finally, iv) there were no statistically significant
effects of any of the genetic modifications on LPS-induced
mortality. Based on these data, we conclude that a deficiency in
any single one of the three major H2S-producing enzymes
only slightly affects the outcome of LPS-induced endotoxemia in
vivo.
As already mentioned in the 'Introduction', the
current body of literature on endotoxemia, endotoxin shock, sepsis,
as well as various other forms of critical illness (e.g., burn
injury, ARDS and hemorrhagic shock) is fairly controversial with
respect to the role of H2S in the pathogenesis of these
diseases; some of the studies have demonstrated that
pharmacological H2S donation is beneficial in some
experimental models (11–16), while other studies have concluded
that pharmacological inhibitors of H2S biosynthesis
(17–21), or the genetic deletion of CSE
(22,23) is beneficial. Moreover, there are
also studies demonstrating that H2S donation can be
detrimental (18–20), and there are even studies
demonstrating that pharmacological inhibitors of H2S
biosynthesis can be detrimental in certain models of critical
illness (31,32). While some of these discrepancies
may be attributable to the differences in the experimental models
used, some of the explanation is likely to be related to the
well-known bell-shaped dose-response character (1–10)
of H2S, where lower concentrations of the mediator exert
distinctly different (often opposite) pharmacological effects than
higher concentrations; indeed, lower concentrations, and delayed
administration of H2S donors are often found protective,
while higher concentrations are often detrimental. It should also
be noted that H2S exerts differential effects on various
functions in different cell types and different organs (e.g.,
vascular functions, pro-inflammatory signaling, redox/oxidant
processes, cell death effector pathways, cellular bioenergetic
pathways); modulation of some of these effects may be ultimately
beneficial for the outcome of critical illness, while modulation of
others may be detrimental. Thus, the ultimate outcome parameters
may depend on the relative importance of the various pathways
affected by H2S in the particular experimental model
studied. The outcome of the experiment may also depend on the
changes in endogenous H2S biosynthesis; for instance, in
some (but not all) models of critical illness, H2S
levels can be elevated; these elevated H2S levels may
serve cytoprotective as well as deleterious roles, dependent on the
type of critical illness, and perhaps the stage and the severity of
the disease as well [reviewed in (6,8,33,34)]. Similarly, H2S has been
demonstrated to affect the production of various pro- and
anti-inflammatory cytokines (6,8,11,12–16,19); both stimulatory and inhibitory
effects have been reported; the direction of the effect is
dependent on the concentration of H2S used, as well as
the experimental model and cell type used, and it has been shown to
involve a variety of signaling pathways including NF-κB, MAP
kinases and histone deacetylases (14,19,20,35–39). Thus, there may be multiple
mechanistic reasons (in addition to model-dependent differences)
why inhibition of H2S biosynthesis or donation of
H2S can affect the outcome of a complex disease like
septic shock in a beneficial or detrimental manner, depending on
the constellation of the multitude of the factors and processes
discussed above.
As regards the effect of genetic deficiency of
H2S-producing enzymes on the outcome of organ injury,
some of the data published in the literature indicate that it can
be detrimental: e.g., CSE deletion in myocardial and hepatic
ischemia-reperfusion models exacerbates organ damage (40). Moreover, CSE or CBS deletion in
renal injury models increases disease severity (41). However, in two recently published
murine models of critical illness, the data indicate that
CSE−/− mice are protected against
LPS/galactosamine-induced hepatic injury (22) and in a model of cecal ligation and
puncture, the specific silencing of CSE in circulating mononuclear
cells was found to improve disease outcomes (23). In the current study, while the
Δ3MST mice tended to exhibit similar patterns to the WT mice for
most of the key parameters studied (organ damage indices, cytokine
profiles, survival), the CBS+/− and CSE−/−
mice tended to exhibit slight trends towards protection such as
lower BUN levels (CBS+/− mice), in several cases lower
cytokine levels (both CBS+/− and CSE−/− mice)
and a trend towards delays in LPS-induced mortality. Based on these
data, and coupled with the fact that we found that LPS induces an
upregulation of various H2S-producing enzymes in various
organs, we conclude that endogenously produced H2S, in
the current model, on the whole, tends to exhibit predominantly a
deleterious overall effect. The reduction in some of the plasma
cytokines in all three groups of mice deficient in
H2S-producing enzymes corresponds to a mixed
pro/anti-inflammatory effect of H2S biosynthesis
inhibition, because both pro-inflammatory (TNFα) and
anti-inflammatory (IL-10) mediator production was suppressed. The
fact that the effects observed in the current study are often
partial (and in many cases do not reach statistical significance)
may be attributed to the fact that each of the genetically modified
animals used in the current study only has a partial defect in the
H2S production; in some cases the deficiency itself is
partial (CBS, 3MST) and even in the animals where the deficiency of
the target (CSE) is complete, the remaining
H2S-producing enzymes continue to synthesize
H2S, which may, in some cases, exert compensatory
effects. Although there are some differences between the various
strains of mice with respect to upregulation of various
H2S-producing enzymes in response to LPS, we do not
suggest that the compensation proposed above occurs because the
genetically modified mice produce more H2S via
upregulation of various alternative H2S-producing
enzymes; we suggest that this compensation is simply the result of
the fact that deletion of either of the three enzymes only reduces
tissue H2S levels to a partial degree.
The purpose of the current study was to determine
the effect of each individual H2S-producing enzyme,
separately, on LPS-induced responses. A mouse that is
simultaneously deficient in all three enzymes is currently not
available - neither in our laboratory nor in other laboratories;
there are no published studies in the literature using such an
approach. It remains, therefore, to be determined, whether the
simultaneous lack of all three H2S-producing enzymes
would change the viability of an animal (under baseline conditions
or under various pathophysiological conditions).
We are aware of several limitations of the current
study. First of all, we did not use littermate controls for the WT
mice. Instead, we used C57/BL6 mice. This is the exactly
appropriate control for the CBS+/− mice, as they were
obtained from Jackson Laboratory, and have the same background, as
well as the 3MST mutant mice which are on the same background as
well. However, the CSE−/− mice were on a mixed
background. This is a limitation of the study. Nevertheless,
genetic background differences tend to pose more of a problem when
there are differences found between the groups of animals compared
(as it remains to be determined whether the differences are due to
the absence of the enzyme studied, or, perhaps due to background
differences). However, in our case, actually, there are no
significant differences between the responses of the WT and the
CSE−/− mice to LPS. This means that neither the
presence/absence of the H2S-producing enzyme, nor the
potential differences due to background make enough difference to
culminate in a significant difference in the outcome variables
studied. We believe that with the additional discussion and caveats
the material presented here continues to contain useful information
for the field. Second, only a single time point (6 h post LPS) was
studied for the various parameters of renal injury, MPO/MDA and
cytokines; since the course of critical illness has several stages,
further studies will be necessary to determine whether the effects
are different, depending on the timing/stage of the illness. Third,
the survival study employed here utilized a severe model, with 100%
mortality. Generally, a severe disease model tends to be harder to
be affected by therapeutic intervention than a milder model;
follow-up studies may employ different models with lower severity.
Fourth, the current model only used one particular model of
sepsis/shock, the one induced by bacterial endotoxin; other models
(e.g., sepsis models induced by live bacteria, or by polymicrobial
sepsis, e.g., the one induced by cecal ligation and puncture) may
yield a more complete picture. Fifth, in the current study some of
the H2S-producing enzymes we sought to study were only
partially downregulated due to technical/practical issues - e.g.,
CBS−/− mice have a very high mortality rate early on
after birth, and the large majority of the animals do not live
until young-adult age to be suitable for the LPS model utilized
here (42); the mutation in 3MST
gene only produced a partial and tissue-dependent reduction in 3MST
levels in the strain of 3MST mice we have had access to. Naturally,
since the strain used in the current study does not have a
downregulation of 3MST in the liver or the kidney, we did not
expect that WT vs. 3MST mutant mice will respond differently to
LPS-induced liver or kidney dysfunction; and, indeed, they did not.
There are other models of CBS deficiency in mice, e.g., a model
where CBS is completely absent, and the mouse is engineered to
contain a deficient human form of CBS (42), that may be better suitable for
future studies; likewise, a group in Japan has created a full 3MST
knockout line (43); these
genetically modified animals may be useful in future studies.
Sixth, in the current study, we did not measure circulating
H2S levels, only the tissue expression of various
H2S-producing enzymes. We do not feel that measurement
of circulating H2S levels would be particularly valuable
in the context of the current study, given the fact that multiple
organ-specific changes were demonstrated in the expression of the
various H2S-producing enzymes after LPS. In addition,
there are many prior studies indicating that the net level of
circulating H2S is not predictable for the outcome of
critical illness, since both H2S donors and
H2S biosynthesis inhibitors have demonstrated beneficial
effects in various models (6,8,11–23,31–23); it has been suggested that the
timing of the donation or inhibition as well as possible regional
(cell- and organ-specific differences likely play a role). In the
current study, we only used mice with global deficiency of the
target enzymes; given the multiple, cell-, tissue- and
organ-specific biological roles of H2S, future studies
with cell-type selective deletion of various
H2S-producing enzymes may also be highly instrumental to
unveil the complex roles of H2S and
H2S-producing enzymes in various forms of critical
illness.
Acknowledgments
This study was supported by the National Institutes
of Health (R01GM107846) to C.S.
Abbreviations:
|
BCA
|
bicinchoninic acid
|
|
BUN
|
blood urea nitro gen
|
|
CBS
|
cystathionine-β-synthase
|
|
CSE
|
cystathionine-γ-lyase
|
|
H2S
|
hydrogen sulfide
|
|
IL
|
interleukin
|
|
LPS
|
lipopolysaccharide
|
|
MDA
|
malondialdehyde
|
|
MPO
|
myeloperoxidase
|
|
3MST
|
3-mercaptopyruvate
sulfurtransferase
|
|
SDS-PAGE
|
sodium dodecyl sulfate-polyacrylamide
gel electrophoresis
|
|
SEM
|
standard error of the mean
|
References
|
1
|
Fiorucci S, Distrutti E, Cirino G and
Wallace JL: The emerging roles of hydrogen sulfide in the
gastrointestinal tract and liver. Gastroenterology. 131:259–271.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Szabó C: Hydrogen sulphide and its
therapeutic potential. Nat Rev Drug Discov. 6:917–235. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang R: Physiological implications of
hydrogen sulfide: A whiff exploration that blossomed. Physiol Rev.
92:791–296. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Predmore BL, Lefer DJ and Gojon G:
Hydrogen sulfide in biochemistry and medicine. Antioxid Redox
Signal. 17:119–240. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vandiver M and Snyder SH: Hydrogen
sulfide: A gasotransmitter of clinical relevance. J Mol Med (Berl).
90:255–263. 2012. View Article : Google Scholar
|
|
6
|
Coletta C and Szabo C: Potential role of
hydrogen sulfide in the pathogenesis of vascular dysfunction in
septic shock. Curr Vasc Pharmacol. 11:208–221. 2013.PubMed/NCBI
|
|
7
|
Szabo C, Ransy C, Módis K, Andriamihaja M,
Murghes B, Coletta C, Olah G, Yanagi K and Bouillaud F: Regulation
of mitochondrial bioenergetic function by hydrogen sulfide. Part I.
Biochemical and physiological mechanisms. Br J Pharmacol.
171:2099–2122. 2014. View Article : Google Scholar :
|
|
8
|
McCook O, Radermacher P, Volani C, Asfar
P, Ignatius A, Kemmler J, Möller P, Szabó C, Whiteman M, Wood ME,
et al: H2S during circulatory shock: Some unresolved
questions. Nitric Oxide. 41:48–21. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kimura H: Signaling molecules: Hydrogen
sulfide and polysulfide. Antioxid Redox Signal. 22:362–276. 2015.
View Article : Google Scholar :
|
|
10
|
Huang CW and Moore PK: 2S synthesizing
enzymes: Biochemistry and molecular aspects. Handb Exp Pharmacol.
230:3–25. 2015. View Article : Google Scholar
|
|
11
|
Li L, Salto-Tellez M, Tan CH, Whiteman M
and Moore PK: GYY4137, a novel hydrogen sulfide-releasing molecule,
protects against endotoxic shock in the rat. Free Radic Biol Med.
47:103–213. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tokuda K, Kida K, Marutani E, Crimi E,
Bougaki M, Khatri A, Kimura H and Ichinose F: Inhaled hydrogen
sulfide prevents endotoxin-induced systemic inflammation and
improves survival by altering sulfide metabolism in mice. Antioxid
Redox Signal. 17:11–21. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Aslami H, Beurskens CJ, de Beer FM,
Kuipers MT, Roelofs JJ, Hegeman MA, Van der Sluijs KF, Schultz MJ
and Juffermans NP: A short course of infusion of a hydrogen
sulfide-donor attenuates endotoxemia induced organ injury via
stimulation of anti-inflammatory pathways, with no additional
protection from prolonged infusion. Cytokine. 61:614–221. 2013.
View Article : Google Scholar
|
|
14
|
Chen X, Xu W, Wang Y, Luo H, Quan S, Zhou
J, Yang N, Zhang T, Wu L, Liu J, et al: Hydrogen sulfide reduces
kidney injury due to urinary-derived sepsis by inhibiting NF-κB
expression, decreasing TNF-α levels and increasing IL-10 levels.
Exp Ther Med. 8:464–270. 2014.PubMed/NCBI
|
|
15
|
Ferlito M, Wang Q, Fulton WB, Colombani
PM, Marchionni L, Fox-Talbot K, Paolocci N and Steenbergen C:
Hydrogen sulfide [corrected] increases survival during sepsis:
Protective effect of CHOP inhibition. J Immunol. 192:1806–2814.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Ahmad A, Druzhyna N and Szabo C: Delayed
treatment with sodium hydrosulfide improves regional blood flow and
alleviates cecal ligation and puncture (CLP)-induced septic shock.
Shock. 46:183–293. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Collin M, Anuar FB, Murch O, Bhatia M,
Moore PK and Thiemermann C: Inhibition of endogenous hydrogen
sulfide formation reduces the organ injury caused by endotoxemia.
Br J Pharmacol. 146:498–205. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Li L, Bhatia M, Zhu YZ, Zhu YC, Ramnath
RD, Wang ZJ, Anuar FB, Whiteman M, Salto-Tellez M and Moore PK:
Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced
inflammation in the mouse. FASEB J. 19:1196–2198. 2005.PubMed/NCBI
|
|
19
|
Zhang H, Zhi L, Moochhala S, Moore PK and
Bhatia M: Hydrogen sulfide acts as an inflammatory mediator in
cecal ligation and puncture-induced sepsis in mice by upregulating
the production of cytokines and chemokines via NF-kappaB. Am J
Physiol Lung Cell Mol Physiol. 292:L960–L971. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Zhang H, Moochhala SM and Bhatia M:
Endogenous hydrogen sulfide regulates inflammatory response by
activating the ERK pathway in polymicrobial sepsis. J Immunol.
181:4320–2331. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yan Y, Chen C, Zhou H, Gao H, Chen L, Chen
L, Gao L, Zhao R and Sun Y: Endogenous hydrogen sulfide formation
mediates the liver damage in endotoxemic rats. Res Vet Sci.
94:590–295. 2013. View Article : Google Scholar
|
|
22
|
Shirozu K, Tokuda K, Marutani E, Lefer D,
Wang R and Ichinose F: Cystathionine γ-lyase deficiency protects
mice from galactosamine/lipopolysaccharide-induced acute liver
failure. Antioxid Redox Signal. 20:204–216. 2014. View Article : Google Scholar :
|
|
23
|
Badiei A, Chambers ST, Gaddam RR and
Bhatia M: Cys tathionine-γ-lyase gene silencing with siRNA in
monocytes/macrophages attenuates inflammation in cecal ligation and
puncture-induced sepsis in the mouse. J Biosci. 41:87–25. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Watanabe M, Osada J, Aratani Y, Kluckman
K, Reddick R, Malinow MR and Maeda N: Mice deficient in
cystathionine beta-synthase: Animal models for mild and severe
homocyst(e) inemia. Proc Natl Acad Sci USA. 92:1585–2589. 1995.
View Article : Google Scholar
|
|
25
|
Yang G, Wu L, Jiang B, Yang W, Qi J, Cao
K, Meng Q, Mustafa AK, Mu W, Zhang S, et al: H2S as a
physiologic vasorelaxant: Hypertension in mice with deletion of
cystathionine gamma-lyase. Science. 322:587–290. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Gero D, Ahmad A, Brunyanszki A, Olah G,
Szczesny B and Szabo C: 3-Mercaptopyruvate sulfurtransferase
deficient mice show accelerated glucose uptake and a dysregulated
metabolic profile. Nitric Oxide. 47:S35–S36. 2015. View Article : Google Scholar
|
|
27
|
Jagtap P, Soriano FG, Virág L, Liaudet L,
Mabley J, Szabó E, Haskó G, Marton A, Lorigados CB, Gallyas F Jr,
et al: Novel phenanthridinone inhibitors of poly (adenosine
5′-diphosphate-ribose) synthetase: Potent cytoprotective and
antishock agents. Crit Care Med. 30:1071–2082. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Namas RA, Bartels J, Hoffman R, Barclay D,
Billiar TR, Zamora R and Vodovotz Y: Combined in silico, in vivo,
and in vitro studies shed insights into the acute inflammatory
response in middle-aged mice. PLoS One. 8:e674192013. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Bhargava R, Altmann CJ, Andres-Hernando A,
Webb RG, Okamura K, Yang Y, Falk S, Schmidt EP and Faubel S: Acute
lung injury and acute kidney injury are established by four hours
in experimental sepsis and are improved with pre, but not post,
sepsis administration of TNF-α antibodies. PLoS One. 8:e790372013.
View Article : Google Scholar
|
|
30
|
Everhardt Queen A, Moerdyk-Schauwecker M,
McKee LM, Leamy LJ and Huet YM: Differential expression of
inflammatory cytokines and stress genes in male and female mice in
response to a lipopolysaccharide challenge. PLoS One.
11:e01522892016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Zhang J, Sio SW, Moochhala S and Bhatia M:
Role of hydrogen sulfide in severe burn injury-induced inflammation
in mice. Mol Med. 16:417–224. 2010.PubMed/NCBI
|
|
32
|
Bekpinar S, Unlucerci Y, Uysal M and
Gurdol F: Propargylglycine aggravates liver damage in LPS-treated
rats: Possible relation of nitrosative stress with the inhibition
of H2S formation. Pharmacol Rep. 66:897–201. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Módis K, Bos EM, Calzia E, van Goor H,
Coletta C, Papapetropoulos A, Hellmich MR, Radermacher P, Bouillaud
F and Szabo C: Regulation of mitochondrial bioenergetic function by
hydrogen sulfide. Part II. Pathophysiological and therapeutic
aspects. Br J Pharmacol. 171:2123–2146. 2014. View Article : Google Scholar :
|
|
34
|
Brunyanszki A, Erdelyi K, Szczesny B, Olah
G, Salomao R, Herndon DN and Szabo C: Upregulation and
mitochondrial sequestration of hemoglobins occurs in circulating
leukocytes during critical illness, conferring a cytoprotective
phenotype. Mol Med. In press.
|
|
35
|
Hu LF, Wong PT, Moore PK and Bian JS:
Hydrogen sulfide attenuates lipopolysaccharide-induced inflammation
by inhibition of p38 mitogen-activated protein kinase in microglia.
J Neurochem. 100:1121–2128. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Chi XP, Ouyang XY and Wang YX: Hydrogen
sulfide synergistically upregulates Porphyromonas gingivalis
lipopolysaccharide-induced expression of IL-6 and IL-8 via NF-κB
signalling in periodontal fibroblasts. Arch Oral Biol. 59:954–261.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Rios EC, Szczesny B, Soriano FG, Olah G
and Szabo C: Hydrogen sulfide attenuates cytokine production
through the modulation of chromatin remodeling. Int J Mol Med.
35:1741–2746. 2015.PubMed/NCBI
|
|
38
|
Lee HH, Han MH, Hwang HJ, Kim GY, Moon SK,
Hyun JW, Kim WJ and Choi YH: Diallyl trisulfide exerts
anti-inflammatory effects in lipopolysaccharide-stimulated RAW
264.7 macrophages by suppressing the Toll-like receptor 4/nuclear
factor-κB pathway. Int J Mol Med. 35:487–295. 2015.
|
|
39
|
Yang H, Wang H, Ju Z, Ragab AA, Lundbäck
P, Long W, Valdes-Ferrer SI, He M, Pribis JP, Li J, et al: MD-2 is
required for disulfide HMGB1-dependent TLR4 signaling. J Exp Med.
212:5–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
King AL, Polhemus DJ, Bhushan S, Otsuka H,
Kondo K, Nicholson CK, Bradley JM, Islam KN, Calvert JW, Tao YX, et
al: Hydrogen sulfide cytoprotective signaling is endothelial nitric
oxide synthase-nitric oxide dependent. Proc Natl Acad Sci USA.
111:3182–2187. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wang P, Isaak CK, Siow YL and O K:
Downregulation of cystathionine β-synthase and cystathionine
γ-lyase expression stimulates inflammation in kidney
ischemia-reperfusion injury. Physiol Rep. 2:e122512014. View Article : Google Scholar
|
|
42
|
Wang L, Chen X, Tang B, Hua X,
Klein-Szanto A and Kruger WD: Expression of mutant human
cystathionine beta-synthase rescues neonatal lethality but not
homocystinuria in a mouse model. Hum Mol Genet. 14:2201–2208. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Nagahara N, Nagano M, Ito T, Shimamura K,
Akimoto T and Suzuki H: Antioxidant enzyme, 3-mercaptopyruvate
sulfurtransferase-knockout mice exhibit increased anxiety-like
behaviors: A model for human mercaptolactate-cysteine disulfiduria.
Sci Rep. 3:19862013. View Article : Google Scholar : PubMed/NCBI
|