Introduction
Type 2 diabetes has become one of the most rapidly
escalating epidemics and debilitating chronic disease, which is a
major public health challenge. Importantly, over 80% of the
individuals diagnosed with type 2 diabetes are obese (1). A major metabolic consequence of
diabetes and obesity is the abnormal deposition of triglycerides in
hepatocytes (hepatic steatosis) and liver injury, which results in
non-alcoholic fatty liver disease (NAFLD). In addition, diabetes
and obesity are comorbid diseases associated with the dysfunction
of lipid metabolism. Moreover, the prevalence of NAFLD in
individuals with diabetes, obesity and dyslipidaemia is higher
(35–80%) than that in individuals without these conditions
(2).
Abnormal lipid accumulation in hepatocytes leading
to hepatic steatosis is due to two major metabolic perturbations:
augmented hepatic de novo lipogenesis (DNL) and reduced fat
combustion. DNL is an essential mechanism for lipid accumulation in
the liver, which is often closely associated with the inhibition of
AMP-activated protein kinase (AMPK) (3). AMPK, a key energy sensor and
regulator of whole-body metabolism, has been demonstrated to be
tightly involved in the development and disease processes of
obesity and related metabolic disorders (4–6).
Moreover, one critical lipogenic transcription factor is sterol
regulatory element-binding protein 1 (SREBP1), which is primarily
responsible for DNL by regulating genes involved in fatty acid and
triglyceride synthesis, including fatty acid synthase (FAS), acetyl
coenzyme A carboxylase [acetyl-CoA carboxylase (ACC)], and stearoyl
CoA desaturase 1 (SCD1) (7,8).
Studies have shown that silent mating type information regulation 2
homolog 1 (Sirt1) plays a central role in modulating hepatic fatty
acid metabolism through AMPK and SREBP1, that is a critical
mediator of fatty acid combustion and synthesis (9,10).
Although considerable progress has been made in
understanding the molecular mechanisms involved in NAFLD,
satisfactory treatment options for this disease remain limited
(11). Tangshen formula (TSF), a
Chinese herbal formula, has been shown to be capable of treating
diabetic nephropathy, a serious complication of diabetes (12,13). However, whether TSF ameliorates
liver injury in diabetes conditions is unclear. C57BL/KsJ-db/db
mice, which have a mutation in the leptin receptor gene, develop
obesity, hyperglycemia, hyperlipidemia and hepatic steatosis and
also develop type 2 diabetes; thus, these animals constitute a
useful animal model for the study of NAFLD (14). In this study, we aimed to
determined whether TSF attenuates hepatic steatosis, and also iamed
to elucidate the underlying mechanisms using db/db mice. Our
findings reveal novel metabolic activities of TSF in the liver,
which point to the potential use of TSF in the treatment of
NAFLD.
Materials and methods
Herbal materials and the preparation of
TSF
TSF granules were composed of the following herbs:
Astragalus membranaceus (Fisch.) Bge. (Leguminosae, voucher
specimen no. 412303), Euonymus alatus (Thunb.) Siebold
(Celastraceae, voucher specimen no. 1412301), Rehmannia
glutinosa Libosch. (Scrophulariaceae, voucher specimen no.
1411616), Citrus aurantium L. (Rutaceae, voucher specimen
no. 1412304), Cornus officinalis Sieb. et Zuce (Cornaceae,
voucher specimen no. 1410652), Rheum palmatum L.
(Polygonaceae, voucher specimen no. 1412302), and Panax
notoginseng (Burk.) F.H. Chen (Araliaceae, voucher specimen no.
1410004) in the ratio of 10:5:4:3.4:3:2:1 (W/W). The herbs were
prepared and standardized by Jiangyin Tianjiang Pharmaceutical
(Jiangyin, Jiangsu, China). The percentage of powered herb was
determined as follows: the herbal drugs were authenticated and
standardized on marker compounds according to the Chinese
Pharmacopoeia (2010 edition). Each gram of each granule was
equivalent to 12.75 g of the raw herbs. The granules were dissolved
in distilled water (0.18 g/ml) for experimental use.
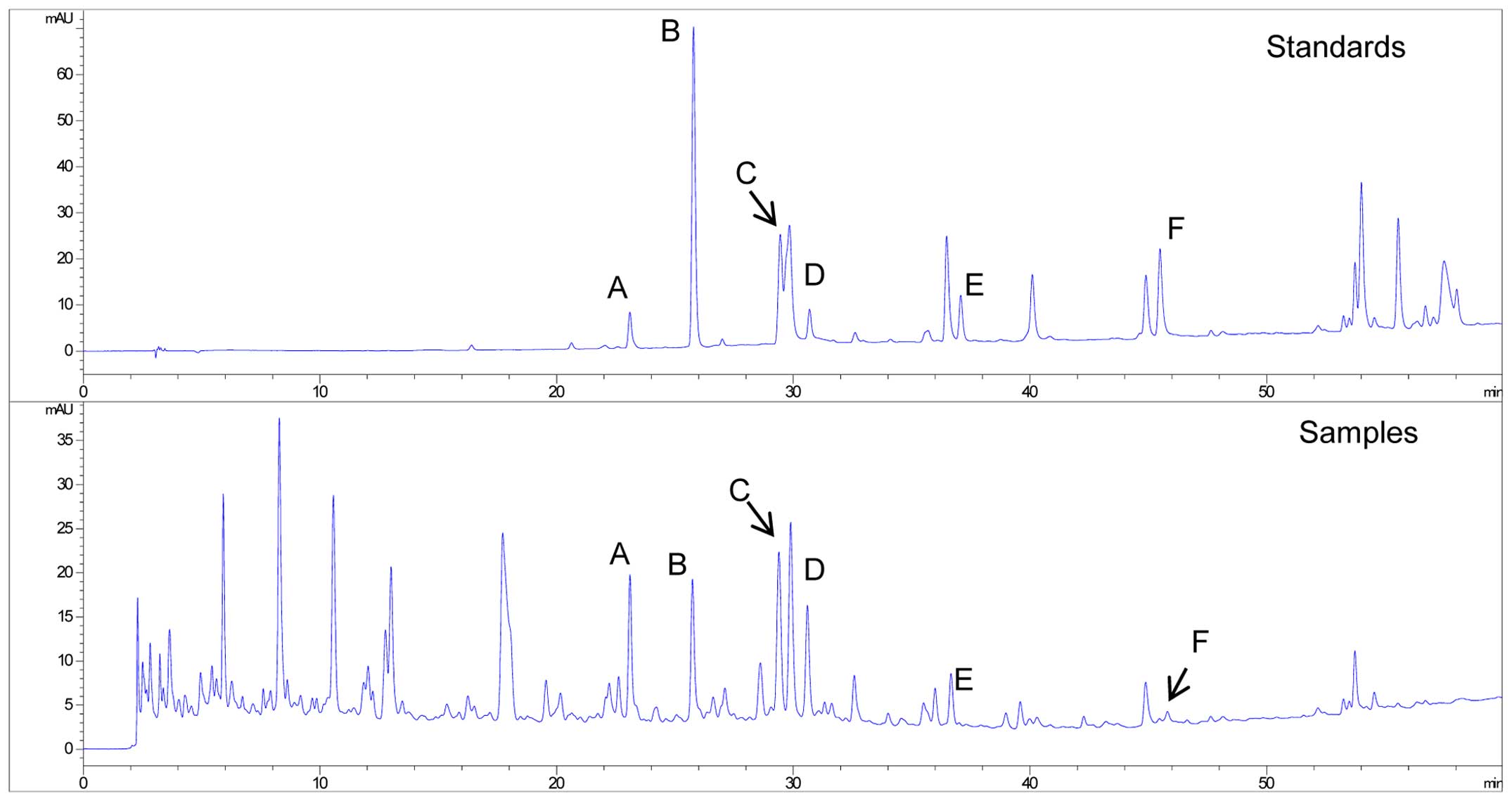
Chromatographic analysis of TSF
TSF was dissolved in distilled water and filtered
through a 0.45 μm filtration membrane prior to being
subjected to high performance liquid chromatography (HPLC)
analyses. HPLC analyses were carried out using an Agilent HPLC
system (Agilent Technologies, Santa Clara, CA, USA).
Chromatographic analysis was performed on an Agilent 1100 system
using a Phenomenex Luna C18 column (4.6×250 mm, 5
μm; Torrance, CA, USA). Oven temperature was maintained at
30°C. Methanol (A) and water containing 0.1% acetic acid (B) served
as the mobile phase. The gradient elution program was as follows:
0–60 min, a linear gradient from 5 to 100% (A). The low rate was
1.0 ml/min and the detection wavelength was set at 254 nm. Pure
standards that were used as external standards during HPLC analysis
(Fig. 1) included loganin
(voucher specimen no. 111640-201005), calycosin-7-O-β-D-glucoside
(voucher specimen no. 111920-201203),
naringenine-7-rhamnosidoglucoside (voucher specimen no.
STA-02106005), neohesperidin (voucher specimen no. 111857-201102),
naringenin (voucher specimen no. STA-02206006) and Aloe-emodin
(voucher specimen no. 110795-201007) and were purchased from the
National Institutes for Food and Drug Control (Beijing, China) and
Shanghai Nature Standard R&D and Biotech (Shanghai, China).
Animals
The study protocol was approved by the Ethics
Committee of the China-Japan Friendship Institute of Clinical
Medical Sciences (Approval no. 13005) and all experiments were
performed in accordance with the NIH Guiding Principles for the
Care and Use of Laboratory Animals. Eight-week-old male C57BLKS/J
db/db and db/m mice (Peking University Laboratory Animal Center,
Beijing, China) were housed at 23±3°C and humidity of 55±15% on a
12-h light-dark cycle and were allowed access to standard chow and
water ad libitum. After 2 weeks of feeding adaptation, the
db/db mice were divided into 2 groups (at 10 weeks of age) as
follows: one that received TSF via intra-gastric gavage (n=9; db/db
+ TSF, 2.4 g/kg/day), another pair-fed group that received saline
instead of TSF (n=9; db/db). The db/m mice were used as controls
(n=9, db/m). The TSF dosage was based on our previous study and
standard conversion formula (body surface areas) (15,16). Body weight was measured weekly
(g). The reducing effects of TSF on body weight are presented as
the 95% confidence level (95% CI) and the top limit/bottom limit of
95% CI = mean ± SE × Tinv (0.05, n-1). The mice were maintained on
TSF treatment for 12 weeks and were then sacrificed under
anesthesia. Animal blood was collected via the tail vein and the
eye socket for further analysis. Liver and skeletal muscles (the
rear part of the left leg) were also collected.
Measurement of serum parameters
All blood samples were collected after overnight
fasting. Serum alanine aminotransferase (ALT), aspartate
transaminase (AST), albumin (ALB), triglycerides and total
cholesterol, low-density lipid cholesterol (LDL-C), high-density
lipid cholesterol (HDL-C) and fasting blood glucose (FBG) levels
were measured using an automatic analyzer (Abbott Diagnostics,
Abbott Park, IL, USA). Fasting blood glucose (FBG) was measured
bi-weekly. Serum free fatty acid (FFA) levels were determined by
enzymatic methods (Meilian Biological, Shanghai, China) according
to the manufacturer's instructions. All measurements were performed
in a blinded manner.
Liver histological and
immunohistochemical analyses
The liver tissues were removed and weighed to
determine the liver index [liver/body weight (LW/BW)]. Liver
samples were fixed in 10% neutral-buffered formalin, embedded in
paraffin, and sectioned into 5-μm-thick slices on slides.
Hematoxylin and eosin (H&E; Beyotime, Jiangsu, China) staining
was performed using standard protocols and scored blindly for the
degree of fatty liver on a scale 0–3, as previously described
(17). For Oil Red O staining,
cryosections of the liver (5-μm-thick) were air-dried for 10
min at room temperature, washed with 60% isopropanol and stained
with fresh Oil Red O working solution (Sigma-Aldrich, St. Louis,
MO, USA). After washing with 60% isopropanol, the sections were
placed under a microscope (Olympus, Tokyo, Japan) to visualize
lipid deposition. All samples were analyzed blindly for overall
pathology using Image-Pro Plus software (Media Cybernetics,
Warrendale, PA, USA). Immunohistochemistry and periodic-acid Schiff
(PAS) staining of liver glycogen was performed as previously
described (18). Briefly, the
livers were fixed in 10% formalin and embedded in paraffin wax.
Paraffin sections were cut and mounted on slides. After
microwave-based heating antigen retrieval, the sections were
stained with antibodies at 4°C overnight, followed by horseradish
peroxidase-conjugated second antibodies (Gene Tech, Shanghai,
China) at room temperature for 1 h and captured using a light
microscope (Olympus). For PAS staining, the sections were stained
with periodic-acid Schiff (Beyotime) for 15 min and hematoxylin for
1 min.
Immunofluorescence staining and confocal
microscopy
Immunofluorescence staining of the liver tissues was
performed under cryoprotection as previously described with minor
modifications (19). The mouse
livers were excised, fixed in 4% paraformaldehyde and 30% sucrose
solution, and processed for embedding with optimal cutting
temperature compound (Sakura Finetek, Tokyo, Japan). Cryosectioning
was performed at −20°C, and the frozen liver tissues were sectioned
into 5-μm-thick slices followed by mounting on glass slides.
For immunofluorescence staining, the sections were air-dried on a
bench for 10 min, and were blocked in phosphate-buffered saline
(PBS) containing 0.1% BSA and incubated overnight at 4°C with an
anti-SREBP1 antibody (ab28481; Abcam, Cambridge, MA, USA) at a
dilution of 1/200 followed by incubation with Alexa-conjugated goat
anti-rabbit secondary antibody (#CA11008s; Invitrogen, Carlsbad,
CA, USA) for 1 h. Following 3 rinses with PBS, the sections were
mounted with PBS with 5 μg/ml of
4′,6-damidino-2-phenylindole dihydrochloride (DAPI; Sigma-Aldrich)
and the specimens were imaged under a Leica DM6000 CS confocal
microscope (Leica Microsystems, Wetzlar, Germany).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA prepared from the mouse tissues was
extracted using TRIzol reagent (Invitrogen) according to the
manufacture's instructions. The RNA concentration of all samples
was quantified using a NanoDrop-1000 spectrophotometer (NanoDrop
Technologies, Wilmington, DE, USA). Total RNA was reverse
transcribed using the Total RevertAid First Strand cDNA synthesis
kit (Thermo Scientific, Waltham, MA, USA) and quantitative PCR
(qPCR) was performed using UltraSYBR Mixture (CWBio, Beijing,
China). The results were normalized to β-actin expression using the
ΔΔC(t) threshold cycle method, as previously described (20). The specific primer sequences are
listed in Table I.
 | Table IList of primers used for RT-qPCR. |
Table I
List of primers used for RT-qPCR.
| Gene | Forward primer | Reverse primer | PCR size (bp) |
|---|
| SREBP1 |
GGAGCCATGGATTGCACATT |
GGCCCGGGAAGTCACTGT | 70 |
| HMGCR |
CTTGTGGAATGCCTTGTGATTG |
AGCCGAAGCAGCACATGAT | 76 |
| Acc1 |
AATGAACGTGCAATCCGATTTG |
ACTCCACATTTGCGTAATTGTTG | 136 |
| FAS |
GCTGCGGAAACTTCAGGAAAT |
AGAGACGTGTCACTCCTGGACTT | 84 |
| SCD1 |
TTCTTCTCTCACGTGGGTTG |
CGGGCTTGTAGTACCTCCTC | 130 |
| Acadm |
AGGGTTTAGTTTTGAGTTGACGG |
CCCCGCTTTTGTCATATTCCG | 110 |
| PGC1α |
TATGGAGTGACATAGAGTGTGCT |
CCACTTCAATCCACCCAGAAAG | 134 |
| PPARα |
AACATCGAGTGTCGAATATGTGG |
CCGAATAGTTCGCCGAAAGAA | 99 |
| Cpt1A |
CTCCGCCTGAGCCATGAAG |
CACCAGTGATGATGCCATTCT | 100 |
| Cpt1B |
GCACACCAGGCAGTAGCTTT |
CAGGAGTTGATTCCAGACAGGTA | 107 |
| Acox1 |
CCGCCACCTTCAATCCAGAG |
CAAGTTCTCGATTTCTCGACGG | 86 |
| Pck1 |
CTGCATAACGGTCTGGACTTC |
GCCTTCCACGAACTTCCTCAC | 87 |
| G6pc |
CGACTCGCTATCTCCAAGTGA |
GTTGAACCAGTCTCCGACCA | 173 |
| LXRα |
ATGTCTTCCCCCACAAGTTCT |
GACCACGATGTAGGCAGAGC | 156 |
| Esrra |
CTCAGCTCTCTACCCAAACGC |
CCGCTTGGTGATCTCACACTC | 168 |
| Tfam |
ATTCCGAAGTGTTTTTCCAGCA |
TCTGAAAGTTTTGCATCTGGGT | 122 |
| LPL |
GGGAGTTTGGCTCCAGAGTTT |
TGTGTCTTCAGGGGTCCTTAG | 115 |
| FAT/CD36 |
AGATGACGTGGCAAAGAACAG |
CCTTGGCTAGATAACGAACTCTG | 83 |
Western blot analysis
The mouse tissues were homogenized in RIPA lysis
buffer supplemented with protease and phosphatase inhibitors
cocktail (Roche Diagnostics, Mannheim, Germany). The protein
concentration was measured using a Bicinchoninic acid assay (BCA)
method (Beijing Solarbio Science and Technology, Beijing, China).
Lysates of 25–50 μg protein were separated by 8–12% sodium
dodecyl sulfate (SDS) gel and transferred onto polyvinylidene
fluoride (PVDF) membranes (Millipore, Billerica, MA, USA). The
membranes were then blocked with 5% BSA in Tris-buffered saline and
Tween-20 (TBST) followed by incubation with primary and secondary
antibodies. The primary antibodies used were against the following
proteins: AMPK (#2532), phosphorylated (p-)AMPK (#2535), ACC
(#3676), p-ACC (#3661), p-Akt (#4060), Akt (#4691),
phosphoinositide 3-kinase (PI3K; #4257), p-mammalian target of
rapamycin (p-mTOR; #2971), mTOR (#2983), Sirt1 (#2028), and SCD1
(#2794) (all from Cell Signaling Technology, Danvers, MA, USA),
SREBP1 (sc8984), peroxisome proliferator-activated receptor (PPAR)α
(sc9000), β-actin (sc69879) (Santa Cruz Biotechnology, Inc., Santa
Cruz, CA, USA), 3′-hydroxylmethyl glutaryl coenzyme A reductase
(HMGCR; ab174830), liver X receptor α (LXRα; ab176323), malonyl-CoA
decarboxylase (MLYCD; ab95945), carnitine palmitoyltransferase 1
(CPT1A; ab128568), PPARγ coactivator 1α (PGC1α; ab54481) (all from
Abcam). The anti-mouse (#115-035-003) or anti-rabbit (#111-035-003)
secondary antibodies were from Jackson ImmunoResearch, West Grove,
PA, USA. Signals were visualized on a ChemiDoc XRS system (Bio-Rad,
Hercules, CA, USA) and the protein bands were quantified by
densitometry using the ImageJ program (National Institutes of
Health, Bethesda, MD, USA).
Statistical analysis
The results are presented as the mean values ± SEM.
Statistical analyses were performed using GraphPad Prism software
version 6.0 (GraphPad Software, La Jolla, CA, USA) by analysis of
one-way variance (ANOVA). Differences were considered significant
at P<0.05.
Results
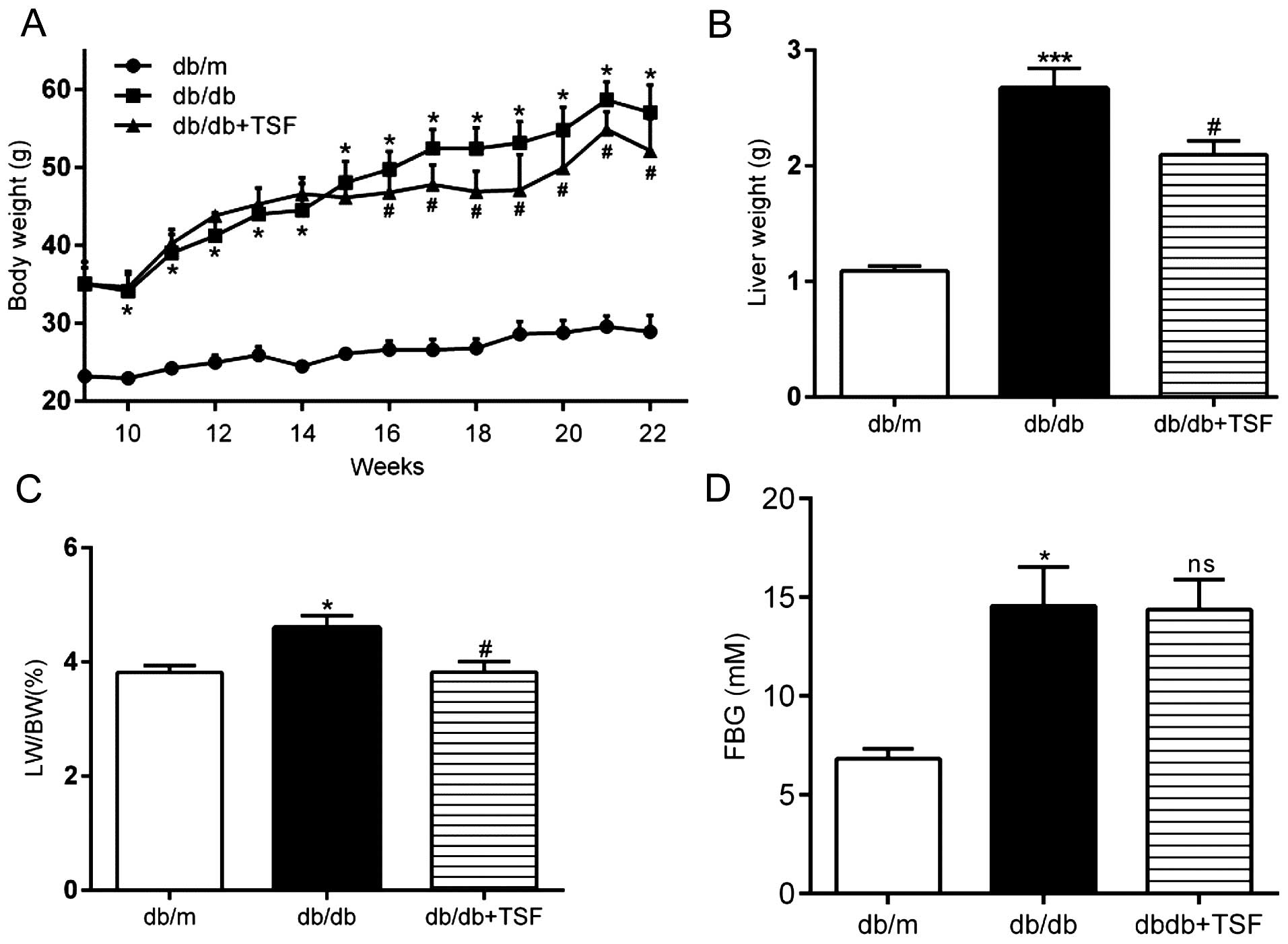
Treatment with TSF diminishes weight gain
and resolves hepatic steatosis in db/db mice
During the experimental period, the db/db mice
exhibited a markedly higher body weight and FBG levels. Treatment
with TSF diminished the increase in body weight, but not in FBG
levels (Fig. 2A and D). An
inhibitory effect of TSF on weight gain in db/db mice was observed
and this reached statistical significant as early as the 16th week
(Fig. 2A). There was a 19.36%
(95% CI, 33.67–5.05%) reduction in weight gain in the db/db mice
treated with TSF at 22 weeks. The increase in liver weight and the
liver index of the db/db mice was reduced in the mice treated with
TSF at the time of sacrifice (Fig. 2B
and C). The serum enzyme, triglycerides, total cholesterol,
LDL-C and FFA levels were also significantly increased in the db/db
mice as compared with the db/m mice, and these effects were
inhibited by TSF. However, HDL-C was modestly increased in the
db/db mice, but was not restored by treatment with TSF (Table II).
| Table IIBlood parameters of the mice in the
study groupsa. |
Table II
Blood parameters of the mice in the
study groupsa.
| db/m | db/db | db/db+TSF |
|---|
| ALT (U/l) | 52.00±4.12 |
192.00±23.87b | 96.57±9.78c |
| AST (U/l) | 143.6±4.18 | 216.2±19.52b | 149.4±5.83c |
| ALB (g/l) | 42.60±0.95 | 42.80±1.36 | 42.73±1.17 |
| Cholesterol
(mmol/l) | 2.60±0.13 | 4.19±0.11b | 3.20±0.17c |
| Triglyceride
(mmol/l) | 0.65±0.03 | 1.05±0.05b | 0.93±0.01c |
| HDL-C (mmol/l) | 2.49±0.16 | 3.43±0.10b | 3.42±0.11 |
| LDL-C (mmol/l) | 0.80±0.05 | 1.39±0.04b | 1.05±0.03c |
| FFA (mmol/l) | 0.74±0.03 | 1.07±0.05b | 0.89±0.03c |
In consonance with the biochemical data, the db/db
mice that received TSF exhibited a significant reduction in
histological steatosis. The histopathological evaluation of
formalin-fixed, H&E stained liver slides revealed clear
vesicular steatosis in the livers of the db/db mice compared with
those of the TSF-treated mice (Fig.
2E). Oil Red O staining performed on the cryosections of liver
tissues demonstrated that treatment with TSF alleviated the
accumulation of triglycerides (Fig.
2E).
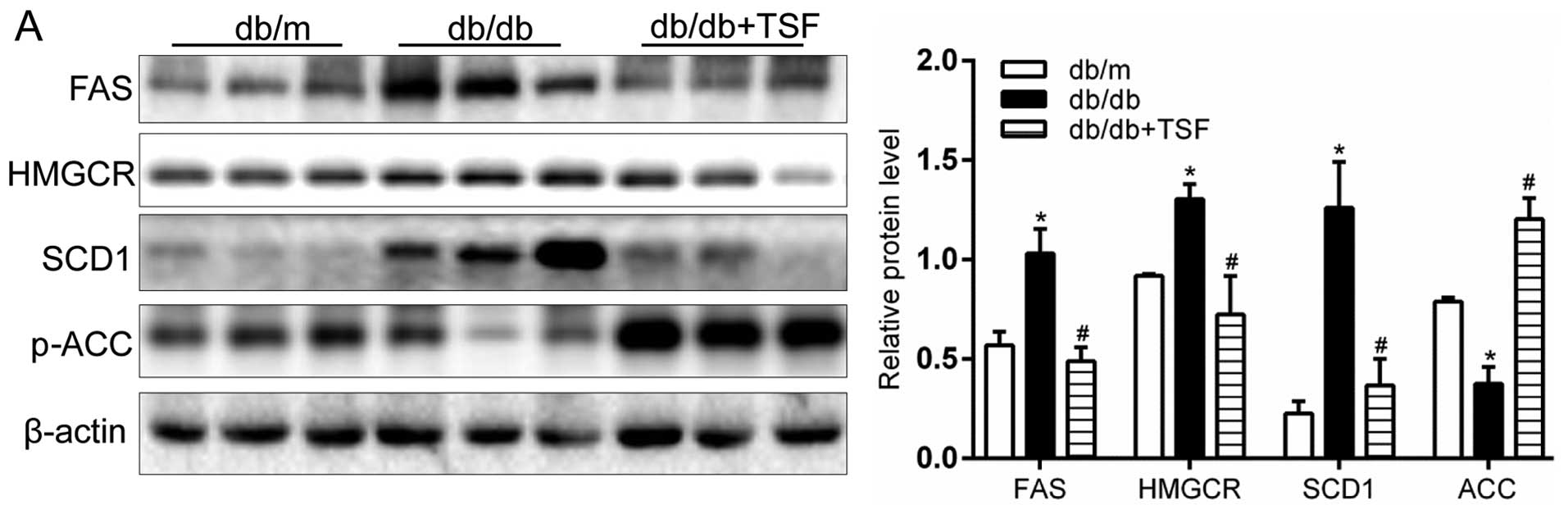
TSF inhibits the expression of hepatic
lipogenic enzymes in the livers of db/db mice and these effects are
mediated by AMPK/SREBP1
FAS, ACC and SCD1, the key targets of SREBP1, are
key enzymes regulating cellular lipogenesis and lipid homeostasis
(21). The results of western
blot analysis and RT-qPCR indicated that the administration TSF
markedly decreased the protein and mRNA levels of FAS and SCD1
(enzymes involved in fatty acid and triglyceride synthesis) in the
livers of db/db mice (Fig. 3A and
C). Similarly, the levels of the key enzyme of cholesterol
biosynthesis, HMGCR, was prominently inhibited by TSF in the livers
of db/db mice (Fig. 3A and C).
Moreover, the decreased protein expression of FAS and HMGCR, the
two essential lipogenic enzymes, was further confirmed by
immunohistochemical analysis of the hepatic sections (Fig. 3B). In addition, the mRNA level and
protein expression of p-ACC, an AMPK downstream target, was
decreased in the db/db mice its levels were and restored by
treatment with TSF (Fig. 3A).
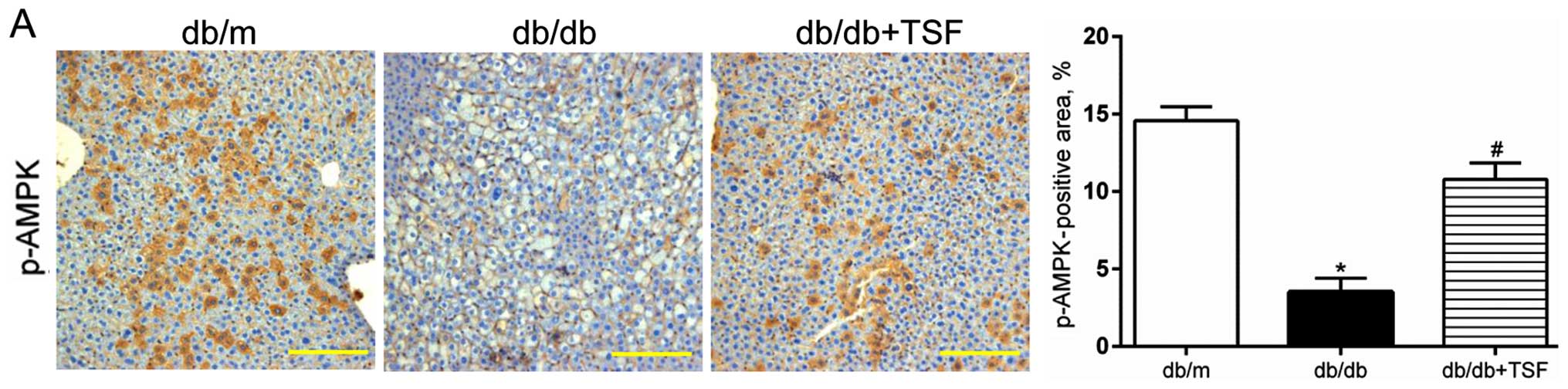
It has been shown that the Thr172 phosphorylation of
AMPK (p-AMPK) blunts the cleavage of the protein of SREBP1 and
inhibits its nuclear translocation, which results in the
amelioration of hepatic steatosis (22,23). In this study, we observed a
decrease in the level of phosphorylation of AMPK in the livers of
db/db mice, and this was substantially restored by treatment with
TSF (Fig. 4B). Moreover, the
increased expression of p-AMPK induced by TSF was further confirmed
by immunohistochemical analysis of the liver sections (Fig. 4A). We then examined the effect of
TSF on SREBP1 activity, which is reflected by the amounts of
cleavage and nuclear translocation. As shown in Fig. 4B and C, the accumulation of
nuclear SREBP1 in the livers of db/db mice was markedly reduced by
TSF. The staining intensity of the nuclear active forms of SREBP1,
in contrast, was largely reduced in the TSF-treated mice (Fig. 4C). The mRNA level of SREBP1 was
further determined by RT-qPCR, and this was also shown to be
significantly increased in the db/db mice and was decreased by TSF
(Fig. 4B). These data indicate
that TSF attenuates hepatic steatosis, at least in part by the
inhibition of SREBP1 and reducing its target gene
transcription.
TSF regulates enzymes and genes involved
in lipid combustion and mitochondrial biogenesis in the liver and
skeletal muscle of db/db mice
We then examined the metabolic genes involved in
lipid combustion and found that TSF significantly increased the
expression of Sirt1 in the livers of db/db mice (Fig. 5A). Existing evidence indicates
that hepatic Sirt1 regulates lipid homeostasis by positively
regulating PPARα (24).
Therefore, we hypothesized that TSF exerts its 'anti-steatosis'
effect in part by augmenting Sirt1-driven PPARα upregulation in the
fatty liver. We found that treatment with TSF increased the
expression of PPARα, as well as that of PGC1α in the livers of
db/db mice. Of note, we observed that the levels of LXRα were
decreased by TSF treatment (Fig.
5A). which was consistent with the findings of previous
studies; namely that the activation of LXRα induces the
transcription of the lipogenic genes, SREBP1, FAS and ACC (25,26).
Malonyl-CoA, the substrate of fatty acid synthesis,
is also an inhibitor of CPT1, which regulates fatty acid oxidation
(27). MLYCD, which generates
acetyl-CoA from malonyl-CoA, has been shown to be activated by AMPK
and PPARα (28). In the present
study, the administration of TSF clearly increased the MLYCD and
CPT1A levels in the livers of db/db mice (Fig. 5B). Moreover, the mRNA expression
levels of genes that were assayed by RT-qPCR were consistent with
the protein expression results (Fig.
5C). Treatment with TSF also increased the expression of genes
involved in fatty acid oxidation, such as medium-chain acyl-CoA
dehydrogenase (Acadm) and acyl-CoA oxidase (Acox1) (Fig. 5C). Peripheral fatty acids
contribute to the accumulation of hepatic triglyceride in NAFLD;
lipoprotein lipase (LPL) and the fatty acid translocase CD36
(FAT/CD36) are involved in the transport of fatty acids to the
liver (29). Accordingly, we
detected a significant downregulation in the levels of both LPL and
FAT/CD36 in the livers of db/db mice treated with TSF (Fig. 5D).
Sirt1 and AMPK are critical regulators in metabolic
tissues, particularly in skeletal muscle as energy sensors
(30). We also found that TSF
increased the expression of hepatic Sirt1 and p-AMPK, as well as
that of their target proteins, PGC1α and MLYCD, that was in keeping
with similar findings in the skeletal muscle of db/db mice
(Fig. 6A and B). In addition, TSF
markedly enhanced the expression of genes involved in fatty acid
oxidation, such as Acadm and CPT1B (Fig. 6C). In addition, mitochondrial
biogenesis was enhanced in skeletal muscle, as reflected by the
upregulation of genes and mitochondrial transcription factor A
(Tfam) and estrogen-related receptor-α (Esrra) (Fig. 6D).
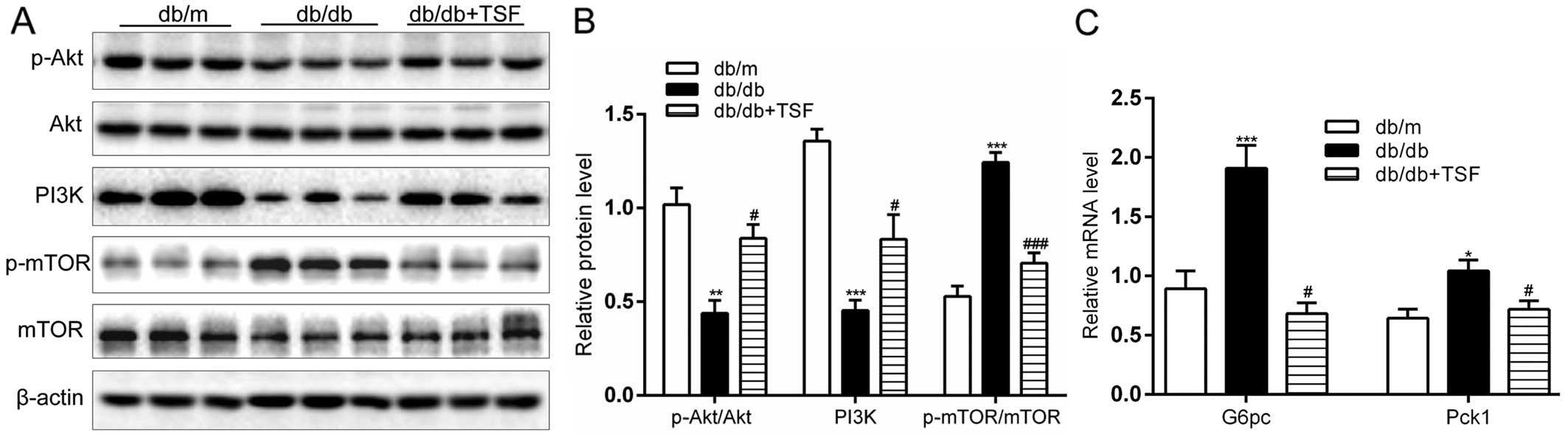
Effect of TSF on the PI3K/Akt/mTOR
cascade and gluconeogenesis in the livers of db/db mice
The expression of PI3K and the phosphorylation of
Akt were significantly enhanced in the db/db mice that were
administered TSF. An important finding is that treatment with TSF
caused a marked decrease in the expression of p-mTOR (Fig. 7A and B). Moreover, the
administration of TSF substantially decreased the expression of
genes involved in hepatic gluconeogenesis, such as
glucose-6-phosphatase (G6pc) and phosphoenolpyruvate carboxykinase
1 (Pck1) (Fig. 7C). In
concordance with these findings, we found that strong hepatic
glycogen accumulated in the db/db mice treated with TSF (Fig. 7D). These results point to a
participation of these genes in the regulation of glucose and lipid
homeostasis by TSF in NAFLD.
Discussion
Our study provides evidence that TSF diminishes body
weight gain, reduces lipid accumulation in the liver and improves
lipid profiles in db/db mice. TSF stimulates AMPK activity and
blocks SREBP1 transcriptional and cleavage activity, which results
in a variety of metabolic effects. These include the reduced
expression of genes involved in fatty acid combustion and the
increased expression of genes involved in lipogenesis. Most
importantly, these data are in keeping with our findings that TSF
prominently blunts hepatic lipogenesis and gluconeogenesis,
enhances fatty acid oxidation and regulates energy balance in
skeletal muscle.
Hepatic lipid accumulation is the most prominent
characteristic of NAFLD in various stages, involving DNL, lipid
transport and oxidation pathways in the liver. In this study, in
both the liver and skeletal tissues, alterations in lipid
metabolism by TSF were accompanied by AMPK activation, which leads
to energy-consuming pathways, such as fatty acid synthesis and
gluconeogenesis, being inhibited, and energy-generating pathways,
such as fatty acid oxidation and glycolysis, being activated.
Additionally, TSF increased the gene expression of AdipoR1 and
AdipoR2 in the liver, reflecting that the level of adiponectin was
elevated. Adiponectin, an 'anti-diabetic' adipokine, has been
reported to have beneficial effects on lipid metabolism via AMPK
and PPARα (31,32).
AMPK also regulates the activity of ACC, a well
characterized target gene of AMPK and SREBP1, by modulating its
inhibitory phosphorylation (33).
ACC is an essential lipogenic enzyme that blunts FFA oxidation and
initiates DNL by increasing the levels of malonyl-CoA, an inhibitor
of CPT1. CPT1 is an crucial enzyme that controls the flow of fatty
acid to mitochondrial β-oxidation (34). Our data demonstrated that TSF
markedly activated the AMPK-mediated the phosphorylation of ACC. In
addition, TSF decreased the expression of target genes of AMPK or
SREBP1 (FAS, SCD1 and HMGCR), which demonstrated a significant
reduction in both the mRNA and protein levels. These data
demonstrate that TSF activated the phosphorylation of AMPK and
suppressed the nuclear translocation of SREBP1 in the liver, thus
blunting the genes involved in lipogenesis by translational
modifications.
AMPK regulates SREBP1 activity and inhibits
lipogenesis in part via the inhibition of mTOR in the fatty liver
(19,35). mTOR signaling has been shown to
regulate lipid metabolic processes, including DNL and triglyceride
synthesis (36,37). With overfeeding and obesity, liver
mTOR activity is elevated, leading to gluconeogenesis and
lipogenesis (38). We found that
the expression of p-mTOR was significantly enhanced in the db/db
mice and was suppressed by the administration of TSF. More
importantly, we demonstrated that impaired hepatic PI3K/Akt
signaling caused by obesity in db/db mice was restored by TSF,
reflecting that TSF enhanced insulin sensitivity in NAFLD.
Increased glycogen accumulation and the decreased gene expression
of gluconeogenesis in the liver were in accordance with this
observation. Notably, the plasma glucose levels were not decreased
in the db/db mice treated with TSF. These results indicate that TSF
promotes hepatic glycogen gathered mainly from other pathways, but
not plasma glucose. Hepatic steatosis is commonly associated with
obesity, insulin resistance and hyperlipidemia (39). We found that treatment with TSF
enhanced insulin sensitivity in fatty liver, which may partially be
due to changes in hepatic lipid profiles. Taken together, these
findings suggest that TSF exerts its beneficial effects on hepatic
steatosis and insulin resistance related to obesity, at least
partially through the regulation of the AMPK/mTOR/SREBP1 axis.
As is known, hepatic lipid accumulation of NAFLD is
closely related to lipid transport. This metabolic process may be
mediated partially by the regulation of FAT/CD36 and LPL (40). The overexpression of FAT/CD36
increases fatty acid and triglyceride storage in the livers of
high-fat induced obesity mice (41). In this study, we found that
treatment with TSF decreased the expression of the FAT/CD36 and LPL
genes, indicating that TSF attenuated hepatic steatosis, in part by
regulating liver lipid transport.
It should be noted that ameliorating hepatic
steatosis by aggravating fatty acid oxidation is an alternative
strategy. In our experiments, we found that several genes involved
in FFA oxidation were mediated in the liver and skeletal muscle by
treatment with TSF. Sirt1 has been implicated in modulating
cellular metabolism and facilitating fatty acid utilization
followed by eliminating fat ectopic accumulation (30). The mechanisms through which Sirt1
activation attenuates hepatic steatosis include the inactivation of
SREBP1 and the activation of PGC1α. The latter pathway may increase
MLYCD expression, a target of PPARα (42). Additionally, PPARα is the common
target gene of AMPK and Sirt1, and is centrally involved in fatty
acid β-oxidation (43). Finally,
Sirt1 leads to decreased hepatic malonyl-CoA, accelerates FFA
oxidation and attenuates hepatic steatosis. Moreover, Sirt1
promotes mitochondrial biogenesis via the activation of PGC1α, by
which FFA oxidation and mitochondrial biogenesis are driven. In
this study, we found that TSF markedly increased Sirt1, PGC1α and
PPARα expression in the liver and skeletal muscle of db/db mice.
Accordingly, mitochondrial biogenesis was enhanced by TSF,
reflected by the upregulated expression of genes involved in
mitochondrial function (Tfam and Esrra). These findings demonstrate
that TSF activated the Sirt1/PPARα/MLYCD pathway and improved CPT1
activity to enhance the mitochondrial oxidation of fatty acid.
In conclusion, the present study demonstrates that
TSF improved lipid profiles and attenuated hepatic steatosis in
db/db mice. The inhibition of lipogenesis and the augmentation of
fatty acid oxidation in the liver and skeletal muscle may be the
underlying mechanisms throught which TSF improves
diabetes-associated liver injury.
Acknowledgments
This study was funded by the National Natural
Science Foundation of China (grant nos. 81130066 and 81473526); the
International S&T Cooperation Program of China (grant no.
2011DFA31860); the Beijing Municipal Science and Technology Program
(grant no. Z151100003815015). The authors would like to thank Ms.
(Pamir Communications, Daly City, CA, USA) Nissi S. Wang for
providing editorial assistance with the manuscript.
References
|
1
|
Smyth S and Heron A: Diabetes and obesity:
The twin epidemics. Nat Med. 12:75–80. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Loomba R and Sanyal AJ: The global NAFLD
epidemic. Nat Rev Gastroenterol Hepatol. 10:686–690. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ruderman NB, Carling D, Prentki M and
Cacicedo JM: AMPK, insulin resistance, and the metabolic syndrome.
J Clin Invest. 123:2764–2772. 2013. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Fu L, Bruckbauer A, Li F, Cao Q, Cui X, Wu
R, Shi H, Zemel MB and Xue B: Interaction between metformin and
leucine in reducing hyperlipidemia and hepatic lipid accumulation
in diet-induced obese mice. Metabolism. 64:1426–1434. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lian Z, Li Y, Gao J, Qu K, Li J, Hao L, Wu
S and Zhu H: A novel AMPK activator, WS070117, improves lipid
metabolism discords in hamsters and HepG2 cells. Lipids Health Dis.
10:672011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lee JH, Jung JY, Jang EJ, Jegal KH, Moon
SY, Ku SK, Kang SH, Cho IJ, Park SJ, Lee JR, et al: Combination of
honokiol and magnolol inhibits hepatic steatosis through
AMPK-SREBP-1 c pathway. Exp Biol Med (Maywood). 240:508–518. 2015.
View Article : Google Scholar
|
|
7
|
Jeon TI and Osborne TF: SREBPs: Metabolic
integrators in physiology and metabolism. Trends Endocrinol Metab.
23:65–72. 2012. View Article : Google Scholar :
|
|
8
|
Han J, Li E, Chen L, Zhang Y, Wei F, Liu
J, Deng H and Wang Y: The CREB coactivator CRTC2 controls hepatic
lipid metabolism by regulating SREBP1. Nature. 524:243–246. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ponugoti B, Kim DH, Xiao Z, Smith Z, Miao
J, Zang M, Wu SY, Chiang CM, Veenstra TD and Kemper JK: SIRT1
deacetylates and inhibits SREBP-1C activity in regulation of
hepatic lipid metabolism. J Biol Chem. 285:33959–33970. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Hou X, Xu S, Maitland-Toolan KA, Sato K,
Jiang B, Ido Y, Lan F, Walsh K, Wierzbicki M, Verbeuren TJ, et al:
SIRT1 regulates hepatocyte lipid metabolism through activating
AMP-activated protein kinase. J Biol Chem. 283:20015–20026. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Musso G, Gambino R and Cassader M:
Emerging molecular targets for the treatment of nonalcoholic fatty
liver disease. Annu Rev Med. 61:375–392. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zhang H, Li P, Burczynski FJ, Gong Y, Choy
P, Sha H and Li J: Attenuation of diabetic nephropathy in Otsuka
Long-Evans Tokushima Fatty (OLETF) rats with a combination of
Chinese Herbs (Tangshen Formula). Evid Based Complement Alternat
Med. 2011:6137372011. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Huang M, Z C, Liang QL, Li P, Li J, Wang
YM and Luo GA: Effect of Tangshen Formula on phospholipids
metabolism in diabetic nephropathy patients. Acta Pharmacol Sin.
46:780–786. 2011.
|
|
14
|
Tiniakos DG, Vos MB and Brunt EM:
Nonalcoholic fatty liver disease: Pathology and pathogenesis. Annu
Rev Pathol. 5:145–171. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li P, Chen Y, Liu J, Hong J, Deng Y, Yang
F, Jin X, Gao J, Li J, Fang H, et al: Efficacy and safety of
tangshen formula on patients with type 2 diabetic kidney disease: A
multicenter double-blinded randomized placebo-controlled trial.
PLoS One. 10:e01260272015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reagan-Shaw S, Nihal M and Ahmad N: Dose
translation from animal to human studies revisited. FASEB J.
22:659–661. 2008. View Article : Google Scholar
|
|
17
|
Brunt EM, Janney CG, Di Bisceglie AM,
Neuschwander-Tetri BA and Bacon BR: Nonalcoholic steatohepatitis: A
proposal for grading and staging the histological lesions. Am J
Gastroenterol. 94:2467–2474. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sun SF, Zhao TT, Zhang HJ, Huang XR, Zhang
WK, Zhang L, Yan MH, Dong X, Wang H, Wen YM, et al: Renoprotective
effect of berberine on type 2 diabetic nephropathy in rats. Clin
Exp Pharmacol Physiol. 42:662–670. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Quan HY, Kim Y, Kim SJ, Jo HK, Kim GW and
Chung SH: Betulinic acid alleviates non-alcoholic fatty liver by
inhibiting SREBP1 activity via the AMPK-mTOR-SREBP signaling
pathway. Biochem Pharmacol. 85:1330–1340. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Schmittgen TD and Livak KJ: Analyzing
real-time PCR data by the comparative C(T) method. Nat Protoc.
3:1101–1108. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Castaño D, Larequi E, Belza I, Astudillo
AM, Martínez-Ansó E, Balsinde J, Argemi J, Aragon T, Moreno-Aliaga
MJ, Muntane J, et al: Cardiotrophin-1 eliminates hepatic steatosis
in obese mice by mechanisms involving AMPK activation. J Hepatol.
60:1017–1025. 2014. View Article : Google Scholar
|
|
22
|
Jung EJ, Kwon SW, Jung BH, Oh SH and Lee
BH: Role of the AMPK/SREBP-1 pathway in the development of orotic
acid-induced fatty liver. J Lipid Res. 52:1617–1625. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Li Y, Xu S, Mihaylova MM, Zheng B, Hou X,
Jiang B, Park O, Luo Z, Lefai E, Shyy JY, et al: AMPK
phosphorylates and inhibits SREBP activity to attenuate hepatic
steatosis and atherosclerosis in diet-induced insulin-resistant
mice. Cell Metab. 13:376–388. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Purushotham A, Schug TT, Xu Q, Surapureddi
S, Guo X and Li X: Hepatocyte-specific deletion of SIRT1 alters
fatty acid metabolism and results in hepatic steatosis and
inflammation. Cell Metab. 9:327–338. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Grefhorst A, Elzinga BM, Voshol PJ, Plösch
T, Kok T, Bloks VW, van der Sluijs FH, Havekes LM, Romijn JA,
Verkade HJ and Kuipers F: Stimulation of lipogenesis by
pharmacological activation of the liver X receptor leads to
production of large, triglyceride-rich very low density lipoprotein
particles. J Biol Chem. 277:34182–34190. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Sim WC, Park S, Lee KY, Je YT, Yin HQ,
Choi YJ, Sung SH, Park SJ, Park HJ, Shin KJ and Lee BH: LXR-α
antagonist meso-dihydroguaiaretic acid attenuates high-fat
diet-induced nonalcoholic fatty liver. Biochem Pharmacol.
90:414–424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Foster DW: Malonyl-CoA: the regulator of
fatty acid synthesis and oxidation. J Clin Invest. 122:1958–1959.
2012. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Derdak Z, Villegas KA, Harb R, Wu AM,
Sousa A and Wands JR: Inhibition of p53 attenuates steatosis and
liver injury in a mouse model of non-alcoholic fatty liver disease.
J Hepatol. 58:785–791. 2013. View Article : Google Scholar :
|
|
29
|
Wang C, Hu L, Zhao L, Yang P, Moorhead JF,
Varghese Z, Chen Y and Ruan XZ: Inflammatory stress increases
hepatic CD36 translational efficiency via activation of the mTOR
signalling pathway. PLoS One. 9:e1030712014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Price NL, Gomes AP, Ling AJ, Duarte FV,
Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro
JS, et al: SIRT1 is required for AMPK activation and the beneficial
effects of resveratrol on mitochondrial function. Cell Metab.
15:675–690. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yamauchi T, Kamon J, Waki H, Imai Y,
Shimozawa N, Hioki K, Uchida S, Ito Y, Takakuwa K, Matsui J, et al:
Globular adiponectin protected ob/ob mice from diabetes and
ApoE-deficient mice from atherosclerosis. J Biol Chem.
278:2461–2468. 2003. View Article : Google Scholar
|
|
32
|
Yamauchi T, Kamon J, Minokoshi Y, Ito Y,
Waki H, Uchida S, Yamashita S, Noda M, Kita S, Ueki K, et al:
Adiponectin stimulates glucose utilization and fatty-acid oxidation
by activating AMP-activated protein kinase. Nat Med. 8:1288–1295.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Moreno-Aliaga MJ, Pérez-Echarri N,
Marcos-Gómez B, Larequi E, Gil-Bea FJ, Viollet B, Gimenez I,
Martínez JA, Prieto J and Bustos M: Cardiotrophin-1 is a key
regulator of glucose and lipid metabolism. Cell Metab. 14:242–253.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
McGarry JD, Mannaerts GP and Foster DW: A
possible role for malonyl-CoA in the regulation of hepatic fatty
acid oxidation and ketogenesis. J Clin Invest. 60:265–270. 1977.
View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Zhang J, Zhang LN, Chen DM, Fu YY, Zhang
F, Yang LL, Xia CM, Jiang HW, Tang CL, Xie ZF, et al:
2-(3-Benzoylthioureido)-4,5,6,7-tetrahydrobenzo[b]thiophene-3-carboxylic
acid ameliorates metabolic disorders in high-fat diet-fed mice.
Acta Pharmacol Sin. 36:483–496. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Lamming DW and Sabatini DM: A Central role
for mTOR in lipid homeostasis. Cell Metab. 18:465–469. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Soliman GA: The integral role of mTOR in
lipid metabolism. Cell Cycle. 10:861–862. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yecies JL, Zhang HH, Menon S, Liu S,
Yecies D, Lipovsky AI, Gorgun C, Kwiatkowski DJ, Hotamisligil GS,
Lee CH and Manning BD: Akt stimulates hepatic SREBP1c and
lipogenesis through parallel mTORC1-dependent and independent
pathways. Cell Metab. 14:21–32. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Birkenfeld AL and Shulman GI: Nonalcoholic
fatty liver disease, hepatic insulin resistance, and type 2
diabetes. Hepatology. 59:713–723. 2014. View Article : Google Scholar :
|
|
40
|
Donnelly KL, Smith CI, Schwarzenberg SJ,
Jessurun J, Boldt MD and Parks EJ: Sources of fatty acids stored in
liver and secreted via lipoproteins in patients with nonalcoholic
fatty liver disease. J Clin Invest. 115:1343–1351. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Koonen DP, Jacobs RL, Febbraio M, Young
ME, Soltys CL, Ong H, Vance DE and Dyck JR: Increased hepatic CD36
expression contributes to dyslipidemia associated with diet-induced
obesity. Diabetes. 56:2863–2871. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Lee GY, Kim NH, Zhao ZS, Cha BS and Kim
YS: Peroxisomal-proliferator-activated receptor alpha activates
transcription of the rat hepatic malonyl-CoA decarboxylase gene: A
key regulation of malonyl-CoA level. Biochem J. 378:983–990. 2004.
View Article : Google Scholar
|
|
43
|
Zhu LH, Wang A, Luo P, Wang X, Jiang DS,
Deng W, Zhang X, Wang T, Liu Y, Gao L, et al: Mindin/Spondin 2
inhibits hepatic steatosis, insulin resistance, and obesity via
interaction with peroxisome proliferator-activated receptor α in
mice. J Hepatol. 60:1046–1054. 2014. View Article : Google Scholar : PubMed/NCBI
|