Introduction
Lung cancer is one of the most malignant cancers,
and the incidence of lung cancer has been rising steadily during
the past few decades (1,2). Despite recent advances in the
treatment of lung cancer, the molecular mechanisms underlying lung
cancer remain unclear and the prognosis for patients with advanced
lung cancer remains dismal.
Increasing lines of evidence suggest that
epithelial-mesenchymal transition (EMT) plays an important role in
cancer progression, invasion and metastasis (3–5).
It is defined by the loss of epithelial characteristics and the
acquisition of a motile, invasive and migratory mesenchymal
phenotype (6). Moreover,
transforming growth factor (TGF)-β1 is a multifunctional cytokine
and is involved in many biological processes including cell
proliferation, differentiation and migration (7). Previous studies have demonstrated
that TGF-β1 induces EMT to promote lung adenocarcinoma invasion and
metastasis. Therefore, inhibition of TGF-β1 signaling
pathway-mediated EMT may yield beneficial therapeutic effects for
cancer patients with advanced metastasis.
Hematopoietic pre-B-cell leukemia transcription
factor (PBX)-interacting protein (HPIP/PBXIP1), a corepressor of
the transcription factor PBX, is involved in organogenesis and
tumorigenesis. Feng et al reported that HPIP is upregulated
in gastric cancer and induces gastric cancer cell migration and
invasion, and modulates EMT (8).
HPIP was also found to be overexpressed in astrocytoma and to
promote proliferation and migration of astrocytoma cells (9). However, the role of HPIP in lung
cancer is unclear. Thus, in the present study, we investigated the
role of HPIP in TGF-β1-induced EMT in A549 lung cancer cells in
vitro.
Materials and methods
Cell culture and treatment
Lung cancer cell lines (A549, 95D and H1299) and one
normal human bronchial epithelial (HBE) cell line were obtained
from the American Type Culture Collection (ATCC; Manassas, VA, USA)
and cultured in Roswell Park Memorial Institute (RPMI)-1640 medium
supplemented with 10% heat-inactivated fetal bovine serum (FBS)
under a humidified incubator that was maintained at 37°C and
supplied with 5% CO2 and 95% air. Then, A549 cells were
treated with TGF-β1 (10 ng/ml) for different periods of time.
RNA preparation and reverse transcription
quantitative PCR (RT-qPCR)
Total RNA was extracted from cells with TRIzol
reagent (Invitrogen, Carlsbad, CA, USA) following the
manufacturer's instructions, and 1 µg of each was used for
reverse transcription using the iScript cDNA Synthesis kit (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The primer sequences used
are as follows: HPIP forward, 5′-GTCCCCTCGAGGAGTTGTGT-3′ and
reverse, 5′-ATCTTCCATCATCTGAGGGC-3′; β-actin forward,
5′-CATGTACGTTGCTATCCAGGC-3′ and reverse,
5′-CTCCTTAATGTCACGCACGAT-3′. The steps for PCR were performed as
follows: 35 cycles of denaturation at 94°C for 30 sec, annealing at
54°C for 30 sec, and polymerization at 72°C for 30 sec. β-actin was
used as a control for normalizing gene expression. The data
obtained were calculated by the 2−ΔΔCt method.
Western blotting
Cells were homogenized and lysed with RIPA lysis
buffer (100 mM NaCl, 50 mM Tris-HCl pH 7.5, 1% Triton X-100, 1 mM
EDTA, 10 mM β-glycerophosphate, 2 mM sodium vanadate and protease
inhibitor). The protein concentration was measured using a BCA
protein assay kit. Equal amount of protein was separated by sodium
dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and
then transferred to a polyvinylidene fluoride (PVDF) membrane. The
membrane was blocked with 5% non-fat milk solution for 1 h, and
then was incubated with a primary mouse monoclonal antibody against
HPIP (1:3,000; H00057326-M02; Abnova, Walnut, CA, USA), E-cadherin
(1:1,500; sc-71008), N-cadherin (1:2,000; sc-53488) (both from
Santa Cruz Biotechnology, Inc., Santa Cruz, CA, USA), and rabbit
monoclonal antibody against Smad2 (1:2,000; sc-101153) or p-Smad2
(1:2,000; sc-101801) (both from Santa Cruz Biotechnology, Inc.) at
4°C overnight. GAPDH (1:1,500; sc-166574; Santa Cruz Biotechnology,
Inc.) was used as the loading control. Following three washes with
TBST buffer, HRP-conjugated secondary antibodies [rabbit anti-mouse
HRP-conjugated secondary antibody (1:3,000; sc-358923) or goat
anti-rabbit horseradish peroxidase conjugated secondary antibody
(1:3,000; sc-2054); both from Santa Cruz Biotechnology, Inc.] were
introduced, and enhanced chemiluminescence (ECL; Amersham
Pharmacia, Piscataway, NJ, USA) was used for detection.
Transfection of siRNA against HPIP
siRNA was transfected into cells using Lipofectamine
2000 transfection reagent (Life Technologies) by following the
manufacturer's instructions. siRNA against HPIP was synthesized by
Shanghai GenePharma Co., Ltd. (Shanghai, China). siRNA knockdown
efficiency was verified by western blotting of HPIP expression.
Cell migration assay
Cell migration was determined using a modified
Boyden chamber model (Transwell apparatus, 8.0-µm pore size;
Costar, Cambridge, MA, USA). Briefly, A549 cells (1×105
cells/well) transfected with siRNA-HPIP or mock were seeded onto
the upper chamber containing serum free culture medium. In
addition, 500 µl of medium supplemented with 10% FBS was
added to the lower compartment. After incubation for 24 h, all
non-migrant cells were removed from the upper faces of the
Transwell membranes with a cotton swab and the migrated cells were
fixed and stained with 0.1% crystal violet. Migration was
quantified by counting the number of stained cells per 100 x field
(high power field) in images captured with a microscope.
Cell invasion assay
The invasiveness of RPE cells was examined using a
Matrigel-coated modified Boyden chamber. Briefly, the A549 cells
(1×105 cells/well) transfected with siRNA-HPIP or mock
were seeded onto the upper chamber containing serum-free culture
medium. The lower compartment of the chamber was filled with
culture medium containing 10% FBS, and a nitrocellulose filter was
placed in it. After incubation for 24 h, the cells on the upper
surface were removed, and cells on the lower filters were fixed
with 95% methanol and stained with 0.1% crystal violet. Then the
number of migrated cells was counted under a microscope.
Statistical analysis
All data are expressed as mean ± SD. One-way
analysis of variance (ANOVA) was used for multiple sample analysis.
P<0.05 was considered to be statistically significant.
Results
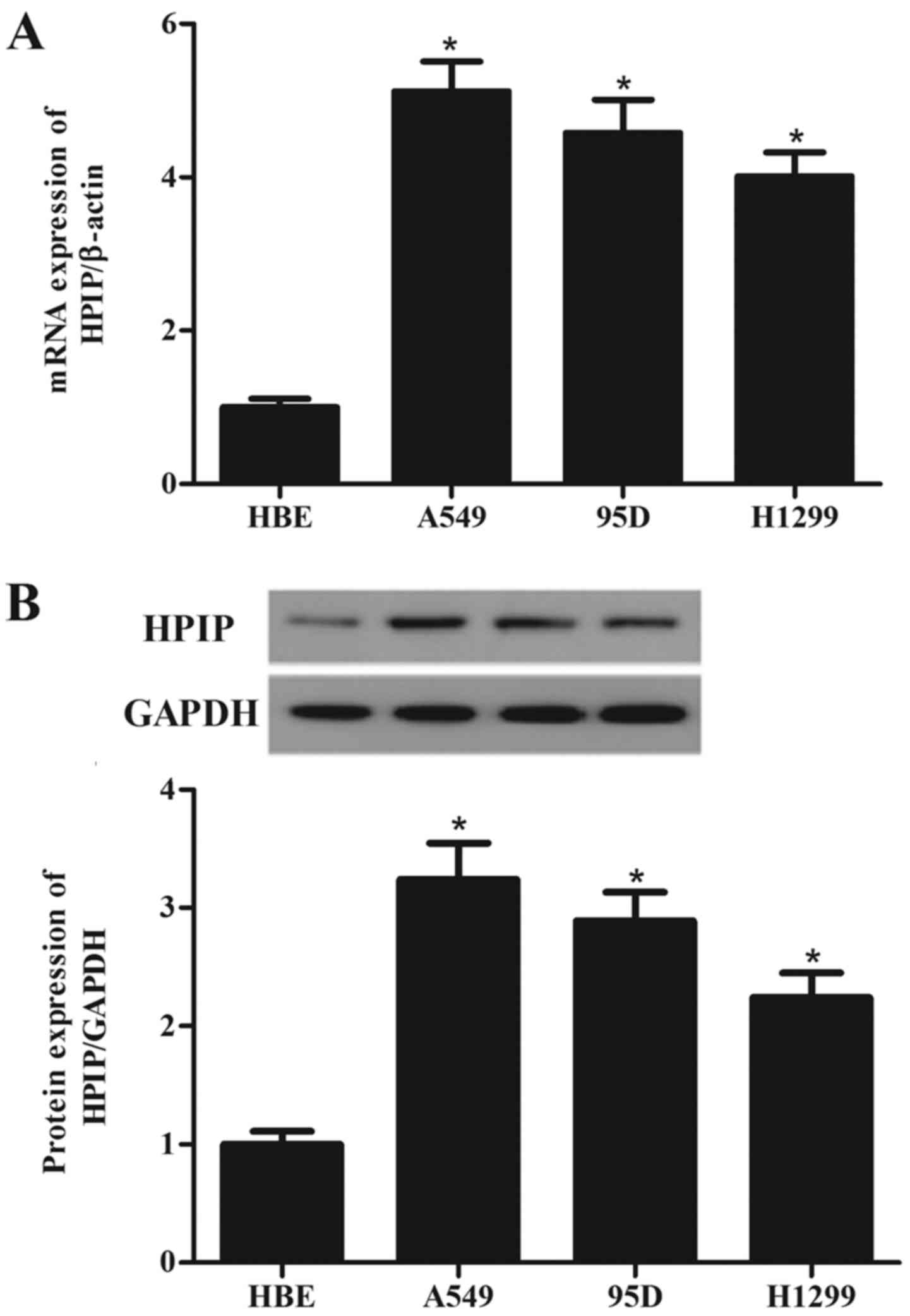
HPIP is highly expressed in lung cancer
cell lines
To investigate the role of HPIP in the tumorigenesis
of lung cancer, we detected the HPIP expression in three lung
cancer cell lines and normal HBE cells. Compared with the HBE
cells, HPIP expression was higher in the A549, 95D and H1299 cells
than the expression noted in the HBE cells (Fig. 1A). Western blotting showed similar
results (Fig. 1B).
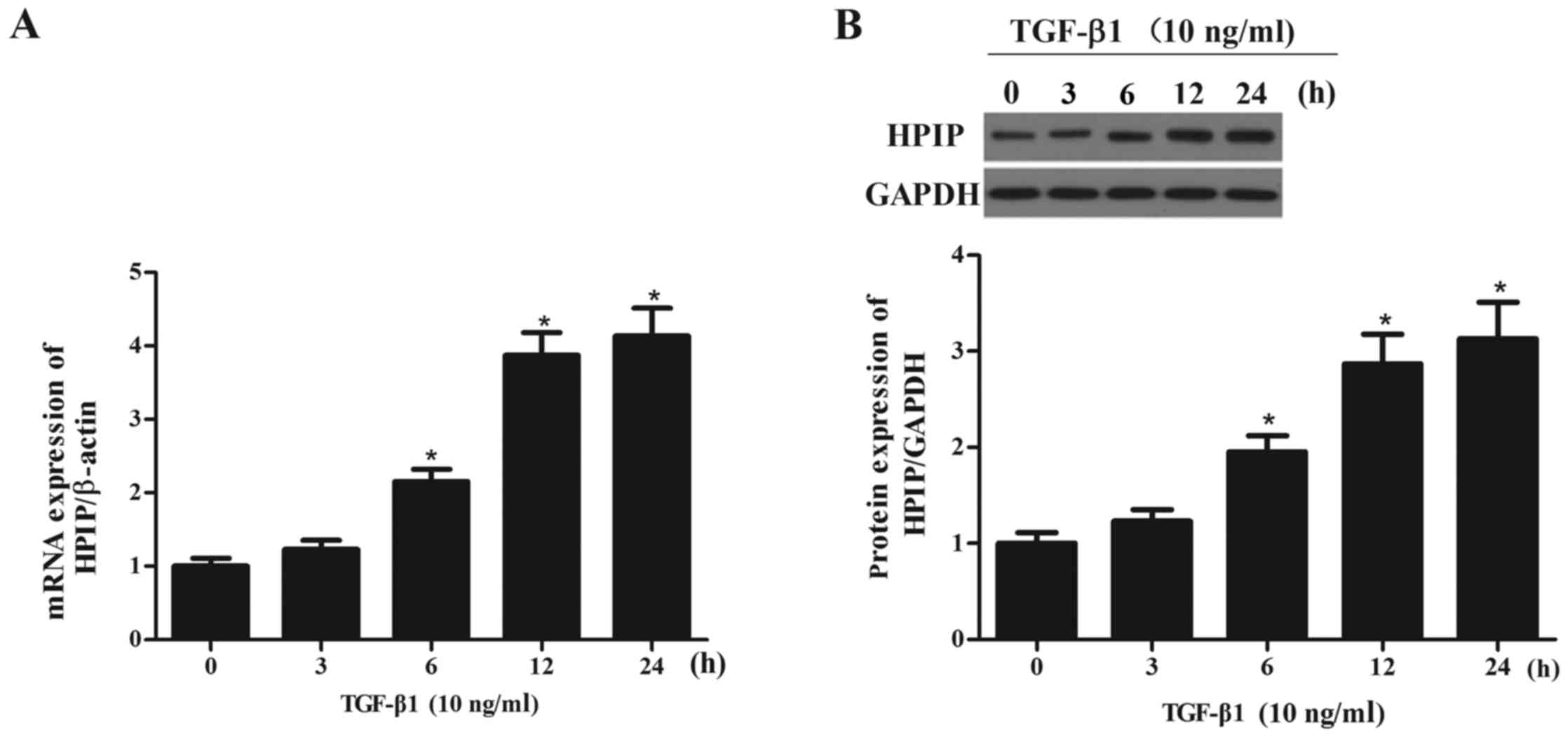
TGF-β1 increases the expression of HPIP
in lung cancer cells
To determine whether the observed changes in HPIP
expression were associated with TGF-β1-mediated EMT, we
investigated the effect of TGF-β1 on the expression levels of HPIP
in the A549 cells. As shown in Fig.
2, TGF-β1 treatment significantly increased the expression of
HPIP at both the mRNA and protein levels in the A549 cells, in a
time-dependent manner.
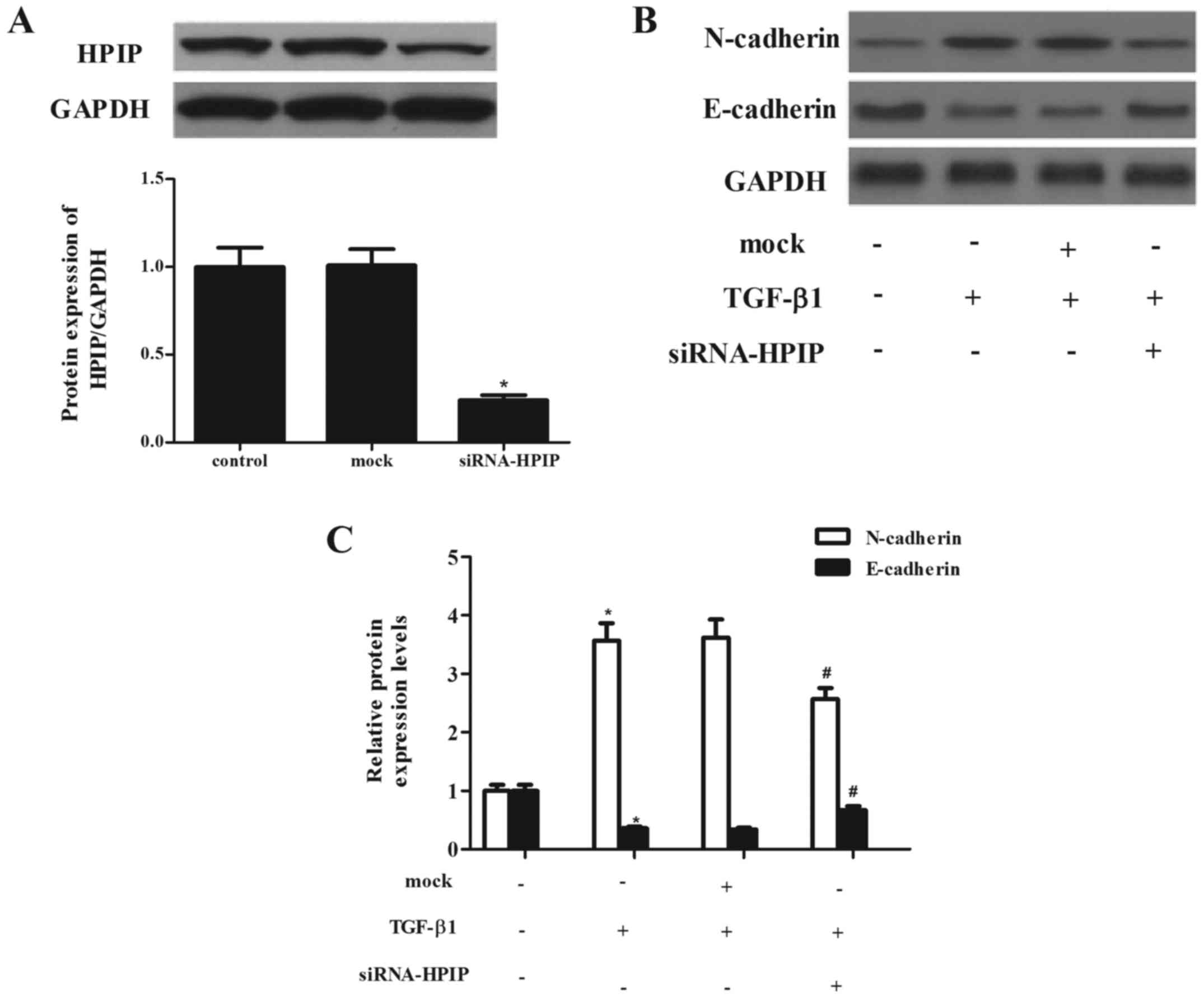
HPIP silencing significantly attenuates
TGF-β1-induced EMT in lung cancer cells
To evaluate the effect of HPIP on lung cancer
carcinogenesis, we detected the EMT phenotype after HPIP was
downregulated. Through RNAi, a stable HPIP-knockdown clone in A549
cells and negative control (mock) clone were generated. As shown in
Fig. 3A, siRNA-HPIP significantly
decreased HPIP expression in the A549 cells, as compared with the
level in the mock group.
TGF-β1 has been reported to induce EMT in A549 lung
cancer cells (10). Thus, we
investigated the effect of HPIP on TGF-β1-induced EMT in A549
cells. As previously reported, treatment with TGF-β1 markedly
induced the expression of N-cadherin and inhibited the expression
of E-cadherin. However, knockdown of HPIP significantly suppressed
TGF-β1-mediated upregulation of N-cadherin and suppression of
E-cadherin in the A549 cells (Fig. 3B
and C).
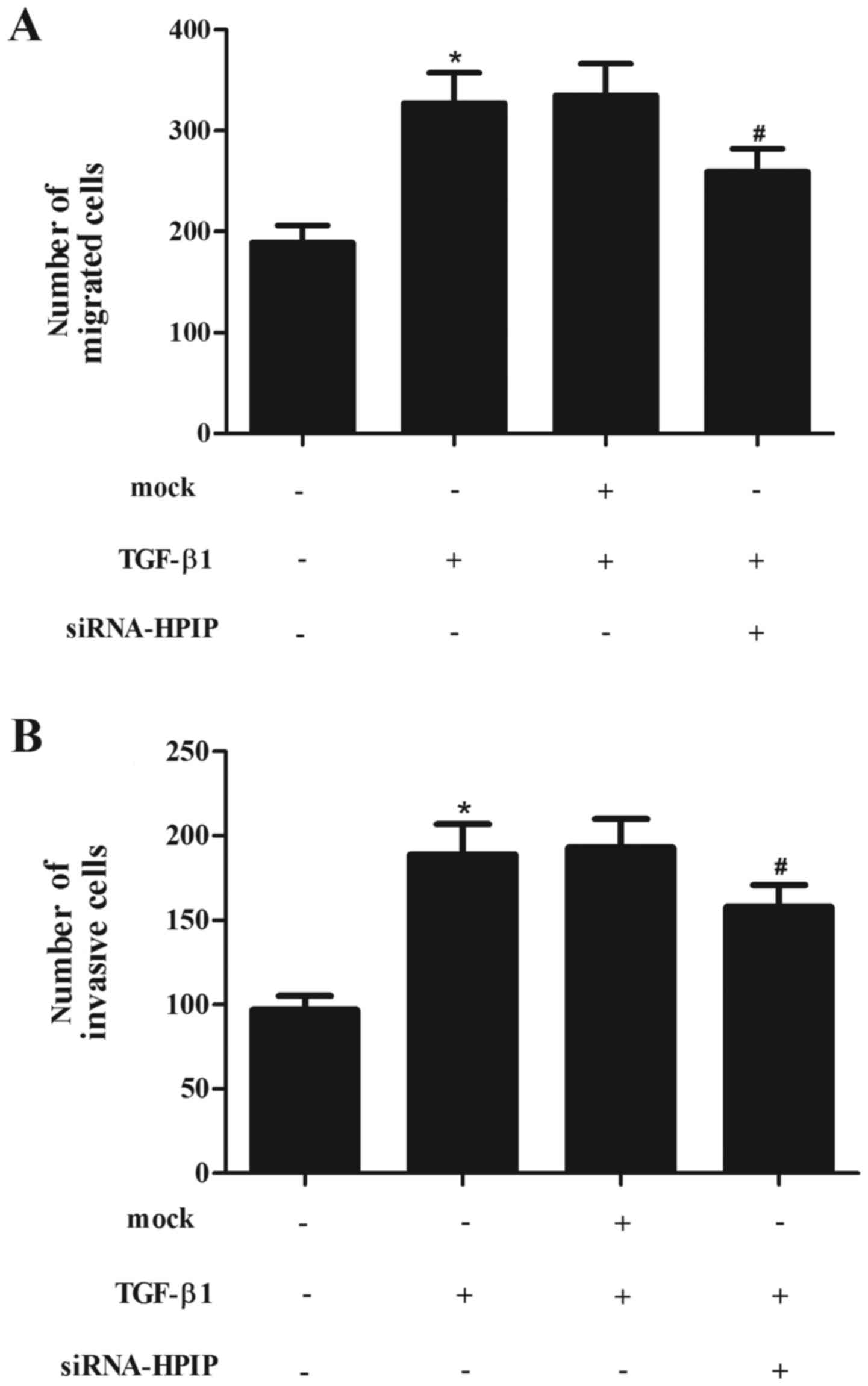
HPIP silencing significantly attenuates
TGF-β1-induced migration and invasion in lung cancer cells
EMT has been indicated as a key step in the
initiation of cancer cell migration. Thus, we evaluated the effect
of HPIP on TGF-β1-induced migration and invasion in A549 cells. As
shown in Fig. 4A, treatment with
TGF-β1 obviously promoted A549 cell migration. However, knockdown
of HPIP significantly inhibited TGF-β1-induced migration.
Consistent with the results of the Transwell assay, knockdown of
HPIP greatly inhibited A549 cell invasion induced by TGF-β1
(Fig. 4B).
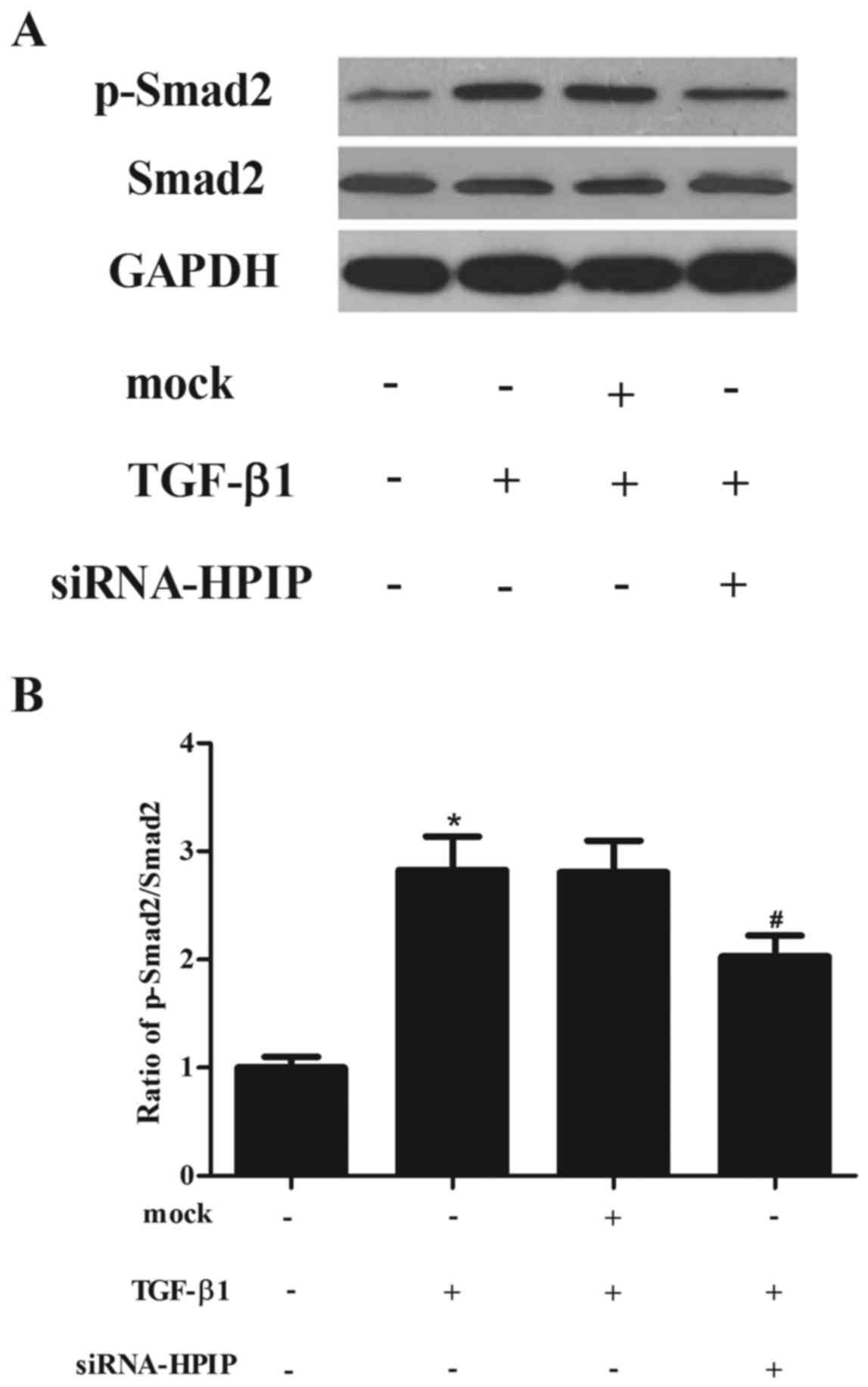
HPIP silencing inhibits the TGF-β1/Smad2
signaling pathway in A549 cells
TGF-β1-induced EMT is mediated through Smad
signaling pathways. Therefore, we examined whether HPIP silencing
suppresses TGF-β1-induced phosphorylation of Smad2. As shown in
Fig. 5, western blotting showed
that TGF-β1 treatment promoted the phosphorylation of Smad2, which
was significantly decreased by siRNA-HPIP in the A549 cells.
Discussion
In the present study, we showed for the first time a
role of HPIP as an oncogene in lung cancer. First, HPIP was
overexpressed in the lung cancer cell lines. Second, TGF-β1
increased the expression of HPIP in the lung cancer cells. Third,
HPIP silencing significantly attenuated TGF-β1-induced EMT and
migration/invasion in the lung cancer cells. Finally, knockdown of
HPIP greatly inhibited the TGF-β1-promoted phosphorylation of Smad2
in the A549 cells.
The expression level of HPIP has been found to be
significantly associated with tumor progression and metastasis in
several malignant tumors including liver, breast and colorectal
cancer (11–13). Recently, Wang et al
demonstrated that the expression of HPIP is upregulated in thyroid
carcinoma cell lines (14).
Consistently, in this study, we observed that HPIP was highly
expressed in the lung cancer cell lines. Furthermore, TGF-β1 is a
multifunctional cytokine that regulates a wide range of cellular
functions. It was reported that blood levels of TGF-β1 are elevated
in patients with lung cancer when compared with normal patients
(15), and increased production
of TGF-β by cancer cells during tumor progression can promote tumor
growth, angiogenesis, and metastasis (16). In the present study, we observed
that TGF-β1 treatment significantly increased the expression of
HPIP in lung cancer cells. Accordingly, these results clearly
suggest that HPIP functions as an oncogene in lung cancer.
It has been reported that EMT contributes to
increased metastatic progression in the development of cancer
(3,17,18). Increasing evidence suggests that
loss of E-cadherin function or high expression of vimentin are
common in lung cancer and are correlated with poor patient
prognosis (19,20). Moreover, in vitro evidence
clearly demonstrates that TGF-β1 stimulates EMT in lung cancer
cells (21). In the present
study, we found that knockdown of HPIP significantly suppressed
TGF-β1-mediated upregulation of N-cadherin and suppression of
E-cadherin in A549 cells. We also found that HPIP silencing
significantly attenuated TGF-β1-induced migration and invasion in
the A549 cells. These results suggest that knockdown of HPIP
inhibits TGF-β1-induced EMT in A549 cells, thus inhibiting lung
cancer cell migration and invasion.
Altered TGF-β signaling has been implicated in tumor
development and progression. It is known that TGF-β1 transmits
intracellular signals via the Smad pathway (22). After TGF-β1 stimulation, Smad2 and
Smad3 are phosphorylated, and then phosphorylated Smad2 and Smad3
form heteromeric complexes with a mediator Smad4. These Smad2/3/4
complexes translocate to the nucleus, where they control specific
target genes by binding with transcription factors, and
subsequently induce development of EMT in cancer cells (23). Smad3 is considered to be the
primary signaling molecule in mediating EMT in many types of tumors
(24,25). However, EMT in several types of
cancer cells has been shown to depend on Smad2 signaling (26,27). In addition, several investigators
have shown that the A549 lung cancer cell line undergoes EMT upon
TGF-β1 treatment (7,28,29). In the present study, we examined
the effect of HPIP on the expression of Smad2 in TGF-β1-induced
A549 cells and found that HPIP silencing inhibited activation of
Smad2 in TGF-β1-induced EMT. These results suggest that HPIP
silencing inhibits TGF-β1-induced EMT in lung cancer cells by
inhibiting Smad2 activation.
In conclusion, we demonstrated that HPIP silencing
suppressed TGF-β1-induced EMT in lung cancer cells by inhibiting
Smad2 activation. Therefore, HPIP may be a new therapeutic target
for the treatment of lung cancer.
References
|
1
|
Greenlee RT, Murray T, Bolden S and Wingo
PA: Cancer statistics, 2000. CA Cancer J Clin. 50:7–33. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wakelee HA, Chang ET, Gomez SL, Keegan TH,
Feskanich D, Clarke CA, Holmberg L, Yong LC, Kolonel LN, Gould MK,
et al: Lung cancer incidence in never smokers. J Clin Oncol.
25:472–478. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Huber MA, Kraut N and Beug H: Molecular
requirements for epithelial-mesenchymal transition during tumor
progression. Curr Opin Cell Biol. 17:548–558. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yang J and Weinberg RA:
Epithelial-mesenchymal transition: at the crossroads of development
and tumor metastasis. Dev Cell. 14:818–829. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Thompson EW, Newgreen DF and Tarin D:
Carcinoma invasion and metastasis: a role for
epithelial-mesenchymal transition? Cancer Res. 65:5991–5995;
discussion 5995. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Iwatsuki M, Mimori K, Yokobori T, Ishi H,
Beppu T, Nakamori S, Baba H and Mori M: Epithelial-mesenchymal
transition in cancer development and its clinical significance.
Cancer Sci. 101:293–299. 2010. View Article : Google Scholar
|
|
7
|
Bierie B and Moses HL: TGF-beta and
cancer. Cytokine Growth Factor Rev. 17:29–40. 2006. View Article : Google Scholar
|
|
8
|
Feng Y, Li L, Zhang X, Zhang Y, Liang Y,
Lv J, Fan Z, Guo J, Hong T, Ji B, et al: HPIP is overexpressed in
gastric cancer and promotes gastric cancer cell proliferation,
migration and invasion. Cancer Sci. 106:1313–1322. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
van Vuurden DG, Aronica E, Hulleman E,
Wedekind LE, Biesmans D, Malekzadeh A, Bugiani M, Geerts D, Noske
DP, Vandertop WP, et al: Pre-B-cell leukemia homeobox interacting
protein 1 is overexpressed in astrocytoma and promotes tumor cell
growth and migration. Neuro Oncol. 16:946–959. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kim AN, Jeon WK, Lim KH, Lee HY, Kim WJ
and Kim BC: Fyn mediates transforming growth factor-beta1-induced
down-regulation of E-cadherin in human A549 lung cancer cells.
Biochem Biophys Res Commun. 407:181–184. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu X, Jiang C, Wang S, Tai Y, Wang T, Kang
L, Fan Z, Li S, Li L, Fu J, et al: HPIP is upregulated in liver
cancer and promotes hepatoma cell proliferation via activation of
G2/M transition. IUBMB Life. 65:873–882. 2013.PubMed/NCBI
|
|
12
|
Bugide S, David D, Nair A, Kannan N,
Samanthapudi VS, Prabhakar J and Manavathi B: Hematopoietic
PBX-interacting protein (HPIP) is over expressed in breast
infiltrative ductal carcinoma and regulates cell adhesion and
migration through modulation of focal adhesion dynamics. Oncogene.
34:4601–4612. 2015. View Article : Google Scholar
|
|
13
|
Feng Y, Xu X, Zhang Y, Ding J, Wang Y,
Zhang X, Wu Z, Kang L, Liang Y, Zhou L, et al: HPIP is upregulated
in colorectal cancer and regulates colorectal cancer cell
proliferation, apoptosis and invasion. Sci Rep. 5:9429–9439. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang SC, Chai DS, Chen CB, Wang ZY and
Wang L: HPIP promotes thyroid cancer cell growth, migration and EMT
through activating PI3K/AKT signaling pathway. Biomed Pharmacother.
75:33–39. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kong FM, Washington MK, Jirtle RL and
Anscher MS: Plasma transforming growth factor-beta 1 reflects
disease status in patients with lung cancer after radiotherapy: a
possible tumor marker. Lung Cancer. 16:47–59. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Akhurst RJ and Derynck R: TGF-beta
signaling in cancer - a double-edged sword. Trends Cell Biol.
11:S44–S51. 2001.PubMed/NCBI
|
|
17
|
Thiery JP: Epithelial-mesenchymal
transitions in tumour progression. Nat Rev Cancer. 2:442–454. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Moustakas A and Heldin CH: Signaling
networks guiding epithelial-mesenchymal transitions during
embryogenesis and cancer progression. Cancer Sci. 98:1512–1520.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Sato M, Shames DS and Hasegawa Y: Emerging
evidence of epithelial-to-mesenchymal transition in lung
carcinogenesis. Respirology. 17:1048–1059. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nakata S, Sugio K, Uramoto H, Oyama T,
Hanagiri T, Morita M and Yasumoto K: The methylation status and
protein expression of CDH1, p16(INK4A), and fragile histidine triad
in nonsmall cell lung carcinoma: epigenetic silencing, clinical
features, and prognostic significance. Cancer. 106:2190–2199. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zavadil J and Böttinger EP: TGF-beta and
epithelial-to-mesenchymal transitions. Oncogene. 24:5764–5774.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Heldin CH, Miyazono K and ten Dijke P:
TGF-beta signalling from cell membrane to nucleus through SMAD
proteins. Nature. 390:465–471. 1997. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dumont N and Arteaga CL: Transforming
growth factor-beta and breast cancer: tumor promoting effects of
transforming growth factor-beta. Breast Cancer Res. 2:125–132.
2000. View Article : Google Scholar
|
|
24
|
Risolino M, Mandia N, Iavarone F, Dardaei
L, Longobardi E, Fernandez S, Talotta F, Bianchi F, Pisati F,
Spaggiari L, et al: Transcription factor PREP1 induces EMT and
metastasis by controlling the TGF-β-SMAD3 pathway in non-small cell
lung adenocarcinoma. Proc Natl Acad Sci USA. 111:E3775–E3784. 2014.
View Article : Google Scholar
|
|
25
|
Yamazaki K, Masugi Y, Effendi K, Tsujikawa
H, Hiraoka N, Kitago M, Shinoda M, Itano O, Tanabe M, Kitagawa Y,
et al: Upregulated SMAD3 promotes epithelial-mesenchymal transition
and predicts poor prognosis in pancreatic ductal adenocarcinoma.
Lab Invest. 94:683–691. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Oft M, Akhurst RJ and Balmain A:
Metastasis is driven by sequential elevation of H-ras and Smad2
levels. Nat Cell Biol. 4:487–494. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lv ZD, Kong B, Li J-G, Qu HL, Wang XG, Cao
WH, Liu XY, Wang Y, Yang ZC, Xu HM, et al: Transforming growth
factor-β 1 enhances the invasiveness of breast cancer cells by
inducing a Smad2-dependent epithelial-to-mesenchymal transition.
Oncol Rep. 29:219–225. 2013.
|
|
28
|
Kasai H, Allen JT, Mason RM, Kamimura T
and Zhang Z: TGF-beta1 induces human alveolar epithelial to
mesenchymal cell transition (EMT). Respir Res. 6:56–70. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Willis BC and Borok Z: TGF-beta-induced
EMT: mechanisms and implications for fibrotic lung disease. Am J
Physiol Lung Cell Mol Physiol. 293:L525–L534. 2007. View Article : Google Scholar : PubMed/NCBI
|