Introduction
Patients with chronic renal failure (CRF) have a
greater risk of developing cardiovascular disease (CVD) than
subjects with normal renal function. Atherosclerosis (AS) is an
important risk factor for the development of CVD in patients with
CRF (1–4). Previous studies have reported that
inflammation plays a key role in the development of AS in patients
with CRF (5–7).
The endoplasmic reticulum (ER) is a multifunctional
organelle that performs protein synthesis, folding and maturation.
An accumulation of unfolded or misfolded proteins in the ER lumen
leads to ER stress, which restores homeostasis in the ER. The
release of ER stress sensors, including inositol-requiring 1α
(IRE1α), double-stranded RNA-dependent protein kinase (PKR)-like ER
kinase (PERK) and activating transcription factor 6 (ATF6)
stimulates three distinct downstream signaling pathways (8,9).
Severe and prolonged ER stress disrupts homeostasis and causes
tissue injury and organ dysfunction. ER stress has been implicated
in various types of CVD. Increasing evidence indicates that ER
stress is the upstream signal for the inflammatory reaction, and is
thus a key player in AS initiation and progression (10–13).
As regards other diseases, the suppression of the
renin-angiotensin system (RAS) can also inhibit ER stress to
decrease angiotensin II (Ang II)-induced inflammation (14,15). Thus, RAS activation promotes ER
stress-related inflammation and may aggravate AS and other
diseases. According to previous studies, RAS inhibition effectively
abolishes the pro-atherogenic effects of uremia, and these effect
are mainly dependent on an anti-inflammatory mechanism (16,17). However, the association between
RAS-mediated ER stress and uremia-associated AS has not yet been
examined to date, at least to the best of our knowledge. In this
study, we investigated the effects of RAS on ER stress-induced
inflammation in uremic apolipoprotein E knockout
(apoE−/−) mice by blocking the Ang II receptor to
further clarify the mechanisms of action of RAS in experimental
uremia-associated AS.
Materials and methods
Serum biochemistry
Serum levels of urea nitrogen (BUN) and creatinine
(CRE) were measured enzymatically with reagents from Nanjing
Jiancheng Bioengineer Institute (Nanjing, China). The serum level
of Ang II was measured by ELISA according to the instructions
provided by the manufacturer (Jianglai Biotechnology Co., Shanghai,
China).
Animal models
Ten-week-old male apoE−/− mice
(homozygous apoE-deficient male mice, back-crossed 20 times from
the C57BL/6 strain; Vital River Laboratory Animal Technology Co.,
Ltd., Beijing, China) were housed in the animal facility of
Chongqing Medical University, Chongqing, China. The animals were
maintained in an air-conditioned, pathogen-free and
light-controlled environment and were fed standard mouse chow
(2018S; Harlan Teklad, Madison, WI, USA) and sterile water.
Experimental mild uremia was induced by 5/6 nephrectomy (5/6 Nx,
termed SNx). In total, 31 apoE−/− mice were randomly
allocated to undergo SNx (n=21) or sham operation (n=10). At 10
weeks of age, both the upper poles of the right kidney were
resected, and electrocoagulation of the incision was performed to
stop bleeding. After 2 weeks, the whole left kidney was removed.
The control animals underwent a sham operation (the bilateral
kidneys were only exposed, without any resection of the kidneys).
Anesthesia was achieved by an intraperitoneal injection of sodium
pentobarbital (Sigma Chemical Co., St. Louis, MO, USA) at a dose of
40 mg/kg.
Four weeks following nephrectomy, the uremic mice
were randomly allocated into 2 subgroups as follows: the mice
treated with the Ang II type 1 (AT1) receptor antagonist, losartan
(Merck Sharp & Dohme Pty. Ltd., Macquarie Park, NSW, Australia)
at a dose of 30 mg/kg in drinking water (n=10) or no medicine
(n=11) for 12 weeks. The sham-operated mice received no medicine
(n=10).
After the 12-week-intervention, each mouse was
anesthetized, and blood was collected from the retro-orbital venous
plexus. The blood was centrifuged at 3,000 rpm, at 4°C, and the
plasma was stored at −80°C for analysis. For each mouse, following
perfusion, the heart and aorta were separated from the iliac
arteries. The heart and aortic root were dissected from the distal
aorta and stored in 4% paraformaldehyde solution for histological
analyses. The remaining aorta was stored at −80°C for gene and
protein evaluations of the atherosclerotic lesions. This study was
performed in strict accordance with the recommendations in the
Guide for the Care and Use of Laboratory Animals of Chongqing
Medical University. The protocols were approved by the
Institutional Ethics Committee for Animal Experiments of Chongqing
Medical University.
Cell culture and treatment
RAW264.7 murine macrophages were obtained from the
Type Culture Collection of the Chinese Academy of Sciences
(Shanghai, China) and grown in Dulbecco's modified Eagle's medium
(DMEM) containing 10% fetal bovine serum (FBS) (both from HyClone,
Logan, UT, USA). The cells were plated on tissue culture dishes
before the experiment and were cultured at 37°C under 5%
CO2 in a humidified incubator. In all the experiments,
the RAW264.7 macrophages were grown to 90% confluence and were
serum-starved for 12 h prior to exposure to to various
concentrations of Ang II (0.01, 0,1, 1, or 10 µg/ml). Prior to
exposure to 1 μg/ml Ang II for 8 h, the cells were treated
with losartan (10 μmol/l) 1 h before.
Infection with lentivirus
To verify whether Ang II-induced inflammation is
associated with ER stress, the RAW264.7 macrophages were
transfected with a specific siRNA-carrying lentiviral vector
directed against IRE1α (forward, 5′-GGAAUUACUGGCUUCUCAUdTdT-3′ and
reverse, 5′-AUGAGAAGCCAGUAAUUCCdTdT-3′) (18). A nonsense sequence served as the
negative control. Following transfection for 48 h, the cells were
exposed to Ang II for 8 h. Target gene silencing was validated by
reverse transcription-quantitative PCR (PCR).
Histopathological analysis
Following fixation, the hearts and aortic roots were
paraffin-embedded and sectioned. The sections were stained with
hematoxylin-eosin or special antibodies, respectively. For
immunohistochemistry, paraffinized sections were deparaffinized.
Endogenous peroxidase activity was blocked with 3%
H2O2 for 20 min, and non-specific staining
was blocked by incubation with goat serum for 15 min. The samples
were stained with anti-CD68 (BA3638; 1:100; Boster, Wuhan, China)
or anti-p-IRE1α (Ser724; ab48187; 1:300; Abcam, Cambridge, UK)
antibodies overnight at 4°C, followed by HRP-conjugated secondary
antibodies (SP-9001; ZSGB-Bio, Beijing, China) at a 1:200 dilution
for 30 min at 37°C. Immunohistochemical staining was performed by
exposure to 3,3′-diaminobenzidine, and counterstaining was
developed with hematoxylin. The sections were examined under a
light microscope (Leica, Wetzlar, Germany) and quantified using
Image-Pro Plus 6.0 software.
RT-qPCR
Total RNA was extracted from the rat aorta tissues
or RAW264.7 macrophages using TRIzol reagent according to the
manufacturer's instructions (Takara Bio, Inc., Otsu, Japan). RNA
was reverse transcribed using the PrimeScript™ RT reagent kit
(Takara Bio, Inc.). qPCR was performed using an Applied Biosystems
StepOnePlus Real-Time PCR system and SYBR®-Green PCR
master mix (both from Applied Biosystems, Foster City, CA, USA).
Glyceraldehyde 3-phosphate dehydrogenase (GADPH) was used as an
internal quantitative control. All cDNA were run twice in separate
runs. The relative mRNA expression of each molecule was calculated
using the 2−ΔΔCt method and normalized to GADPH. The
primer sequences are listed in Table
I.
 | Table ISequences of primers for the detected
genes. |
Table I
Sequences of primers for the detected
genes.
| Gene | Primer sequences
(5′→3′) |
|---|
| IL-6 | F:
GAGGATACCACTCCCAACAGACC |
| R:
AAGTGCATCATCGTTGTTCATACA |
| TNF-α | F:
CATGAGCACAGAAAGCATGATCCG |
| R:
AAGCAGGAATGAGAAGAGGCTGAG |
| CCL2/MCP-1 | F:
CTTCCTCCACCACCATGCA |
| R:
CCAGCCGGCAACTGTGA |
| CX3CL1 | F:
GTGCTGACCCGAAGGAGAAA |
| R:
CACCCGCTTCTCAAACTTGC |
| GADPH | F:
TGCTGAGTATGTCGTGGAGTCTA |
| R:
AGTGGGAGTTGCTGTTGAAATC |
Western blot analysis
The aorta tissue or RAW264.7 macrophages were lysed
for 30 min at 4°C in lysis buffer. Total protein concentrations
were determined using bicinchoninic acid reagent. Total protein
(50–100 μg) was separated by sodium dodecyl
sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then
transferred onto polyvinylidene difluoride membranes. After
blocking, the membranes were incubated with anti-IRE1α (ab37073;
1:800; Abcam), anti-glucose-regulated protein 78 (GRP78;
11587-1-AP; 1:500; ProteinTech, Wuhan, China), anti-p-IRE1α
(Ser724; ab48187; 1:800; Abcam), IκB kinase α (IKKα; 2682S; Cell
Signaling Technology, Danvers, MA, USA), IκB kinase β (IKKβ; 2684S;
Cell Signaling Technology), anti-p-IκB kinase α/β (p-IKK;
Ser176/180; 2697T; 1:800; Cell Signaling Technology), IκB (YM3718;
1:500; ImmunoWay, Newark, DE, USA), anti-NF-κB p65 (YT5339; 1:500;
ImmunoWay), anti-β-actin (6008-1-Ig; 1:750; ProteinTech) and
anti-LaminB (sc-6216; 1:500; ZSGB-Bio) primary antibodies overnight
at 4°C. Horseradish peroxidase-conjugated secondary antibodies
(ZB-2301; ZSGB-Bio) were applied the following day for 2 h at 37°C.
The bands were visualized using chemiluminescence (Beyotime,
Shanghai, China). Band density was analyzed using fusion software
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) and normalized to
β-actin or Lamin B density.
Statistical analyses
Statistical analyses were performed using SPSS
software version 19.0. The data are presented as the means ± SEM
for in vivo experiments and as the means ± SD for in
vitro experiments. Analyses of variance (one-way ANOVA)
followed by Bonferroni's or Dunnett's tests and the Kruskal-Wallis
test followed by post-hoc tests were performed to evaluate group
differences.
Results
Effect of losartan treatment on serum
biochemistry and the development of AS in uremic mice
As shown in Fig.
1, 16 weeks after nephrectomy, the serum BUN and CRE
concentrations were markedly increased in the uremic mice compared
to the control mice (P<0.01; Fig.
1A and B). These increases indicated that the model was
successfully developed. In addition, the serum Ang II levels was
significantly increased in the uremic mice compared to the controls
(P<0.01; Fig. 1C). Treatment
with losartan treatment improved renal function in the uremic mice
(BUN, P<0.05; CRE, P<0.01; Fig.
1A and B). The level of Ang II was also decreased in the
losartan treatment group (P<0.01; Fig. 1C). At 16 weeks after nephrectomy,
the uremic mice displayed a significant increase in the aortic root
plaque area compared with the control mice (282,293±12,375 vs.
146,627±10,411 μm2, P<0.01) (Fig. 1D and E). Treatment with losartan
treatment markedly decreased the lesion area in the uremic mice
(222,703±9,857, P<0.05) (Fig. 1D
and E). These results indicate RAS inhibition ameliorated the
development of accelerated AS in uremia mice.
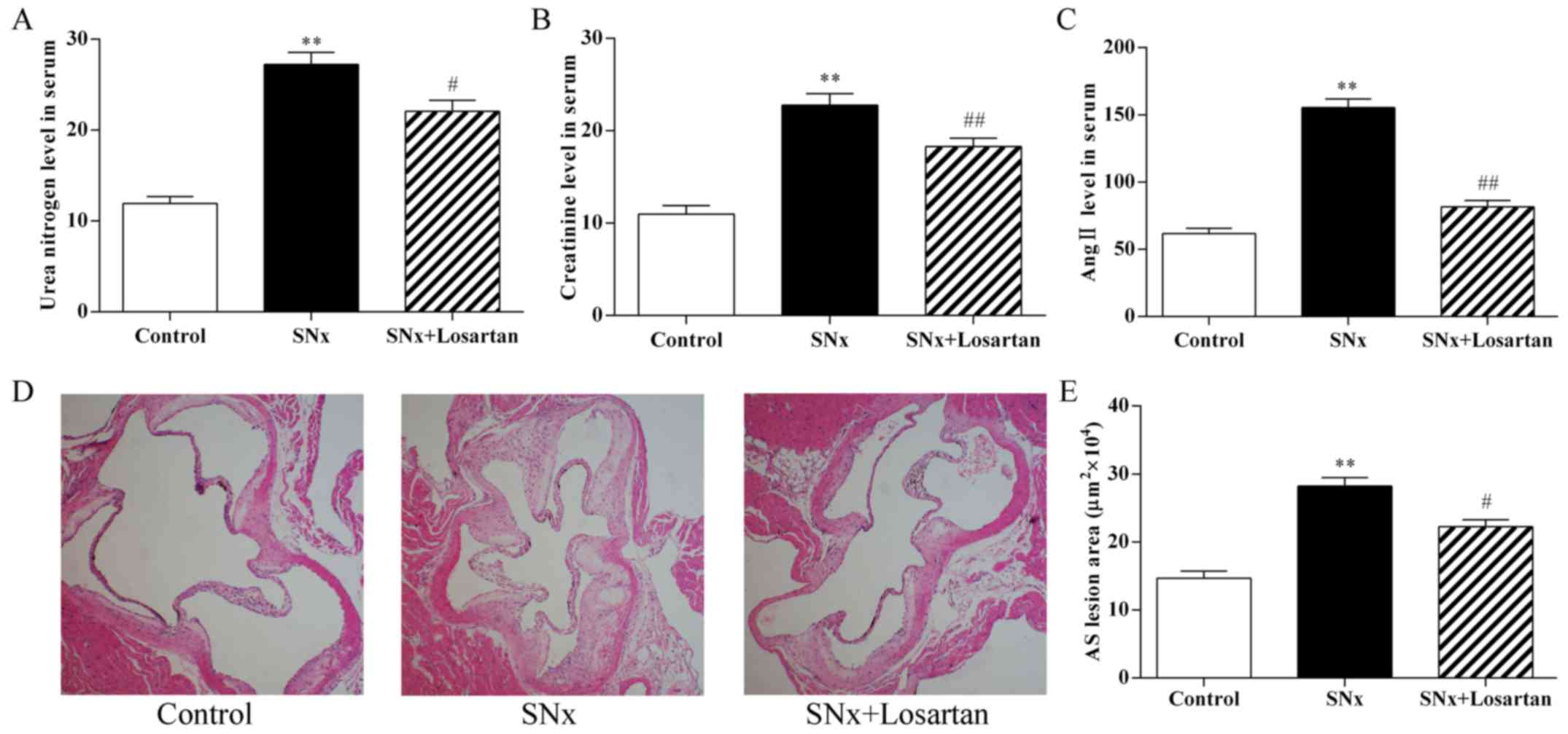 | Figure 1Effects of losartan treatment on
serum biochemistry and the development of atherosclerosis in uremic
mice. (A–C) Quantitative analysis of the level of urea nitrogen,
creatinine and angiotensin II (Ang II) in serum. n=10, 11, 10 for
the control, SNx and SNx + losartan groups, respectively. (D and E)
Quantitative analysis of aortic root AS lesion area. Hematoxylin
and eosin staining; original magnification, ×40. n=10, 11, 10 for
the control, SNx and SNx + losartan groups, respectively. SNx,
subtotal nephrectomized apolipoprotein E knockout
(apoE−/−) mice. Losartan was administered at the doses
of 30 mg/kg for 12 weeks from 4 weeks after SNx. Data are the means
± SEM. **P<0.01 vs. control group;
#P<0.05 and ##P<0.01 vs. SNx group. |
Effect of losartan treatment on vascular
inflammation and ER stress in uremic mice
As shown in Fig.
2, macrophage inflammation plays an important role in the
development of AS and ER stress is the upstream signal for the
inflammatory reaction. Using immunohistochemistry on adjacent
sections, we stained macrophages with anti-CD68 antibodies and
using p-IRE1α, a typical ER stress sensor, to evaluate the state of
ER stress in the aortic root sections. The uremic mice had a
significantly increased macrophage content within the aortic root
plaques compared with the control mice (29.64±1.59 vs. 13.27±1.11%,
P<0.01) (Fig. 2A and B). The
uremic mice also exhibited a significant increase in p-IRE1α
expression (24.40±1.28 vs. 11.58±0.86%, P<0.01) (Fig. 2C and D). The co-localization of
CD68 and p-IRE1α on adjacent sections indicated that p-IRE1α was
mainly expressed in macrophages within the plaques. The results of
western blot analysis revealed that the phosphorylation of IRE1α
was significantly increased in the aortas of the uremic mice
(P<0.01) (Fig. 2E and F).
Treatment with losartan significantly reduced the atherosclerotic
plaque macrophage content in the uremic mice (17.62±1.71%,
P<0.01) (Fig. 2A and B).
Treatment with losartan also markedly decreased p-IRE1α expression
(16.05±1.29, P<0.01) (Fig. 2C and
D) and markedly inhibited IRE1α phosphorylation in the uremic
mice (P<0.01; Fig. 2E and F).
We also observed the upregulation of the common ER stress maker,
GRP78, in the aortas of the uremic mice (P<0.01; Fig. 2E). The upregulation of GRP78 was
significantly inhibited in the aortas of the losartan-treated
uremic mice (P<0.01; Fig. 2E).
In addition, we compared the mRNA expression levels of
pro-inflammatory cytokines, such as tumor necrosis factor-α (TNF-α)
and interleukin-6 (IL-6), and chemokines, such as chemokine (C-C
motif) ligand 2 (CCL2)/monocyte chemoattractant protein-1 (MCP-1)
and chemokine (C-X3-C motif) ligand 1 (CX3CL1), in the mouse
aortas. The mRNA expression levels of the pro-inflammatory
cytokines and chemokines were higher in the uremic mice than in the
controls (P<0.01; Fig. 2G).
Treatment with losartan markedly decreased the mRNA expression
levels of the pro-inflammatory cytokines and chemokines in the
uremic mice (P<0.05; Fig.
2G).
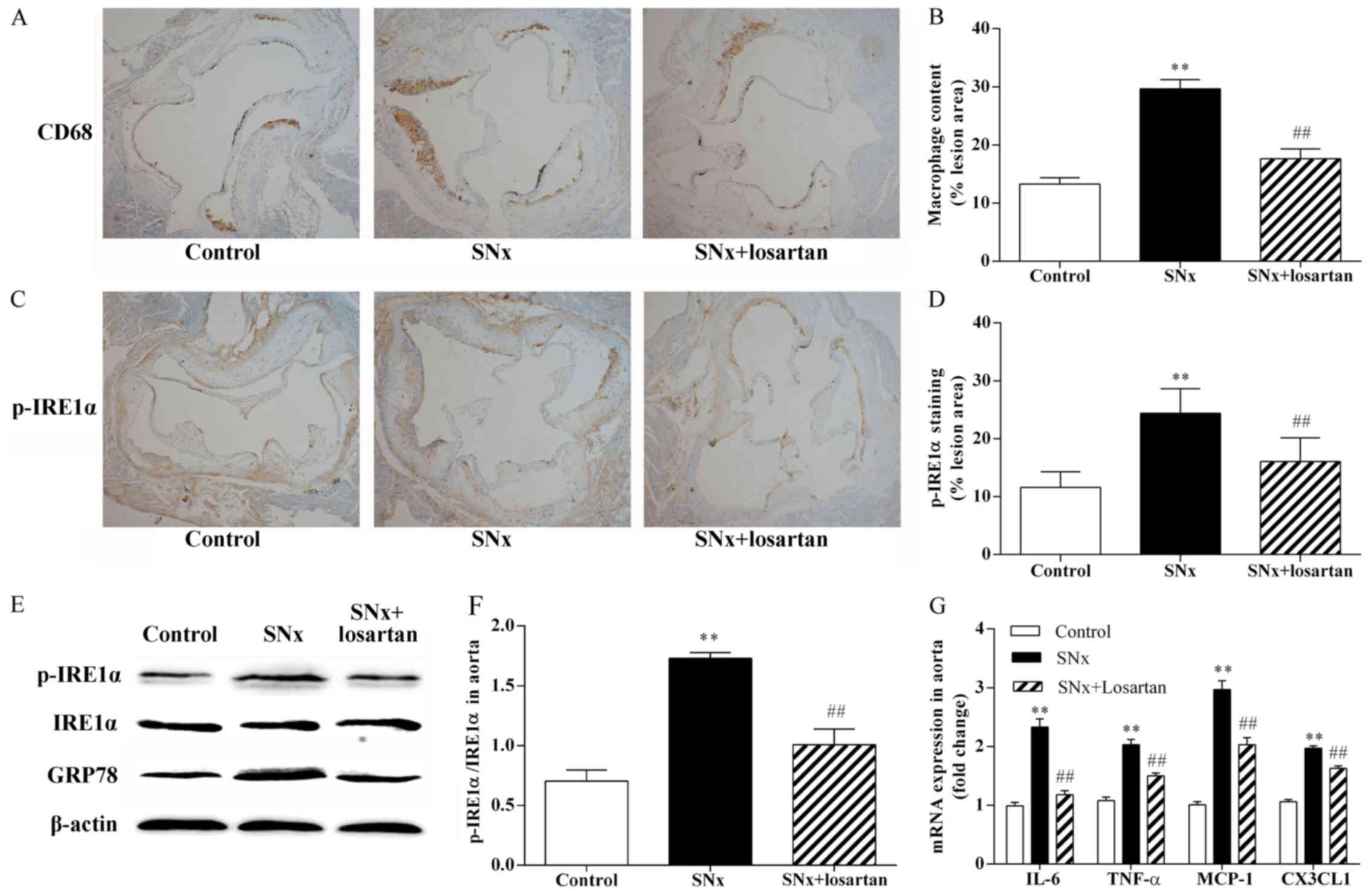 | Figure 2Effects of losartan treatment on
vascular inflammation and ER stress in uremic mice. The
co-localization of CD68 and p-inositol-requiring 1α (IRE1α) was
examined by immunohistochemistry using adjacent sections. (A and B)
Quantitative analysis of macrophages content in lesion plaques.
Immunohistochemistry with CD68, original magnification, ×40. n=10,
11, 10 for the control, SNx and SNx + losartan groups,
respectively. (C and D) Quantitative analysis of p-IRE1α in lesion
plaques in mice. Immunohistochemistry with p-IRE1α; original
magnification, ×40. (E and F) Western blot analysis of the
activation of IRE1α and the expression of glucose-regulated protein
78 (GRP78) in aortas of mice. n=5, 6, 5 for the control, SNx and
SNx + losartan groups, respectively. (G) RT-qPCR analysis of
pro-inflammatory cytokine and chemokine gene expression in aortas.
n=5, 5, 5 for the control, SNx and SNx + losartan groups,
respectively. Data are the means ± SEM. **P<0.01 vs.
control group; ##P<0.01 vs. SNx group. |
Effect of losartan on the expression of
ER stress marker proteins and the NF-κB inhibitor protein IκB in
Ang II-stimulated RAW264.7 macrophages
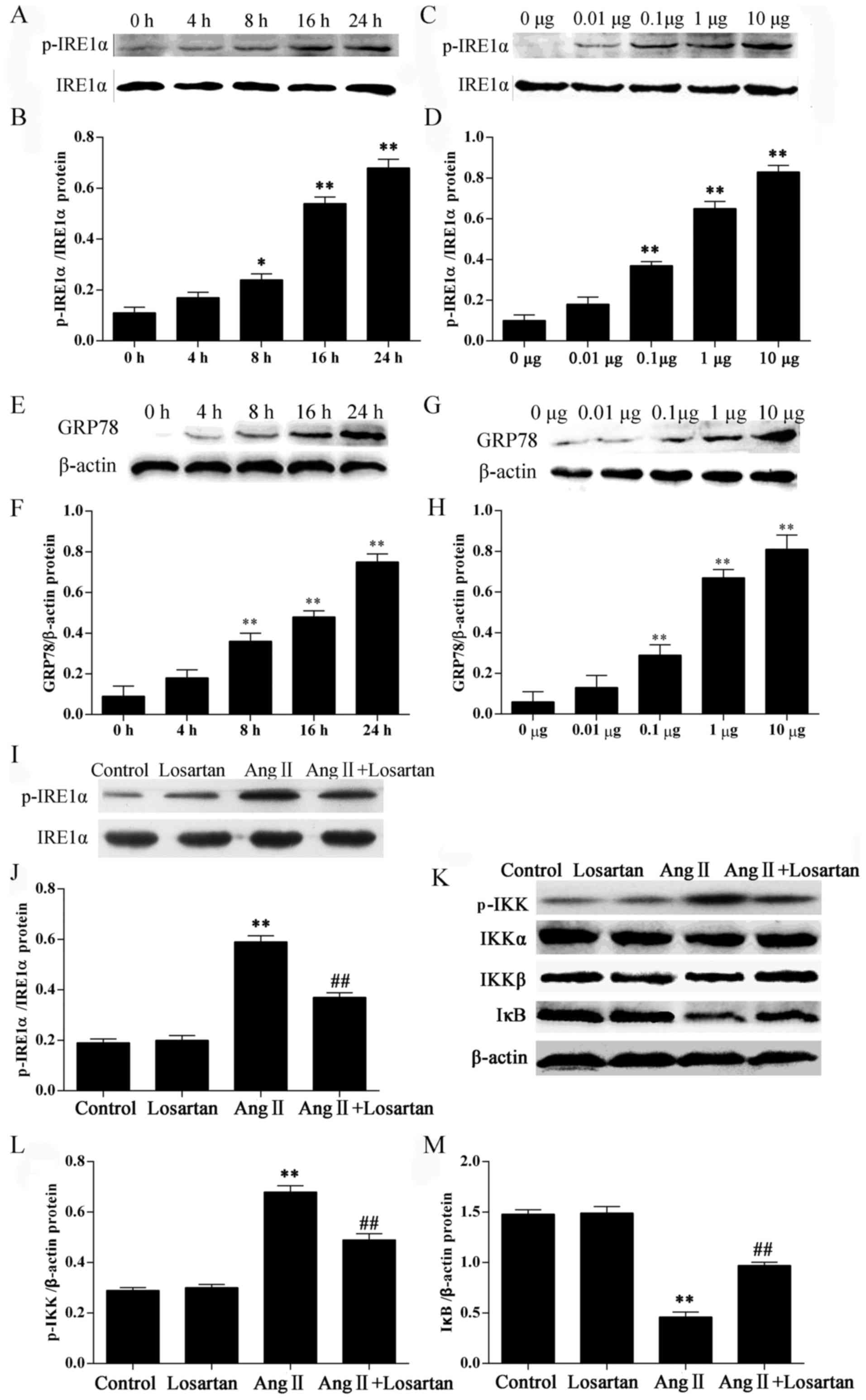
In this study, we examined the ER stress response in
RAW264.7 macrophages following treatment with Ang II by measuring
the protein levels of ER stress markers. Stimulation of the
RAW264.7 macrophages with Ang II significantly increased the
phosphorylation of IRE1α in a time− and dose-dependent
manner (P<0.05; Fig. 3A–D).
Stimulation of the RAW264.7 macrophages with Ang II also
significantly increased the GRP78 levels in a time- and
dose-dependent manner (P<0.05; Fig. 3E–H). Stimulation with Ang II also
increased the phosphorylation of IKK (P<0.05; Fig. 3K and L) in the RAW264.7
macrophages. Losartan is a common AT1 receptor antagonist. Losartan
significantly inhibited the phosphorylation of IRE1α and IKK
(P<0.01; Fig. 3I–L) compared
with the Ang II-stimulated RAW264.7 macrophages not treated with
losartan. IκB is a regulatory protein binding to the transcription
factor, NF-κB, retaining it in the cytoplasm. Losartan
significantly enhanced the expression of IκB in the Ang
II-stimulated RAW264.7 macrophages (P<0.01; Fig. 3K and M), suggesting the inhibition
of NF-κB by losartan in the Ang II-stimulated RAW264.7
macrophages.
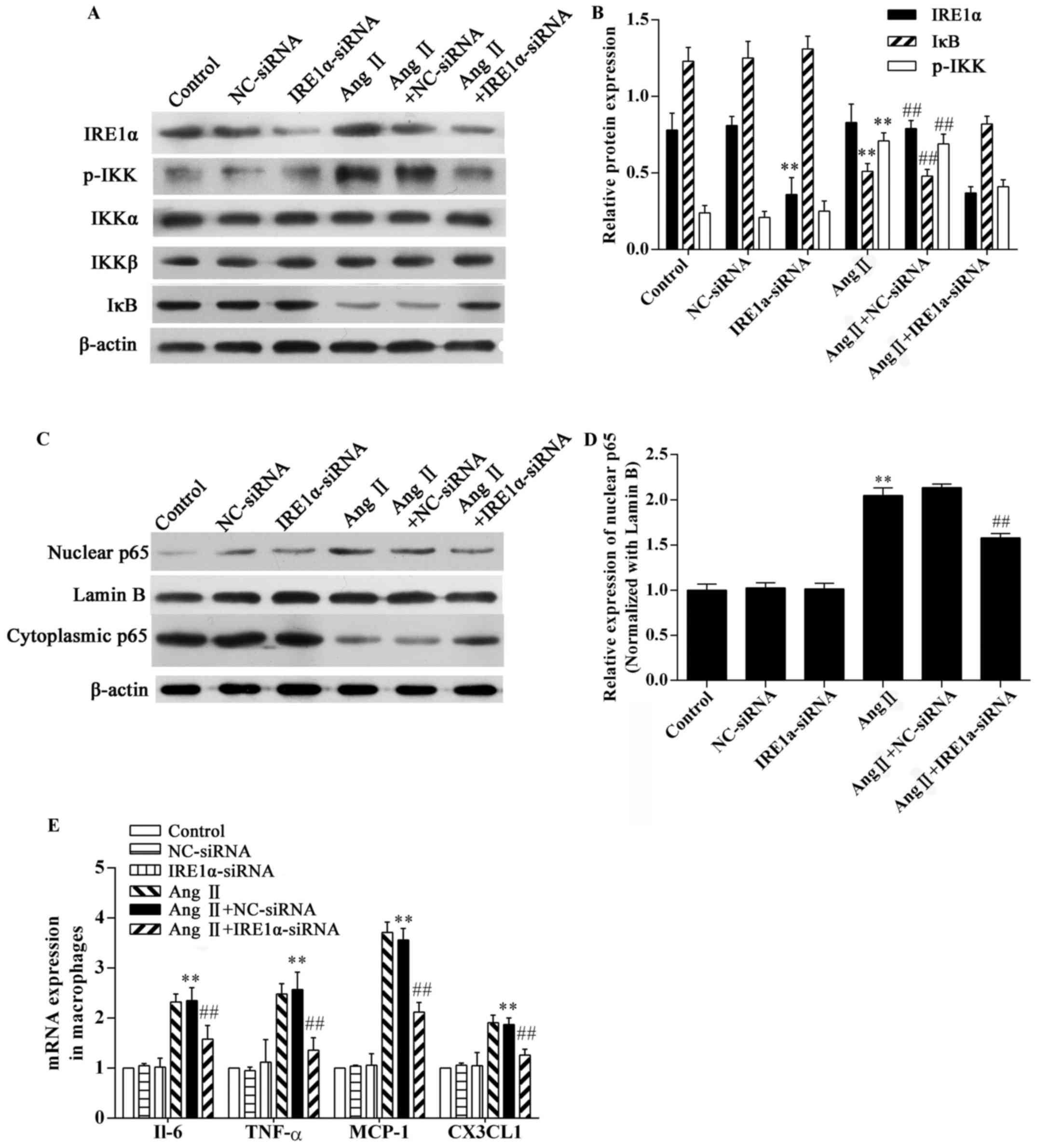
Effect of IRE1α-siRNA on the Ang
II-induced inflammatory response in RAW264.7 macrophages
To confirm the role of ER stress in Ang II-induced
inflammation, RAW264.7 macrophages were transfected with
IRE1α-specific siRNA or control-siRNA 48 h prior to exposure to Ang
II. The expression of the ER stress sensor, IRE1α, was
downregulated in the IRE1α-siRNA group (Fig. 4A and B). The results of western
blot analysis demonstrated that stimulation with Ang II
significantly increased IKK phosphorylation (P<0.01; Fig. 4A and B) and caused a marked
degradation of IκB in the RAW264.7 macrophages (P<0.01; Fig. 4A and B). Ang II stimulation
significantly increased NF-κB p65 in nuclear extracts from
macrophages, and the cytoplasmic NF-κB p65 content decreased
(Fig. 4C). Compared with the Ang
II-exposed group, IRE1α-siRNA decreased IKK phosphorylation and
inhibited IκB degradation (P<0.01; Fig. 4A and B) and inhibited NF-κB p65
nuclear translocation (P<0.01; Fig. 4C and D). In addition, Ang II
stimulation significantly upregulated the mRNA expression of
pro-inflammatory cytokines and chemokines in the RAW264.7
macrophages (P<0.01; Fig. 4E).
The depletion of IRE1α expression markedly downregulated the mRNA
expression of pro-inflammatory cytokines and chemokines (Fig. 4E). Without the presence of Ang II,
the IRE1α-specific siRNA did not affect IKK phosphorylation, IκB
degradation, or NF-κB p65 nuclear translocation (Fig. 4). The negative control (NC-siRNA)
did not influence the effects of Ang II (Fig. 4). Thus, Ang II-mediated
inflammation in RAW264.7 macrophages is partially dependent on the
activation of ER stress.
Discussion
A number of experimental and clinical studies have
demonstrated that RAS activation occurs during uremia, and RAS
plays a key role in the development and progression of uremic AS
(16,17,19). Similar to these previous findings
(16), our study found that the
Ang II serum concentration was significantly increased in uremic
mice, and that RAS blockade with losartan significantly abolished
the pro-atherogenic effects in apoE−/− mice subjected to
SNx. However, the exact mechanisms through which RAS promotes large
vessel disease in patients with CRF were not fully explained.
Atherosclerotic plaques, particularly advanced
lesions, contain abundant pro-inflammatory cytokines and chemokines
(20,21). Pedersen et al reported that
the pro-inflammatory response overrules the pro-atherogenic
potential of traditional factors during uremia-induced AS (6). In the present study, we induced
experimental mild uremia by subjecting apoE−/− mice to
SNx. The levels of Ang II were found to increase after SNx. It has
been demonstrated that the anti-atherogenic effects of losartan are
due to the direct inhibition of Ang II activity (22). Other events that occur due to Ang
II, such as hypertension (15),
may also be regulated by losartan. Furthermore, treatment with
losartan has been shown to prevent both coronary vascular injury
and myocyte damage induced by continuous Ang II infusion in rats
(23). We aim to investigate the
signaling pathways mediating the effects of losartan in the
future.
Chronic inflammation is an important risk factor for
the progression of AS in patients with CRF. According to our study,
macrophage accumulation was increased, and pro-inflammatory
cytokine and chemokine gene expression was upregulated in the
aortas from uremic mice. Treatment with losartan, an AT1 receptor
blocker, reduced macrophage accumulation and inhibited aortic
inflammation in the uremic mice. These molecular and cellular
changes were associated with accelerated AS. These findings are
consistent with those of previous studies (16,17). RAS inhibition abolished the
pro-atherogenic effecta of uremia, which was mainly dependent on
its anti-inflammatory mechanisms (16).
A recent study demonstrated that ER stress is a key
player during the development of AS. Prolonged ER stress triggers
macrophage and endothelial cell apoptosis, which in turn leads to
plaque necrosis. Another important pro-atherogenic effect of
prolonged ER stress is the activation of inflammatory pathways in
cells within the plaques, especially macrophages (11). To determine the association
between RAS and ER stress in uremic mice, we assessed the
expression and activation of the ER stress response signaling
proteins. GRP78 is a prominent ER-resident chaperone, which binds
to the three ER stress sensors. During ER stress, GRP78 stably
interacts with misfolded or unfolded proteins, and the three
previously mentioned GRP78-bound proteins, PERK, IRE1α and ATF6 are
free. The upregulation of GRP78 is a common ER stress marker
(10). In this study, we observed
that GRP78 upregulation was significantly inhibited in the aortas
of losartan-treated uremic mice. The three free proteins can
activate their respective downstream signaling pathways, and
prolong the activation of ER stress, causing disease. In this
study, among the three branches of ER stress signaling, we focused
on IRE1α to address the inflammation caused by uremia. In addition,
the effects of Ang II and losartan upon endothelial and vascular
smooth cells in the pathogenesis of AS cannot be neglected. A
previous in vivo study demonstrated that IRE1 signaling is
essential for ischemia-induced vascular endothelial growth factor
(VEGF)-A expression and contributes to angiogenesis (24). Exogenous Ang II can activate IRE1
protein expression in a dose-dependent manner in endothelial cells,
and the activation of IRE1 is necessary for both VEGF production
and capillary tube formation from endothelial cells induced by Ang
II (25).
IRE1α activates both the NF-κB and AP-1
transcriptional pathways, which are crucial regulators of the
inflammatory response (26–29). It is widely accepted that IRE1 is
involved in the activation of NF-κB induced by ER stress. In a
previous study, the activation of NF-κB by the ER stress-inducing
agents, thapsigargin and tunicamycin, was inhibited by a
dominant-negative IRE1 (27). Our
results also suggested that the NF-κB activation from the ER is
transduced by IRE1. Moreover, the study by Miyazaki-Anzai et
al demonstrated that CHOP inhibition alone was not sufficient
to inhibit CKD-dependent AS. The IRE1α-NF-κB axis of the ER stress
signal may be more important in the development of CKD-dependent AS
(30). In this study, losartan
treatment decreased IRE1α activation in atherosclerotic plaques.
From this perspective, we suggest that RAS activation may be
associated with ER stress-induced inflammation during uremic
AS.
To further explore the association between RAS
activation and ER stress-related inflammation, we performed in
vitro experiments. In the in vivo experiments, we
discovered the co-localization of CD68 and p-IRE1α by
immunohistochemistry using adjacent sections, showing that p-IRE1α
was mainly expressed in macrophages within the plaques. ER
stress-induced inflammation in macrophages is an important
contributor to AS (11). During
early development of AS, monocytes migrate into the arterial intima
by chemotaxis and then mature into macrophages. Macrophage
infiltration into the arteries is a key factor for atherosclerotic
plaque initiation. Macrophages in plaques produce pro-inflammatory
cytokines to promote AS progression (31,32). In our in vitro experiment,
Ang II was used to stimulate RAW264.7 macrophages as Ang II is the
primary effector of RAS. The results demonstrated that ER stress
was induced by Ang II stimulation in RAW264.7 macrophages. GRP78
expression was significantly upregulated, and IRE1α phosphorylation
was increased in the Ang II-stimulated macrophages. During ER
stress, IRE1α is activated by auto-phosphorylation at S724, and
IRE1α autophosphorylation induces a conformational change in its
cytosolic domain, which can subsequently bind to the adaptor
protein TNF-α-receptor associated factor 2 (TRAF2). The IRE1α-TRAF2
complex can phosphorylate IKK, which leads to IκB degradation and
the nuclear translocation of NF-κB (26–28). In this study, we determined that
stimulation with Ang II significantly upregulated IKK
phosphorylation, IκB degradation and NF-κB nuclear translocation.
More importantly, IRE1α depletion effectively downregulated IKK
phosphorylation and inhibited IκB degradation and NF-κB nuclear
translocation.
In previous studies, Ang II stimulation was shown to
effectively activate an inflammatory response. Guo et al
indicated that NF-κB is a critical inflammatory transcription
factor for Ang II-induced inflammation in RAW264.7 macrophages
(33). NF-κB is a pro-atherogenic
factor, mainly due to its regulation of many pro-inflammatory genes
that are linked to AS (34,35). This study indicated that ER stress
is an upstream signal of the NF-κB pathway. Ang II stimulation
significantly elevated pro-inflammatory cytokine and chemokine gene
expression in RAW264.7 macrophages, which was mostly dependent on
the activation of the IRE1α/IKK/NF-κB pathway. In this study,
losartan significantly enhanced the expression of IκB in Ang
II-stimulated RAW264.7 macrophages, suggesting the inhibition of
NF-κB by losartan in Ang II-stimulated RAW264.7 macrophages. This
further confirmed the effect of losartan on the IRE1α/IKK/NF-κB
pathway in vitro. In addition, it has been demonstrated that
phosphorylated forms of IRE1α could be detected by slower migration
upon stimulation with lipopolysaccharide (36). The knockdown of IRE1α in J774
macrophages resulted in reduced IL-6 induction by tunicamycin and
lipopolysaccharide co-treatment (36). Similarly, the effect of
IRE1α-siRNA on the Ang II-induced inflammatory response is not only
due to the reduced macrophage number.
Furthermore, MCP-1 has been detected in
atherosclerotic lesions (37,38). Blocking the expression of MCP-1 or
its receptor CCR2 decreases atheroma formation in
hypercholesterolemic mice (39,40). In future studies we aim to explore
the influence of MCP-1 and its receptor, CCR2, on ER stress-induced
AS.
In conclusion, our findings demonstrated that the
blockade of RAS by losartan almost completely prevented the
acceleration of atherosclerotic lesion formation in uremic
apoE−/− mice. This effect was associated with the
modulation of chronic inflammation via ER stress-dependent
mechanisms. This study further clarified the mechanism through
which RAS aggravates and accelerates atherogenesis in uremic
mice.
Acknowledgments
We are grateful to Mr. Zhengyi Wang, Ms. Jingmei Xie
and Ms. Yao Xiao for providing experiment technical support.
References
|
1
|
Go AS, Chertow GM, Fan D, McCulloch CE and
Hsu CY: Chronic kidney disease and the risks of death,
cardiovascular events, and hospitalization. N Engl J Med.
351:1296–1305. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Amann K, Gross ML and Ritz E:
Pathophysiology underlying accelerated atherogenesis in renal
disease: Closing in on the target. J Am Soc Nephrol. 15:1664–1666.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Foley RN, Parfrey PS and Sarnak MJ:
Clinical epidemiology of cardiovascular disease in chronic renal
disease. Am J Kidney Dis. 32(Suppl 3): S112–S119. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Seliger SL, Gillen DL, Tirschwell D, Wasse
H, Kestenbaum BR and Stehman-Breen CO: Risk factors for incident
stroke among patients with end-stage renal disease. J Am Soc
Nephrol. 14:2623–2631. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Banerjee D, Recio-Mayoral A, Chitalia N
and Kaski JC: Insulin resistance, inflammation, and vascular
disease in nondiabetic predialysis chronic kidney disease patients.
Clin Cardiol. 34:360–365. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Pedersen TX, Binder CJ, Fredrikson GN,
Nilsson J, Bro S and Nielsen LB: The pro-inflammatory effect of
uraemia overrules the anti-atherogenic potential of immunization
with oxidized LDL in apoE−/− mice. Nephrol Dial
Transplant. 25:2486–2491. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yilmaz MI, Stenvinkel P, Sonmez A, Saglam
M, Yaman H, Kilic S, Eyileten T, Caglar K, Oguz Y, Vural A, et al:
Vascular health, systemic inflammation and progressive reduction in
kidney function; clinical determinants and impact on cardiovascular
outcomes. Nephrol Dial Transplant. 26:3537–3543. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pluquet O, Pourtier A and Abbadie C: The
unfolded protein response and cellular senescence. A review in the
theme: Cellular mechanisms of endoplasmic reticulum stress
signaling in health and disease. Am J Physiol Cell Physiol.
308:C415–C425. 2015. View Article : Google Scholar
|
|
9
|
Ron D and Walter P: Signal integration in
the endoplasmic reticulum unfolded protein response. Nat Rev Mol
Cell Biol. 8:519–529. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sozen E, Karademir B and Ozer NK: Basic
mechanisms in endoplasmic reticulum stress and relation to
cardiovascular diseases. Free Radic Biol Med. 78:30–41. 2015.
View Article : Google Scholar
|
|
11
|
Gotoh T, Endo M and Oike Y: Endoplasmic
reticulum stress-related inflammation and cardiovascular diseases.
Int J Inflamm. 2011:2594622011. View Article : Google Scholar
|
|
12
|
Tabas I: The role of endoplasmic reticulum
stress in the progression of atherosclerosis. Circ Res.
107:839–850. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhou J, Lhoták S, Hilditch BA and Austin
RC: Activation of the unfolded protein response occurs at all
stages of atherosclerotic lesion development in apolipoprotein
E-deficient mice. Circulation. 111:1814–1821. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang J, Wen Y, Lv LL, Liu H, Tang RN, Ma
KL and Liu BC: Involvement of endoplasmic reticulum stress in
angiotensin II-induced LRP3 inflammasome activation in human renal
proximal tubular cells in vitro. Acta Pharmacol Sin. 36:821–830.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Young CN, Li A, Dong FN, Horwath JA, Clark
CG and Davisson RL: Endoplasmic reticulum and oxidant stress
mediate nuclear factor-κB activation in the subfornical organ
during angiotensin II hypertension. Am J Physiol Cell Physiol.
308:C803–C812. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bernardi S, Candido R, Toffoli B, Carretta
R and Fabris B: Prevention of accelerated atherosclerosis by AT1
receptor blockade in experimental renal failure. Nephrol Dial
Transplant. 26:832–838. 2011. View Article : Google Scholar
|
|
17
|
Bro S, Binder CJ, Witztum JL, Olgaard K
and Nielsen LB: Inhibition of the renin-angiotensin system
abolishes the proatherogenic effect of uremia in apolipoprotein
E-deficient mice. Arterioscler Thromb Vasc Biol. 27:1080–1086.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Yao S, Miao C, Tian H, Sang H, Yang N,
Jiao P, Han J, Zong C and Qin S: Endoplasmic reticulum stress
promotes macrophage-derived foam cell formation by up-regulating
cluster of differentiation 36 (CD36) expression. J Biol Chen.
289:4032–4042. 2014. View Article : Google Scholar
|
|
19
|
Berger AK, Duval S and Krumholz HM:
Aspirin, beta-blocker, and angiotensin-converting enzyme inhibitor
therapy in patients with end-stage renal disease and an acute
myocardial infarction. J Am Coll Cardiol. 42:201–208. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Mach F: The role of chemokines in
atherosclerosis. Curr Atheroscler Rep. 3:243–251. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Hansson GK: Inflammatory mechanisms in
atherosclerosis. J Thromb Haemost. 7(Suppl 1): 328–331. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Liang C, Wu ZG, Ding J, Jiang JF, Huang
GZ, Du RZ and Ge JB: Losartan inhibited expression of matrix
metalloproteinases in rat atherosclerotic lesions and angiotensin
II-stimulated macrophages. Acta Pharmacol Sin. 25:1426–1432.
2004.PubMed/NCBI
|
|
23
|
Kabour A, Henegar JR, Devineni VR and
Janicki JS: Prevention of angiotensin II induced myocyte necrosis
and coronary vascular damage by lisinopril and losartan in the rat.
Cardiovasc Res. 29:543–548. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Drogat B, Auguste P, Nguyen DT,
Bouchecareilh M, Pineau R, Nalbantoglu J, Kaufman RJ, Chevet E,
Bikfalvi A and Moenner M: IRE1 signaling is essential for
ischemia-induced vascular endothelial growth factor-A expression
and contributes to angiogenesis and tumor growth in vivo. Cancer
Res. 67:6700–6707. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wang X, Bai YP, Hong D, Gao HC, Li LF, Li
CC, Zhu LP, Sun Q and Zhang GG: Ang II induces capillary formation
from endothelial cells via the AT1R-dependent inositol requiring
enzyme 1 pathway. Biochem Biophys Res Commun. 434:552–558. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hu P, Han Z, Couvillon AD, Kaufman RJ and
Exton JH: Autocrine tumor necrosis factor alpha links endoplasmic
reticulum stress to the membrane death receptor pathway through
IRE1alpha-mediated NF-kappaB activation and downregulation of TRAF2
expression. Mol Cell Biol. 26:3071–3084. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Kaneko M, Niinuma Y and Nomura Y:
Activation signal of nuclear factor-kappa B in response to
endoplasmic reticulum stress is transduced via IRE1 and tumor
necrosis factor receptor-associated factor 2. Biol Pharm Bull.
26:931–935. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wu L, Wang D, Xiao Y, Zhou X, Wang L, Chen
B, Li Q, Guo X and Huang Q: Endoplasmic reticulum stress plays a
role in the advanced glycation end product-induced inflammatory
response in endothelial cells. Life Sci. 110:44–51. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Urano F, Wang X, Bertolotti A, Zhang Y,
Chung P, Harding HP and Ron D: Coupling of stress in the ER to
activation of JNK protein kinases by transmembrane protein kinase
IRE1. Science. 287:664–666. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Miyazaki-Anzai S, Masuda M, Demos-Davies
KM, Keenan AL, Saunders SJ, Masuda R, Jablonski K, Cavasin MA,
Kendrick J, Chonchol M, et al: Endoplasmic reticulum stress
effector CCAAT/enhancer-binding protein homologous protein (CHOP)
regulates chronic kidney disease-induced vascular calcification. J
Am Heart Assoc. 3:e0009492014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Galkina E and Ley K: Immune and
inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol.
27:165–197. 2009. View Article : Google Scholar
|
|
32
|
Libby P: Inflammation in atherosclerosis.
Nature. 420:868–874. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Guo F, Chen XL, Wang F, Liang X, Sun YX
and Wang YJ: Role of angiotensin II type 1 receptor in angiotensin
II-induced cytokine production in macrophages. J Interferon
Cytokine Res. 31:351–361. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Xanthoulea S, Curfs DM, Hofker MH and de
Winther MP: Nuclear factor kappa B signaling in macrophage function
and atherogenesis. Curr Opin Lipidol. 16:536–542. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
de Winther MPJ, Kanters E, Kraal G and
Hofker MH: Nuclear factor kappaB signaling in atherogenesis.
Arterioscler Thromb Vasc Biol. 25:904–914. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Martinon F, Chen X, Lee AH and Glimcher
LH: TLR activation of the transcription factor XBP1 regulates
innate immune responses in macrophages. Nat Immunol. 11:411–418.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Takeya M, Yoshimura T, Leonard EJ and
Takahashi K: Detection of monocyte chemoattractant protein-1 in
human atherosclerotic lesions by an anti-monocyte chemoattractant
protein-1 monoclonal antibody. Hum Pathol. 24:534–539. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Ylä-Herttuala S, Lipton BA, Rosenfeld ME,
Särkioja T, Yoshimura T, Leonard EJ, Witztum JL and Steinberg D:
Expression of monocyte chemoattractant protein 1 in macrophage-rich
areas of human and rabbit atherosclerotic lesions. Proc Natl Acad
Sci USA. 88:5252–5256. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Boring L, Gosling J, Cleary M and Charo
IF: Decreased lesion formation in CCR2−/− mice reveals a
role for chemokines in the initiation of atherosclerosis. Nature.
394:894–897. 1998. View
Article : Google Scholar : PubMed/NCBI
|
|
40
|
Gu L, Okada Y, Clinton SK, Gerard C,
Sukhova GK, Libby P and Rollins BJ: Absence of monocyte
chemoattractant protein-1 reduces atherosclerosis in low density
lipoprotein receptor-deficient mice. Mol Cell. 2:275–281. 1998.
View Article : Google Scholar : PubMed/NCBI
|