Introduction
Inherited hypophosphatemic rickets, characterized by
bone mineralization disorders due to hypophosphatemia, secondary to
a leak of phosphate from the kidneys, was first introduced by
Albright et al (1). The
clinical characteristics of the disease include growth retardation,
bending of the weight-bearing extremities, and the resistance to
vitamin D therapy during early childhood. Subsequently, patients
suffer from spontaneous dental abscesses, extensive bone pain and
arthropathy with aging. X-linked hypophosphatemic rickets (XLH;
OMIM 307800), with an occurrence of appproximately 1 in 20,000 live
births, is known as the most prevalent form of inherited
hypophosphatemic rickets (2,3).
The mutation in the phosphate-regulating gene with homologies to
endopeptidase on the X chromosome (PHEX; MIM 300550) was
identified in 1995 as the causative gene for the disease (4). Thereafter, the genetic basis of
other less prevalent types of inherited hypophosphatemic rickets,
such as autosomal dominant hypophosphatemic rickets (ADHR; OMIM
193100) and autosomal recessive hypophosphatemic rickets (ARHR1;
OMIM 241520), was successively clarified (5,6).
Nevertheless, the underlying pathogenesis for some sporadic cases
without known genetic mutations remains to be determined and the
significant findings of key roles of microRNAs (miRNAs of miRs)
involved in bone formation may provide potential research issues
(7–9).
The PHEX gene, located on X chromosome
Xp22.1, is composed of 22 exons spanning 243 kb and encodes for 749
amino acid proteins (10). It
exhibits great homology to the M13 zinc metallopeptidases, a class
of type 2 integral membrane glycoproteins that includes neprilysin
(NEP), endothelin-converting enzymes 1 and 2 (ECE-1
and ECE-2), as well as the Kell antigen (KELL)
(11). These proteins show the
common structural traits of a short N-terminal cytoplasmic domain,
a single transmembrane hydrophobic region and a large extracellular
domain. The latter contains 10 highly conserved cysteine residues
and two zinc-binding motifs and is involved in the secondary
structure conformation or catalytic activity of the protein
(11). It has been demonstrated
that the tissue-specific expression of the PHEX gene and its
mRNA expression is predominantly detected in bone lineage cells;
for instance, osteoblasts, osteocytes and odontoblasts (10,12). However, the exact mechanisms
through which inactivated PHEX leads to abnormal skeletal
and renal manifestations in XLH have not yet been fully clarified.
Studies have suggested the existence of phosphatonin that is
responsible for the regulation of phosphate homeostasis in the
Hyp mouse (13), an animal
model of XLH with a 3′ deletion of the PHEX gene. Based on
the finding of increased fibroblast growth factor 23 (FGF23)
transcripts in the Hyp mouse, FGF23 was then regarded as a
leading candidate for phosphatonin downstream of PHEX
(14).
An extensive mutation analysis of the genetic
defects of the PHEX gene only revealed a few cases in China
(15–21). In the present study, we screened a
total of 18 affected families for mutations in the PHEX gene
in order to interpret the mutation traits in Chinese patients and
potentially provide evidence of a critical domain in PHEX
protein. Moreover, the serum FGF23 levels in affected individuals
were also measured as this may also contribute to our understanding
of the molecular basis of XLH.
Materials and methods
Ethics statement and study subjects
This study was approved by the Ethics Committee of
the Shanghai Jiao Tong University Affiliated Sixth People's
Hospital (Shanghai, China) and all subjects or their guardians (for
the under-aged participants) provided written informed consent
prior to enrollment. The subjects enrolled in this study were from
the Department of Osteoporosis and Bone Diseases in Shanghai Jiao
Tong University Affiliated Sixth People's Hospital, Shanghai, China
over the past 3 years. A total of 18 unrelated Chinese families
with hypophosphatemic rickets was examined and all subjects were of
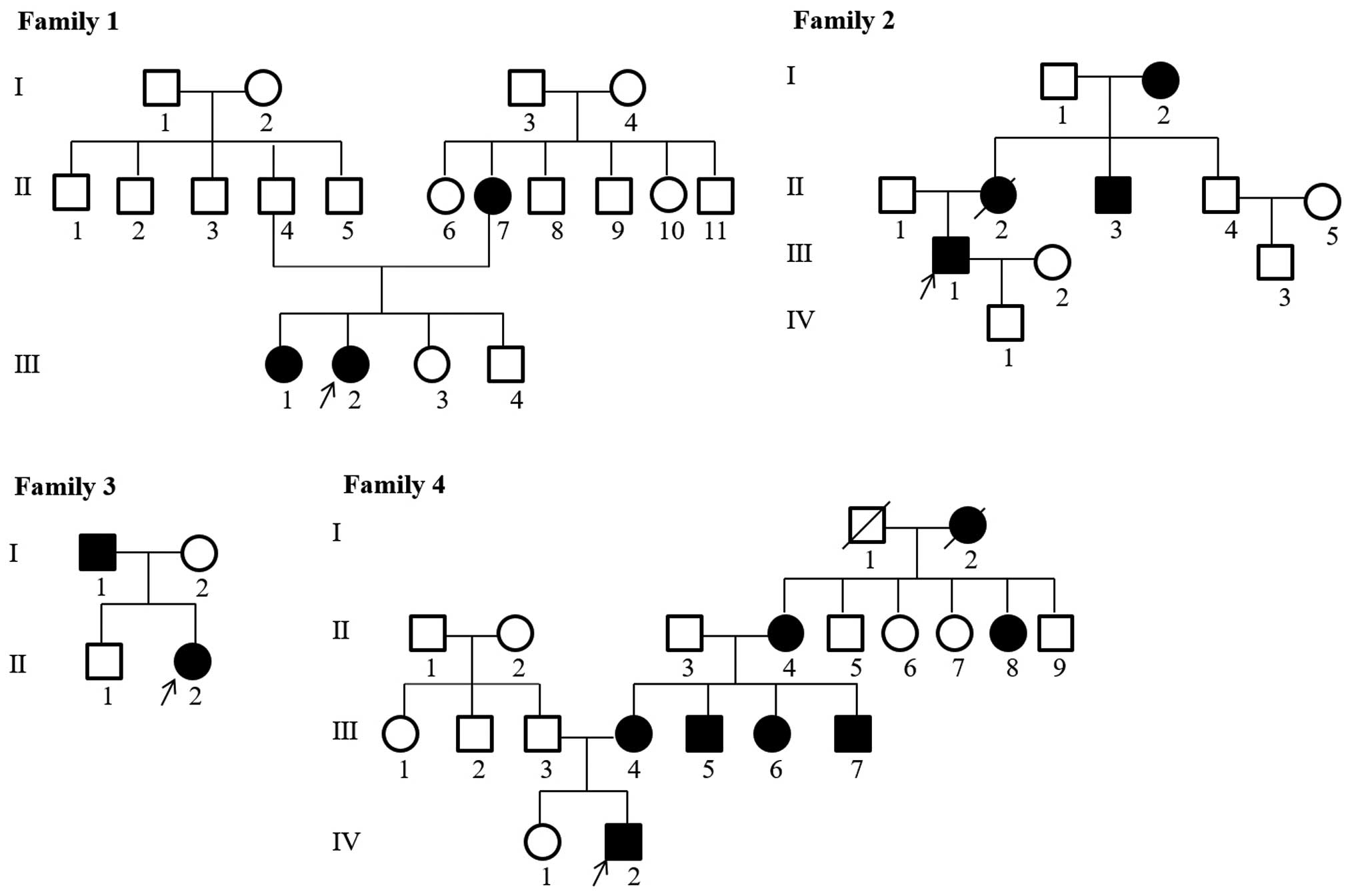
Han ethnicity. The pedigrees of the families with hypophosphatemic
rickets are shown in Fig. 1. The
diagnosis of hypophosphatemic rickets was mainly based on a history
of childhood rickets, clinical manifestations and biochemical tests
indicating hypophosphatemia along with elevated levels of serum
alkaline phosphatases. Patients with secondary rickets due to
malnutrition, medication or tumor-induced osteomalacia were
excluded. Finally, 65 individuals, including 43 patients, and 250
unrelated healthy controls were recruited and subjected to blood
sampling followed by DNA analyses. In addition, another 95 healthy
controls with normal serum phosphate and calcium levels and normal
renal function were randomly selected to carry out measurements of
serum levels of FGF23 in order to derive a reference range.
Mutation screening
To detect mutations in the PHEX gene, all DNA
samples from the probands were initially analyzed. When a
PHEX mutation was confirmed, DNA from first-degree
relatives, as well as the symptomatic individuals was then screened
for the detected mutation. A proband was identified as a sporadic
case if the PHEX mutation was not detected in the parents,
namely if both were asymptomatic.
Mutation analyses of the affected individuals were
performed by the direct sequencing of polymerase chain reaction
(PCR) products amplified from genomic DNA. The DNA was extracted
from peripheral white blood cells by proteinase K digestion
followed by purification with phenol/chloroform and isopropyl
alcohol precipitation. The DNA sequence for the PHEX gene
was obtained from the available online database (NCBI Reference
Sequence Accession no. NG_007563.2). All the 22 exons with their
adjacent intronic sequences of the PHEX gene were amplified
by PCR with 21 pairs of sequencing primers designed using Primer 3
software (http://bioinfo.ut.ee/primer3-0.4.0/). The primer
sequences are presented in Table
I. Direct sequencing was performed using the BigDye Terminator
Cycle Sequencing Ready Reaction kit, version 3.1 and the PCR
products were directly sequenced using an automated ABI PRISM 3130
sequencer (both from Applied Biosystems, Foster City, CA, USA).
Simultaneously, single-nucleotide polymorphisms (SNPs) were
identified using PolyPhred (http://droog.gs.washington.edu/polyphred/) and novel
mutations were identified using HGMD (http://www.hgmd.cf.ac.uk/). Mutations were checked
using Mutalyzer 2.0 (http://mutalyzer.nl/check). The DNA sequences obtained
were aligned with homologous sequences that had been deposited into
GenBank using the CluxtalX 1.83 algorithm. To predict the impact of
missense mutations on protein structure and function, Polymorphism
Phenotyping v2 (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2) (22) and Sorting Tolerant from Intolerant
(SIFT; http://sift.jcvi.org) (23) were used based on sequence
alignments.
 | Table IList of primer sequences used for PCR
amplification of the PHEX gene. |
Table I
List of primer sequences used for PCR
amplification of the PHEX gene.
| Exons | Forward primer
(5′→3′) | Reverse primer
(5′→3′) |
|---|
| 1 |
AGGGACTTTGCTGAGGGAGAG |
CCACTCGAAGCCACTTACACC |
| 2 |
TGGGTTTTGGAATACCGTGTC |
AAGAGAGGCCATTCAGCCTTC |
| 3 |
CAAGGCTTGGAAACTGGTTGA |
TTATGTTGAGATCTGGGAGTCCA |
| 4 |
GGCACCATATGTGGGTGGATA |
GTTTGCCCTGCTGACTTTGTC |
| 5 |
CACATTGAAGCGTGGATCGTA |
CGGGAGAAGGGAATATTCTGG |
| 6 |
GCTCTGCCCAATCATGTTACC |
GCAGCCTGGTAAGGCACATAG |
| 7 |
GGGTGCCTGGTATTGCATAAT |
CCAATGGGCAATGACACAAA |
| 8 |
ACCACACCAAAGCCTTGAAAA |
GAGCCAATGCCAACAATTACC |
| 9 |
GGATGGCAATGATCAGGAGTT |
GACAGTGCTTTTGGCCAGTTC |
| 10 |
ATGTTCACTCTGAGGGCTGGA |
GGCTACAAACTCCCCCTGTCT |
| 11 |
CAGCCATGGGTTTTATCCAAA |
CCCACTCCCCTGGAAAACTAC |
| 12 |
AGTGTTGCCAGAGCATGGAGT |
AGGAAAGGCCGAATTACAAGG |
| 13 |
TCGATTCAGTCACCTTCTCCA |
GAAAGGCACAAGGCCAGTAAA |
| 14 |
TGACTGATGCAGCTTCTCTGC |
ATGCTAGAAATGGGGGACCTG |
| 15 |
GCAGGGACAGCCCTTTAGATT |
GCCACTTTTGGGGGAAATAAG |
| 16 |
GTGCAAAATGGTTTCCCTGAA |
GTCCAGCCATACACCCTGGTA |
| 17 |
AAGCAGTTTATCTTGGCTTTCCA |
CAAGCCATCACAGCAAGACAC |
| 18 |
CTGCTTTTTGAAGGCTTGTCG |
ATGCCTGGTTAAGGGATGACC |
| 19 |
TTGATGCCTCTTGCTGAATGA |
AAATGAACCTAGCCCCAAGGA |
| 20 |
TGGTAAGCAACAGGACATGGA |
AGGGCTGCTAACCCATTTGAT |
| 21 |
TTCCTGGGCACATATACGATTC |
TTTTGGCTGCAAAATGGAAAT |
| 22 |
CAGAACCTGTTGATGTGCAAGA |
GCCAACACCCTAAAATGGACA |
Measurement of circulating intact FGF23
levels
Serum FGF23 leels were measured using an intact
human FGF23 enzyme-linked immunosorbent assay (ELISA) kit obtained
from Kainos Laboratories Inc. (Tokyo, Japan). The detectable
concentration ranged from 3 to 800 pg/ml. This assay has been
validated in previous studies (16,24).
Statistical analyses
Normally distributed variables are presented as the
means ± SD, and non-normally distributed variables as the median
(2.5th and 97.5th percentiles). Comparisons between groups were
made using unpaired Student's t-tests for normal data, and the
Wilcoxon rank sum test for non-normal data. Values of P<0.05
were considered to indicate statistically significant differences.
Statistical analyses were performed using SPSS 13.0 software.
Results
The baseline clinical data and PHEX gene
mutation analyses of 65 participants originating from 18 families
are summarized in Table II. A
total of 43 patients was confirmed to have XLH according to their
clinical and genetic evidence. All the patients exhibited varying
degrees of growth retardation, bending legs, dental anomalies,
hypophosphatemia, markedly elevated serum alkaline phosphatase
levels and normal serum calcium. The median (25th and 75th
percentiles) age of the patients was 22.0 (8.3, 29.5) years and the
median serum phosphate levels were 0.70 (0.57, 0.80) mmol/l.
| Table IIClinical findings and PHEX
mutations identified in families affected by XLH. |
Table II
Clinical findings and PHEX
mutations identified in families affected by XLH.
| Family no. | Patienta | Gender | Age (years) | Clinical
characteristics | Serum P-value
(mmol/l)b | Mutation site | Mutation type | DNA level | Protein level |
|---|
| 1 | III-2 | F | 21 | Growth retardation;
teeth falling out; genu valgum | 0.68 | Exon 15 | Missense | c.1601C>T | p.P534L |
| III-1 | F | 22 | Growth retardation;
genu valgum | 0.73 | Exon 15 | Missense | c.1601C>T | p.P534L |
| II-7 | F | 48 | Short stature;
teeth falling out; genu valgum | 0.75 | Exon 15 | Missense | c.1601C>T | p.P534L |
| 2 | III-1 | M | 30 | Growth retardation;
teeth falling out; genu varum | NA | Exon 11 | Nonsense | c.1294A>T | p.K432X |
| I-2 | F | 78 | Short stature | 0.56 | Exon 11 | Nonsense | c.1294A>T | p.K432X |
| II-3 | M | 51 | Short stature;
failure to walk; teeth falling out; genu varum | 0.64 | Exon 11 | Nonsense | c.1294A>T | p.K432X |
| 3 | II-2 | F | 23 | Growth retardation;
genu varum | 0.86 | Exon 22 | Missense | c.2192T>C | p.F731S |
| I-1 | M | 53 | Short stature; genu
varum | 0.72 | Exon 22 | Missense | c.2192T>C | p.F731S |
| 4 | IV-2 | M | 5 | Growth retardation;
genu varum | 0.84 | Intron13 | Alternative
splicing | c.1483-1G>C | p.? |
| III-4 | F | 39 | Short stature | 0.54 | Intron 13 | Alternative
splicing | c.1483-1G>C | p.? |
| 5 | II-2 | F | 28 | Short stature; genu
varum | 0.60 | Intron 10 | Alternative
splicing | c.1174-1G>A | p.? |
| III-1 | F | 4 | Growth retardation;
genu varum | 0.67 | Intron 10 | Alternative
splicing | c.1174-1G>A | p.? |
| 6 | III-15 | F | 11 | Growth retardation;
teeth falling out; genu varum | 0.75 | Exon 11 | Deletion | c.1234delA | p.S412VfsX12 |
| II-12 | F | 36 | Short stature;
teeth falling out; genu varum | 0.60 | Exon 11 | Deletion | c.1234delA | p.S412VfsX12 |
| II-8 | F | 43 | Short stature;
teeth falling out; genu varum | 0.77 | Exon 11 | Deletion | c.1234delA | p.S412VfsX12 |
| III-11 | M | 11 | Growth retardation;
genu varum | 0.68 | Exon 11 | Deletion | c.1234delA | p.S412VfsX12 |
| III-12 | F | 12 | Growth retardation;
genu varum | 0.77 | Exon 11 | Deletion | c.1234delA | p.S412VfsX12 |
| 7 | III-3 | F | 28 | Short stature; genu
varum | 0.69 | Intron 4 | Alternative
splicing |
c.436_436+1delAG | p.? |
| II-4 | F | 59 | Short stature; genu
varum | NA | Intron 4 | Alternative
splicing |
c.436_436+1delAG | p.? |
| II-6 | F | 56 | Short stature; genu
varum | 0.54 | Intron 4 | Alternative
splicing |
c.436_436+1delAG | p.? |
| II-8 | F | 52 | Short stature; genu
varum | NA | Intron 4 | Alternative
splicing |
c.436_436+1delAG | p.? |
| 8 | III-3 | F | 27 | Short stature; genu
varum | 0.55 | Exon 7 | Missense | c.824T>C | p.L275P |
| III-2 | F | 30 | Short stature; genu
varum | 0.42 | Exon 7 | Missense | c.824T>C | p.L275P |
| IV-1 | F | 8 | Growth retardation;
genu varum | 0.90 | Exon 7 | Missense | c.824T>C | p.L275P |
| II-4 | F | 54 | Growth retardation;
genu varum; teeth falling out | 0.62 | Exon 7 | Missense | c.824T>C | p.L275P |
| 9 | IV-3 | F | 19 | Growth retardation;
genu valgum | 0.50 | Exon 8 | Nonsense | c.931C>T | p.E311X |
| 10 | III-1 | F | 2 | Genu varum; rib
eversion; pectus carinatum | 0.75 | Intron 7 | Alternative
splicing | c.849+1G>C | p.? |
| 11 | III-1 | F | 10 | Growth retardation;
genu varum | 0.70 | Intron 4 | Alternative
splicing | c.436+1G>C | p.? |
| 12 | IV-10 | F | 24 | Short stature; genu
varum; thoracic deformity | 0.55 | Exon 3 | Missense | c.304G>A | p.G102R |
| III-5 | M | 53 | Short stature; genu
varum; spinal deformity; teeth falling out | 0.32 | Exon 3 | Missense | c.304G>A | p.G102R |
| IV-8 | F | 27 | Short stature; genu
varum; loose teeth | 0.64 | Exon 3 | Missense | c.304G>A | p.G102R |
| 13 | II-4 | F | 27 | Short stature; genu
varum; teeth falling out | 0.56 | Exon 3 | Missense | c.229T>C | p.C77R |
| III-2 | F | 1.5 | Genu varum; pillow
bald | 0.88 | Exon 3 | Missense | c.229T>C | p.C77R |
| 14 | III-6 | M | 30 | Short stature;
teeth falling out; lower limb deformity; limited mobility | 0.53 | Exon 18 | Insertion | c.1843dupA | p.T615nfsX6 |
| II-10 | F | 63 | Short stature;
lower limb deformity | 0.40 | Exon 18 | Insertion | c.1843dupA | p.T615nfsX6 |
| III-1 | M | 36 | Short stature;
lower limb deformity | 0.52 | Exon 18 | Insertion | c.1843dupA | p.T615nfsX6 |
| III-5 | F | 31 | Short stature;
lower limb deformity | 0.75 | Exon 18 | Insertion | c.1843dupA | p.T615nfsX6 |
| 15 | III-2 | M | 8 | Growth retardation;
genu varum; pectus carinatum; rib eversion | 0.87 | Intron 14 | Alternative
splicing |
c.1586_1586+1delAG | p.? |
| III-1 | F | 10 | Growth retardation;
genu varum | 0.54 | Intron 14 | Alternative
splicing |
c.1586_1586+1delAG | p.? |
| II-5 | F | 37 | Short stature; genu
varum | 0.65 | Intron 14 | Alternative
splicing |
c.1586_1586+1delAG | p.? |
| 16 | III-1 | F | 5 | Genu varum | 0.78 | Exon 5 | Deletion | c.528delT | p.E177KfsX44 |
| 17 | III-1 | F | 4 | Genu varum | 1.02 | Intron 14 | Alternative
splicing |
c.1586_1586+1delAG | p.? |
| 18 | III-1 | F | 15 | Genu varum; teeth
falling out | 0.54 | Exon 8 | Nonsense | c.871C>T | p.R291X |
We identified 17 different mutations in the
PHEX gene from 18 unrelated families and the distribution of
PHEX mutations were 6 alternative splicing mutations
(35.3%), 5 missense mutations (29.4%), 3 nonsense mutations
(17.6%), 2 deletion mutations (11.8%) and 1 insertion mutation
(5.9%) (Fig. 2). Among these, 7
mutation sites were identified as novel and they were respectively
detected from families 4, 6, 7, 8, 12, 13 and 16: c.1483-1G>C in
intron 13 at splicing acceptor sites leading to truncated protein,
c.1234delA (p.Ser412ValfsX12) in exon 11, c.436_436+1delAG in
intron 4 at splicing donor sites leading to truncated protein,
c.824T>C (p.Leu275Pro) in exon 7, c.304G>A (p.Gly102Arg) in
exon 3, c.229T>C (p.Cys77Arg) in exon 3 and c.528delT
(p.Glu177LysfsX44) in exon 5. PolyPhen−2 and SIFT were performed to
assess the missense mutational consequence of PHEX, and all
3 missense mutations were predicted to be probably damaging with a
score of 0.930 for L275R, 0.917 for G102R and 1.000 for C77R, and a
SIFT score of <0.05. Notably, the amino acid residues at p.275,
p.102 and p.77 were evolutionarily highly conserved across 12
different species as shown in Fig.
3. In addition, the probands from families 9, 10, 11, 16, 17
and 18 were identified as sporadic cases and the detected
PHEX mutations were likely to be de novo, and the 6
probands were all female patients.
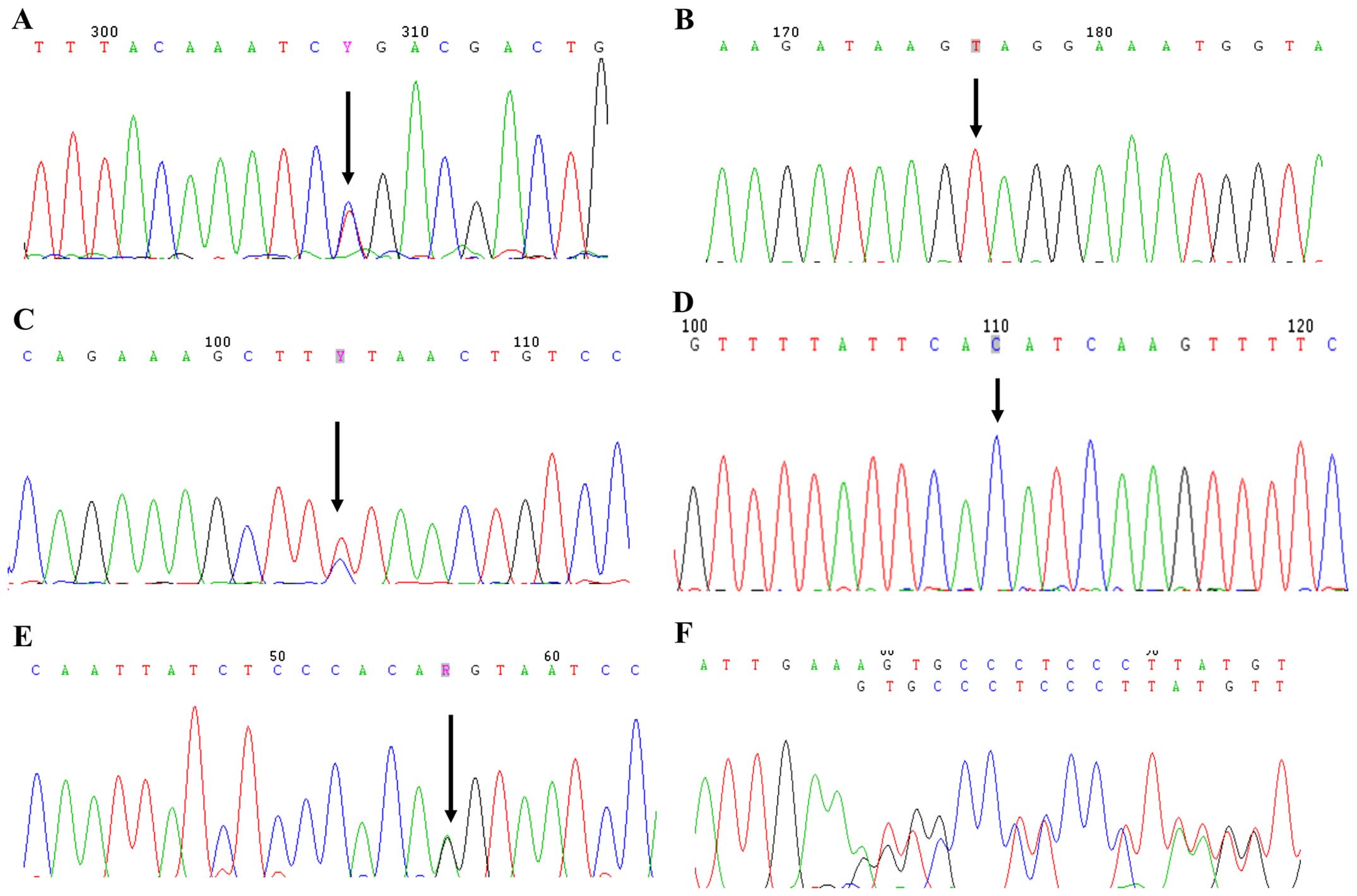 | Figure 2Mutational analyses of
phosphate-regulating gene with homology to endopeptidase on the X
chromosome (PHEX) gene in patients with hypophosphatemic
rickets. (A) c.1601C>T in exon 15, (B) c.1294A>T in exon 11,
(C) c.2192T>C in exon 22, (D) c.1483-1G>C in intron 13, (E)
c.1174−1G>A in intron 11, (F) c.1234delA in exon 11, (G)
c.436_436+1delAG in intron 4, (H) c.824T>C in exon 7, (I)
c.931C>T in exon 8, (J) c.849+1G>C in intron 7, (K)
c.436+1G>C in intron 4, (L) c.229T>C in exon 3. Mutational
analyses of PHEX gene in patients with hypophosphatemic
rickets. (M) c.304G>A in exon 3, (N) c.1843dupA in exon 18, (O)
c.1586_1586+1delAG in intron 14, (P) c.528delT in exon 5 and (Q)
c.871C>T in exon 8. |
Serum FGF23 levels were not normally distributed in
either the normal controls or the XLH patients. The median value
for intact serum FGF23 levels from the 95 healthy individuals aged
between 22 to 77 years was 40.6 pg/ml and the reference range
(2.5th and 97.5th percentiles) was from 24.6 to 136.8 pg/ml
(Table III). For the patients
with XLH, as shown in Table IV,
the serum FGF23 levels were below the reference range in 4 of 11
subjects, within the range in 4 subjects, and mildly elevated in 3
subjects. The serum FGF23 levels exhibited a wide variation in the
patients with XLH, and no significant differences were found when
compared with those of the normal controls (p>0.05).
| Table IIISerum intact human FGF23 levels in
healthy controls. |
Table III
Serum intact human FGF23 levels in
healthy controls.
| Subjects | FGF23 (pg/ml) | Subjects | FGF23 (pg/ml) | Subjects | FGF23 (pg/ml) | Subjects | FGF23 (pg/ml) | Subjects | FGF23 (pg/ml) |
|---|
| 1 | 49.3 | 20 | 45.7 | 39 | 26.8 | 58 | 47.2 | 77 | 53.9 |
| 2 | 62.0 | 21 | 20.7 | 40 | 61.0 | 59 | 50.3 | 78 | 56.9 |
| 3 | 37.0 | 22 | 31.9 | 41 | 47.2 | 60 | 29.9 | 79 | 25.8 |
| 4 | 33.0 | 23 | 37.0 | 42 | 60.0 | 61 | 30.4 | 80 | 29.6 |
| 5 | 33.5 | 24 | 34.5 | 43 | 49.3 | 62 | 43.2 | 81 | 40.2 |
| 6 | 26.3 | 25 | 51.3 | 44 | 40.6 | 63 | 40.1 | 82 | 34.0 |
| 7 | 37.0 | 26 | 34.0 | 45 | 47.7 | 64 | 60.5 | 83 | 37.8 |
| 8 | 41.6 | 27 | 93.1 | 46 | 40.1 | 65 | 37.6 | 84 | 41.4 |
| 9 | 153.8 | 28 | 42.1 | 47 | 56.9 | 66 | 53.9 | 85 | 34.1 |
| 10 | 31.9 | 29 | 31.4 | 48 | 35.0 | 67 | 51.3 | 86 | 38.3 |
| 11 | 23.8 | 30 | 68.1 | 49 | 59.0 | 68 | 50.3 | 87 | 47.6 |
| 12 | 44.2 | 31 | 71.7 | 50 | 28.4 | 69 | 30.9 | 88 | 49.0 |
| 13 | 35.0 | 32 | 75.3 | 51 | 136.0 | 70 | 48.3 | 89 | 39.3 |
| 14 | 44.7 | 33 | 65.6 | 52 | 26.3 | 71 | 41.1 | 90 | 47.5 |
| 15 | 116.6 | 34 | 50.3 | 53 | 38.1 | 72 | 64.6 | 91 | 137.4 |
| 16 | 116.1 | 35 | 39.1 | 54 | 35.5 | 73 | 35.0 | 92 | 28.7 |
| 17 | 51.8 | 36 | 39.6 | 55 | 27.4 | 74 | 41.1 | 93 | 35.1 |
| 18 | 33.5 | 37 | 82.4 | 56 | 67.6 | 75 | 30.9 | 94 | 39.6 |
| 19 | 30.9 | 38 | 37.6 | 57 | 33.5 | 76 | 38.1 | 95 | 54.6 |
| Table IVSerum intact human FGF23 in patients
with XLH. |
Table IV
Serum intact human FGF23 in patients
with XLH.
| Family no.-
Patienta | Gender | Age (years) | Serum P-value
(mmol/l)b | Serum FGF23
(pg/ml)c | Mutations in
PHEX gene |
|---|
| F2-III1 | M | 30 | NA | 38.7 | c.1294A>T (Exon
11) |
| F2-II3 | M | 51 | 0.64 | 40.4 | c.1294A>T (Exon
11) |
| F3-II2 | F | 23 | 0.86 | 21.1 | c.2192T>C (Exon
22) |
| F5-II2 | F | 28 | 0.60 | 7.8 | c.1174-1G>A
(Intron 10) |
| F6-III15 | F | 11 | 0.75 | 171.5 | c.1234delA (Exon
11) |
| F9-IV3 | F | 19 | 0.50 | 29.7 | c.931C>T (Exon
8) |
| F10-III1 | F | 2 | 0.75 | 111.4 | c.849+1G>C
(Intron 7) |
| F11-III1 | F | 10 | 0.70 | 15.7 | c.436+1G>C
(Intron 4) |
| F12-III5 | M | 53 | 0.32 | 24.0 | c.304G>A (Exon
3) |
| F12-IV10 | F | 24 | 0.55 | 162.2 | c.304G>A (Exon
3) |
| F12-IV8 | F | 27 | 0.64 | 143.2 | c.304G>A (Exon
3) |
Discussion
Based on the PHEX mutation database
(http://www.PHEXdb.mcgill.ca), the
frequencies of different mutation types were 25% frameshifts, 23%
alternative splicing, 22% missense, 18% nonsense, 8% deletion and
4% polymorphisms. The mutations were scattered throughout the gene
and the majority would potentially influence the pattern of
post-translational modification of the protein and alter its
secondary structure, resulting in the loss of PHEX function
(25,26). In this study, we identified 17
different PHEX mutations from 18 unrelated Chinese families,
and to the best of our knowledge, 7 were novel.
Three novel missense mutations, namely, Cys77Arg,
Gly102Arg and Leu275Pro, were detected. The residue p.77 was
amongst the 10 highly conserved cysteine residues that were
critical for disulfide-bond formation and protein folding (25,26). Thus, the single base change at
this position would likely alter the secondary structure of the
protein and render it out of action. Gly102 has been found to be
conserved among NEP, PHEX and ECE-1 according
to the multiple sequence alignment analysis in molecular research
(27). Additionally, the glycine
to arginine exchange was likely to increase the local charge of the
PHEX gene product as arginine was positively charged, while
glycine had an uncharged polar group. Therefore, it is possible
that this base alteration plays a role in a different spatial
conformation. The novel missense mutation detected in exon 7
involved a substitution of proline for leucine at residue 275. To
date, there are only 14 different mutations confirmed in exon 7 and
it is among the rarest mutant exons in line with the PHEX
mutation database. Moreover, this site was estimated to be
conserved with the replaced leucine occurred in both ECE-1
and PHEX. The 3 novel missense mutations identified in this
study were shown to be highly conserved under the protein alignment
of the PHEX gene from 12 different species and were verified
to be pathogenic by bioinformatics tools (PolyPhen-2 and SIFT),
which also rendered the evidence of their potential value for the
phenotype in patients with XLH.
Three nonsense mutations: Lys432X in exon 11,
Arg291X and Gln311X in exon 8 were detected in this study, all of
which would cause the translation of truncated protein with
accidental loss of C-terminal region. It was demonstrated that the
C-terminal region in the large extracellular domain was abundant in
conserved cysteine residues and contained the zinc-binding motifs
in exons 17 and 19 (10,11). The cysteine residues are
responsible for the secondary structure formation and contribute to
conformation integrity. The highly conserved zinc-binding motifs
among NEP, PHEX, ECE-1 and KELL, are
essential for the catalytic activity of the protein. Therefore,
these 2 nonsense mutations would inevitably lead to impaired PHEX
protein function.
Six of the 17 PHEX mutations were identified
as alternative splicing mutations, including 3 splice acceptor
mutations and 3 splice donor mutations. Among these, 2 were novel
mutations: 436_436+1delAG in the splice donor site of exon 4 and an
alteration of C to G in intronic sequence 5' to initiate exon 11,
-1 bp upstream. These 2 splicing mutations were predicted to result
in the skipping of exons 4 and 11, respectively. Notably, both of
the 2 affected exons comprised one conserved cyteine residue that
was relevant for secondary structure transformation and thus
protein function. That is, the newly identified 2≈splicing
mutations were associated with the onset of XLH.
The remaining 3 mutations were 1234delA in exon 11,
c.528delT in exon 5 and 1843dupA in exon 18, characterized as
frameshift mutations. The mutation occurring in exon 18 was first
reported by Gauche et al (28). It caused the replacement of
aspartic acid for threonine at residue 615 and opened a new reading
frame for 6 amino acids and deemed to disrupt the overall integrity
of the PHEX protein. Moreover, the hetero-PHEX protein. Moreover,
the heteroprotein. Moreover, the hetero zygous deletion of one
adenine nucleotide and one thymine nucleotide in exons 11 and 5
were 2 novel mutations identified in our study. These mutations
would result in premature termination of the PHEX protein.
No hotspot mutations were found in XLH according to
the present study in accordance with previous studies on
PHEX mutations in Chinese patients (15–21). However, it was valued that
exceeding 50% mutations occurred around exons 18 to 22 in the
C-terminal region based on the mutation analyses in PHEXdb and it
was speculated that this region may be the critical domain for PHEX
function (29). We failed to
determine the gene dosage effect on disease severity by comparing
the phenotypes of hemizygous males to those of heterozygous females
from the same family. An evidence-based study also indicated that
there was no difference in severity of the disease between genders
in mutant Hyp mouse (30).
Theoretically, heterozygous females should have a less severe
phenotype due to random X-inactivation, the process of
transcriptional silencing of one of the X chromosomes bringing
about half of the normal alleles, while males have none. Sabbagh
et al owed the absence of gene dose effect to a threshold of
PHEX activity that was required for maintaining normal protein
function (25). In addition to
the gender impact, questions remain as to the possibility of
correlations among either mutation location or type with phenotype
severity. In the present study, the association between genotype
and phenotype in patients with XLH was not observed. It has been
proposed that there is a trend towards a more severe phenotype with
mutations located in the C-terminal region or with truncating
mutations (31); however, this
has yet to be verified in a larger sample size.
The aberrant activity of FGF23 was revealed as a
common fundamental mechanism for the development of defects in
phosphate and vitamin D metabolism in several hypophosphatemic
diseases, including ADHR, XLH and tumor-induced osteomalacia (TIO)
(5,32,33). The Hyp mouse, an animal
model of XLH, provided evidence of increased levels of FGF23
transcripts due to the inactivated mutations of PHEX.
Furthermore, an injection of FGF23 antibodies or the deletion of
Fgf23 from the Hyp mouse has been shown to ameliorate
or reverse the phosphate metabolic disorders (14,34). These findings strongly indicate
the essential role of FGF23 in the regulation of systemic phosphate
homeostasis in the Hyp mouse. However, we failed to observe
a significant increase in serum FGF23 levels in all affected
individuals compared with the healthy controls. Moreover, 4 of 11
patients with XLH even exhibited serum FGF23 levels below the
reference range. Despite of the effect of confounding factors, the
wide variation in serum FGF23 levels in XLH, as well as the
overlapping FGF23 levels in XLH and healthy controls revealed its
limited diagnostic value in patients with suspected XLH. Moreover,
it was speculated that other factors, such as parathyroid hormone
and 1,25-dihydroxyvitamin-D, were responsible for hypophosphatemia
in patients with XLH with inappropriate low to normal serum FGF23
levels, and for the normal serum phosphorus in healthy subjects
with high serum FGF23 levels (33,34). Additionally, it was suggested that
hypophosphatemia alone was not entirely responsible for the
skeletal phenotype in XLH and the potential direct local effects of
FGF23 may be involved in bone mineralization independent of its
systematic action in phosphate homeostasis (35,36).
In conclusion, we identified 17 different mutations
in the PHEX gene in 18 unrelated Chinese families with
hypophosphatemic rickets and 7 of these were novel. It should be
noted that that 6 of the 17 PHEX mutations have been proven
to be de novo, which suggests the frequent occurrence of
sporadic cases of XLH in the Chinese population. The findings of
the present study highlight the major role of PHEX gene
mutations in hypophosphatemic rickets and emphasize the
significance of genetic diagnosis in suspected cases to ascertain
the clinical diagnosis of XLH, enabling timely intervention.
Further studies are warranted in order to perform more extensive
mutation analyses in affected individuals in order to elaborate the
key domain in PHEX and the identification of unequivocal
FGF23 function in the pathogenesis of XLH, subsequently, exploring
for more effective treatments, not only for XLH, but also for
associated diseases sharing similar molecular mechanism.
Acknowledgments
This study was supported by grants from the National
Natural Science Foundation of China (nos. 81370978 and 81170803 to
Z.-L.Z., and no. 81200646 to J.-M.G.), the National Basic Research
Program of China (no. 2014CB942903), the Shanghai Leading Talents
Award (051) to Z.-L.Z., the Science and Technology Commission of
Shanghai Municipality (no. 14JC140500) to Z.-L.Z., the Shanghai
Municipal Commission of Health and Family Planning (no.
2014ZYJB0009), the Science and Technology Commission of Chongqing
Municipality (no. CSTC2013jcyjC00009) to Z.-L.Z.
References
|
1
|
Albright F, Butler A and Bloomberg E:
Rickets resistant to vitamin D therapy. Am J Dis Child. 54:529–547.
1937.
|
|
2
|
Beck-Nielsen SS, Brock-Jacobsen B, Gram J,
Brixen K and Jensen TK: Incidence and prevalence of nutritional and
hereditary rickets in southern Denmark. Eur J Endocrinol.
160:491–497. 2009. View Article : Google Scholar
|
|
3
|
Carpenter TO: New perspectives on the
biology and treatment of X-linked hypophosphatemic rickets. Pediatr
Clin North Am. 44:443–466. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Francis F, Hennig S, Korn B, Reinhardt R,
De Jong P, Poustka A, Lehrach H, Rowe PSN, Goulding JN, Summerfield
T, et al: The HYP Consortium: A gene (PEX) with homologies to
endopeptidases is mutated in patients with X-linked
hypophosphatemic rickets. Nat Genet. 11:130–136. 1995. View Article : Google Scholar
|
|
5
|
White KE, Evans WE, O'Riordan JLH, Speer
MC, Econs MJ, Lorenz-Depiereux B, Grabowski M, Meitinger T and
Strom TM: ADHR Consortium: Autosomal dominant hypophosphataemic
rickets is associated with mutations in FGF23. Nat Genet.
26:345–348. 2000. View
Article : Google Scholar
|
|
6
|
Feng JQ, Ward LM, Liu S, Lu Y, Xie Y, Yuan
B, Yu X, Rauch F, Davis SI, Zhang S, et al: Loss of DMP1 causes
rickets and osteomalacia and identifies a role for osteocytes in
mineral metabolism. Nat Genet. 38:1310–1315. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li H, Xie H, Liu W, Hu R, Huang B, Tan YF,
Xu K, Sheng ZF, Zhou HD, Wu XP and Luo XH: A novel microRNA
targeting HDAC5 regulates osteoblast differentiation in mice and
contributes to primary osteoporosis in humans. J Clin Invest.
119:3666–3677. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hu R, Liu W, Li H, Yang L, Chen C, Xia ZY,
Guo LJ, Xie H, Zhou HD, Wu XP and Luo XH: A Runx2/miR-3960/miR-2861
regulatory feedback loop during mouse osteoblast differentiation. J
Biol Chem. 286:12328–12339. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li CJ, Cheng P, Liang MK, Chen YS, Lu Q,
Wang JY, Xia ZY, Zhou HD, Cao X, Xie H, et al: MicroRNA-188
regulates age-related switch between osteoblast and adipocyte
differentiation. J Clin Invest. 125:1509–1522. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Du L, Desbarats M, Viel J, Glorieux FH,
Cawthorn C and Ecarot B: cDNA cloning of the murine Pex gene
implicated in X-linked hypophosphatemia and evidence for expression
in bone. Genomics. 36:22–28. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Francis F, Strom TM, Hennig S, Böddrich A,
Lorenz B, Brandau O, Mohnike KL, Cagnoli M, Steffens C, Klages S,
et al: Genomic organization of the human PEX gene mutated in
X-linked dominant hypophosphatemic rickets. Genome Res. 7:573–585.
1997.PubMed/NCBI
|
|
12
|
Ruchon AF, Tenenhouse HS, Marcinkiewicz M,
Siegfried G, Aubin JE, DesGroseillers L, Crine P and Boileau G:
Developmental expression and tissue distribution of Phex protein:
Effect of the Hyp mutation and relationship to bone markers. J Bone
Miner Res. 15:1440–1450. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Nesbitt T, Fujiwara I, Thomas R, Xiao ZS,
Quarles LD and Drezner MK: Coordinated maturational regulation of
PHEX and renal phosphate transport inhibitory activity: Evidence
for the pathophysiological role of PHEX in X-linked
hypophosphatemia. J Bone Miner Res. 14:2027–2035. 1999. View Article : Google Scholar
|
|
14
|
Liu S, Zhou J, Tang W, Jiang X, Rowe DW
and Quarles LD: Pathogenic role of Fgf23 in Hyp mice. Am J Physiol
Endocrinol Metab. 291:E38–E49. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jap TS, Chiu CY, Niu DM and Levine MA:
Three novel mutations in the PHEX gene in Chinese subjects with
hypophosphatemic rickets extends genotypic variability. Calcif
Tissue Int. 88:370–377. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Xia W, Meng X, Jiang Y, Li M, Xing X, Pang
L, Wang O, Pei Y, Yu LY, Sun Y, et al: Three novel mutations of the
PHEX gene in three Chinese families with X-linked dominant
hypophosphatemic rickets. Calcif Tissue Int. 81:415–420. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kang QL, Xu J, Zhang Z, He JW, Lu LS, Fu
WZ and Zhang ZL: Three novel PHEX gene mutations in four Chinese
families with X-linked dominant hypophosphatemic rickets. Biochem
Biophys Res Commun. 423:793–798. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yue H, Yu JB, He JW, Zhang Z, Fu WZ, Zhang
H, Wang C, Hu WW, Gu JM, Hu YQ, et al: Identification of two novel
mutations in the PHEX gene in Chinese patients with
hypophosphatemic rickets/osteomalacia. PLoS One. 9:e978302014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu S, Wei M, Xiao J, Wang CY and Qiu ZQ:
Three PHEX gene mutations in Chinese subjects with hypophosphatemic
rickets and literature review. Zhongguo Dang Dai Er Ke Za Zhi.
16:518–523. 2014.In Chinese. PubMed/NCBI
|
|
20
|
Yang L, Yang J and Huang X: PHEX gene
mutation in a Chinese family with six cases of X-linked
hypophosphatemic rickets. J Pediatr Endocrinol Metab. 26:1179–1183.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yuan L, Wu S, Xu H, Xiao J, Yang Z, Xia H,
Liu A, Hu P, Lu A, Chen Y, et al: Identification of a novel PHEX
mutation in a Chinese family with X-linked hypophosphatemic rickets
using exome sequencing. Biol Chem. 396:27–33. 2015. View Article : Google Scholar
|
|
22
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ng PC and Henikoff S: SIFT: Predicting
amino acid changes that affect protein function. Nucleic Acids Res.
31:3812–3814. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Smith ER, McMahon LP and Holt SG:
Method-specific differences in plasma fibroblast growth factor 23
measurement using four commercial ELISAs. Clin Chem Lab Med.
51:1971–1981. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Sabbagh Y, Boileau G, Campos M, Carmona AK
and Tenenhouse HS: Structure and function of disease-causing
missense mutations in the PHEX gene. J Clin Endocrinol Metab.
88:2213–2222. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Beck-Nielsen SS, Brixen K, Gram J and
Brusgaard K: Mutational analysis of PHEX, FGF23, DMP1, SLC34A3 and
CLCN5 in patients with hypophosphatemic rickets. J Hum Genet.
57:453–458. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rowe PS, Oudet CL, Francis F, Sinding C,
Pannetier S, Econs MJ, Strom TM, Meitinger T, Garabedian M, David
A, et al: Distribution of mutations in the PEX gene in families
with X-linked hypophosphataemic rickets (HYP). Hum Mol Genet.
6:539–549. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Gaucher C, Walrant-Debray O, Nguyen TM,
Esterle L, Garabédian M and Jehan F: PHEX analysis in 118 pedigrees
reveals new genetic clues in hypophosphatemic rickets. Hum Genet.
125:401–411. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Filisetti D, Ostermann G, von Bredow M,
Strom T, Filler G, Ehrich J, Pannetier S, Garnier JM, Rowe P,
Francis F, et al: Non-random distribution of mutations in the PHEX
gene, and under-detected missense mutations at non-conserved
residues. Eur J Hum Genet. 7:615–619. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Qiu ZQ, Tenenhouse HS and Scriver CR:
Parental origin of mutant allele does not explain absence of gene
dose in X-linked Hyp mice. Genet Res. 62:39–43. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Holm IA, Nelson AE, Robinson BG, Mason RS,
Marsh DJ, Cowell CT and Carpenter TO: Mutational analysis and
genotype-phenotype correlation of the PHEX gene in X-linked
hypophosphatemic rickets. J Clin Endocrinol Metab. 86:3889–3899.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Jonsson KB, Zahradnik R, Larsson T, White
KE, Sugimoto T, Imanishi Y, Yamamoto T, Hampson G, Koshiyama H,
Ljunggren O, et al: Fibroblast growth factor 23 in oncogenic
osteomalacia and X-linked hypophosphatemia. N Engl J Med.
348:1656–1663. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Weber TJ, Liu S, Indridason OS and Quarles
LD: Serum FGF23 levels in normal and disordered phosphorus
homeostasis. J Bone Miner Res. 18:1227–1234. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Aono Y, Yamazaki Y, Yasutake J, Kawata T,
Hasegawa H, Urakawa I, Fujita T, Wada M, Yamashita T, Fukumoto S
and Shimada T: Therapeutic effects of anti-FGF23 antibodies in
hypophosphatemic rickets/osteomalacia. J Bone Miner Res.
24:1879–1888. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sapir-Koren R and Livshits G: Bone
mineralization is regulated by signaling cross talk between
molecular factors of local and systemic origin: The role of
fibroblast growth factor 23. Biofactors. 40:555–568. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Sitara D, Kim S, Razzaque MS, Bergwitz C,
Taguchi T, Schüler C, Erben RG and Lanske B: Genetic evidence of
serum phosphate-independent functions of FGF-23 on bone. PLoS
Genet. 4:e10001542008. View Article : Google Scholar : PubMed/NCBI
|