Introduction
The temporomandibular joint (TMJ) is a synovial
joint that participates in very complicated movement. Excessive
mechanical stress caused by severe malocclusion, jaw asymmetry, and
muscle overuse during mastication against the TMJ contribute to
osteoarthritis (OA) pathogenesis (1–3).
Patients suffering from TMJ-OA show various symptoms such as
cartilage degeneration, bone spur formation, and fibrosis
accompanied by chronic inflammation in the synovial tissue,
resulting in TMJ pain and stiffness (1,4,5).
The pathogenesis of fibrosis results from excess
connective tissue deposition, which causes failure of various
organs including scleroderma (6).
A typical characteristic of fibrosis is the transition of
fibroblasts into myofibroblasts (MFs) (7–9).
MFs vigorously secrete large amounts of extracellular matrix (ECM)
proteins such as type I collagen, fibronectin, tenascin C, and
metalloproteinases, resulting in fibrosis (10). MFs are also recognized as smooth
muscle cell (SMC)-like contractile cells (9): MFs vigorously express contractile
proteins such as α-smooth muscle actin (α-SMA) and SM22, resulting
in enhanced MF migratory activity and the progression of wound
healing, especially wound closure (10). Thus, MFs play important roles in
the progression of wound healing. However, abnormally elevated
fibrogenic activity of MFs causes pathological organ fibrosis and
excessive scar formation (11).
α-SMA expression has been identified in fibroblastic cells in
fibrosis lesions (12).
Transforming growth factor-β (TGF-β) is a key inducer of fibrosis
in various organs in humans (6).
Fahlgren et al previously reported that TGF-β1 secretion
into synovial fluid was increased in patients suffering from OA in
the knee (13). Another study
found that TGF-β1 induced osteophyte formation at characteristic OA
sites in an experimental model of mouse OA (14). In addition, TGF-β is well known to
be a potent inducer of MF differentiation in cells derived from
mesenchymal origin, which are typically characterized by the
expression of α-SMA and type I collagen (15).
The fibroblast growth factor (FGF) family consists
of 18 members including FGF-1 to -10 and FGF-16 to -23 which are
classified into 6 subfamilies (16). It is generally known that FGF-11
to -15 are homologs of the FGF family, but do not activate any FGF
receptors (FGFRs). Therefore, FGF-11 to -15 are not recognized as
members of the FGF family. FGF family proteins affect various
cellular responses such as cell growth, migration, differentiation,
and apoptosis (16). These FGF
ligands specifically bind to FGFRs, consisting of FGFR1-4, which
belong to the receptor tyrosine kinase (RTK) family (17). FGF-1 binds to FGFR1-4 (18) to activate various intracellular
signaling molecules including mitogen-activated protein kinases
(MAPKs) and phosphoinositide 3-kinase (PI3K)/Akt (19). Shimbori et al demonstrated
that FGF-1 attenuates TGF-β1-induced lung fibrosis by inhibiting
TGF-β1-induced MF differentiation of lung fibroblasts (20). In addition, Maltseva et al
reported that FGF-1 and FGF-2 reversed TGF-β1-induced MF
differentiation of corneal fibroblastic cells (21). Takahashi et al also
reported that FGF-1 suppressed the expression of SMC
differentiation markers including α-SMA in periodontal ligament
(PDL)-derived endothelial progenitor cells in a MAPK/extracellular
signal-regulated kinase (ERK)-dependent manner (22). We previously demonstrated that
epidermal growth factor (EGF) attenuates the MF differentiation of
PDL-derived endothelial progenitor cells in a MAPK/ERK-dependent
manner (23). However, it remains
to be clarified whether FGF-1-induced intracellular signals affect
the MF differentiation status of fibroblast-like synoviocytes
(FLSs) derived from the mouse TMJ.
On the other hand, RhoA, belonging to the Rho family
of GTPases, is activated by various cytokines including TGF-β
(24). Activated RhoA
sequentially activates Rho-associated coiled-coil forming kinase
(ROCK) to promote actin dynamics and cytoskeletal reorganization by
incorporating globular actin (G-actin) into growing filamentous
actin (F-actin) stress fibers. Myocardin-related transcription
factor (MRTF) is a key molecule for promoting gene expression of MF
differentiation markers including α-SMA and type I collagen in
cooperation with serum response factor (SRF). MRTFs bind to G-actin
and are sequestered in the cytoplasm. F-actin stress fiber
formation allows the MRTFs to be released from G-actin, allowing
MRTFs to enter the nucleus and promote the expression of MF
differentiation markers (11).
Thus, TGF-β induces myofibroblastic differentiation
of fibroblasts through RhoA/ROCK/actin/MRTF signaling. However, it
remains to be clarified how this pathway affects the status of
myofibroblastic characters of synoviocytes derived from the TMJ.
Here, we established a FLS cell line derived from the mouse TMJ.
Then, we examined the effects of i) the MF differentiation-inducer
TGF-β1; ii) the MF differentiation-attenuator FGF-1; iii) an
inhibitor of the actin-polymerizing agent ROCK, Y-27632; iv) the
actin-depolymerizing agent, cytochalasin B (CytB); and v) an
inhibitor of MRTF/SRF-regulated transcription, CCG-100602, on the
MF differentiation status of FLSs.
Materials and methods
Reagents
Recombinant human TGF-β1 and recombinant mouse FGF-1
were purchased from PeproTech Inc. (Rocky Hill, NJ, USA). The ROCK
inhibitor Y-27632, the actin-depolymerizing agent CytB, and the
FGFR1 inhibitor SU-5402 were purchased from Wako Pure Chemical
Industries Ltd. (Osaka, Japan). The inhibitor of MRTF/SRF-regulated
transcription CCG-100602 was purchased from Cayman Chemical Inc.
(Ann Arbor, MI, USA).
Establishment of an FLS cell line and
cell culture
To prepare FLSs derived from the mouse TMJ, TMJ
synovial tissue was obtained from seven 8-week-old female mice
(C57BL/6J) got from got from CLEA Japan, Inc. (Tokyo, Japan). Then,
the tissue was rinsed once in Nutrient Mixture F-12 Ham (Ham's
F-12; Sigma, St. Louis, Mo, USA) medium supplemented with kanamycin
(100 µg/ml). The tissue was then immersed in digestion
solution composed of 20 ml of Ham's F-12 containing 2 mg/ml
collagenases consisting of class I and class II collagenases
(Collagenase NB4; Wako, Tokyo, Japan), at 37°C for 30 min with
continuous vigorous rocking. The solution was centrifuged to
collect the digested tissue released from synovial tissue of TMJ.
The released tissue was then treated with 0.1% trypsin (Difco
Laboratories Inc., Detroit, MI, USA) in phosphate-buffered saline
(PBS) at 37°C for 10 min with continuous vigorous rocking. The
solution was then centrifuged to collect the cells released from
the tissue. The isolated cells were maintained in growth medium,
Ham's F-12 supplemented with 10% fetal bovine serum (FBS) and
penicillin-streptomycin (Invitrogen Life Technologies, Carlsbad,
CA, USA), for the subsequent plasmid transfection. To establish a
FLS cell line, FLSs were transfected with pBABE-puro-simian virus
40 large T antigen (SV40LT) expression plasmid (no. 13970) obtained
from Addgene, Inc. (Cambridge, MA, USA) with Lipofectamine LTX
reagent (Thermo Fisher Scientific Inc., Waltham, MA, USA) according
to the manufacturer's protocol. The transfected FLSs were selected
with 2 µg/ml puromycin (Clontech Laboratories, Mountain
View, CA, USA). After 14 days, SV40LT-transfected cells were
maintained in serial culture with Ham's F-12 supplemented with 2 mM
glutamine, 10% FBS, and penicillin-streptomycin (Invitrogen Life
Technologies), but without puromycin. Then, the SV40LT-transfected
cells were subcultured at a 1:4 ratio when the cells reached
subconfluency. For the following experiments, the cells at 50
passages (immortalized FLSs) were used unless otherwise indicated.
Apart from the immortalized FLSs, primary FLS cultures were
maintained in the previously mentioned growth medium. NIH3T3 mouse
embryonic fibroblasts and RAW264.7 mouse monocytes/macrophages were
obtained from Riken Cell Bank (Tsukuba Science City, Japan) and
maintained in a growth medium consisting of minimum essential
medium Eagle's α-modification (α-MEM) (Wako Pure Chemical
Industries Ltd.) supplemented with 10% FBS and
penicillin-streptomycin (Invitrogen Life Technologies).
Mice
This study was approved by the Ethics Committee for
Animal Research of Iwate Medical University (approval no. 27-001).
Breeding and care of mice, and all animal experiments were carried
out in accordance with the Guidelines for the Animal Experiments of
Iwate Medical University and Act on Welfare and Managements of
Animals of Japan.
Cell viability assay
The status of cell viability was evaluated using an
alamarBlue® assay (AbDSerotec, Oxon, UK) according to
the manufacturer's protocol. This assay reagent includes an
indicator that fluoresces and undergoes a colorimetric change when
reduced by mitochondrial respiration, which is proportional to the
number of living cells. For the viability assay, the cells were
seeded into 96-well plates at a density of 1.0×104 cells
per well and cultured for 24 h in a medium containing 10% FBS.
Then, the cells were subsequently treated with Y-27632 (10
µM), CytB (5 µM), CCG-100602 (10 µM), or FGF-1
(10 ng/ml) in medium containing 10% FBS for the indicated times.
For FGF-1 stimulation, some cells were pre-treated with SU-5402 30
min before FGF-1 administration. Then, the medium was replaced with
Ham's F-12 containing 10% alamarBlue solution to evaluate cell
viability, and the cells were cultured for an additional 4.5 h. The
absorbance (Abs) for each well was measured using an ELISA plate
reader (Tosoh Corp., Tokyo, Japan). The data were shown as values
of Abs570-Abs600. Each experiment was repeated 3 times, with
8-wells dedicated for each time point.
RNA isolation and RT-qPCR
Total RNA was isolated from FLSs, NIH3T3 cells and
RAW264.7 cells with Isogen reagent (Nippon Gene, Toyama, Japan)
according to the manufacturer's protocol. First-strand cDNA was
synthesized from total RNA using the PrimeScript RT reagent kit
(Takara Bio, Inc., Shiga, Japan). PCR was subsequently performed on
a Thermal Cycler Dice Real Time System (Takara Bio, Inc.) using
SYBR Premix Ex Taq II (Takara Bio, Inc.) with specific
oligonucleotide primers [mouse α-SMA, 5′-CAGATGTGGATCAGCAAACAGGA-3′
(forward) and 5′-GACTTAGAAGCATTTGCGGTGGA-3′ (reverse); mouse α1
chain of collagen type I (colIα1), 5′-GACATGTTCAGCTTTGTGGACCTC-3′
(forward) and 5′-GGGACCCTTAGGCCATTGTGTA-3′ (reverse); mouse
vimentin, 5′-ACCGCTTTGCCAACTACAT-3′ (forward) and
5′-TTGTCCCGCTCCACCTC-3′ (reverse); mouse CD45,
5′-GAACATGCTGCCAATGGTTCT-3′ (forward) and
5′-TGTCCCACATGACTCCTTTCC-3′ (reverse); mouse FGFR1-IIIb,
5′-TAACCGAAGGCAACCTCTGCTC-3′ (forward) and
5′-TGGTACCAGGCAGGTATTTGGTC-3′ (reverse); mouse FGFR2-IIIc,
5′-TGTTTCAACTCTGCTGTCCGATG-3′ (forward) and
5′-ATCTTGGGATGAGGATGCTGGTA-3′ (reverse); mouse FGFR3-IIIb,
5′-CTTCCAGGGACCATTGTGGAG-3′ (forward); 5′-AGAGGTTTGCCACACAGAGCAG-3′
(reverse); mouse FGFR4, 5′-CGAGGCATGCAGTATCTGG-3′ (forward) and
5′-CAAAGTCAGCGATCTTCATCACA-3′ (reverse); and mouse GAPDH,
5′-TGTGTCCGTCGTGGATCTGA-3′ (forward) and
5′-TTGCTGTTGAAGTCGCAGGAG-3′ (reverse)]. mRNA levels of α-SMA,
colIα1, vimentin, FGFR1-IIIb, FGFR2-IIIc, FGFR3-IIIb, FGFR4 and
CD45 were normalized to the level of GAPDH, and the relative
expression levels were shown as the fold increase or decrease
relative to the control.
Western blot analysis
Cells were lysed in RIPA buffer (Sigma) [50 mM
Tris-HCl (pH 7.2), 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate
and 0.1% SDS] or lysis buffer [20 mM HEPES (pH 7.5), 150 mM NaCl, 1
mM EDTA, 1% Triton X-100] containing protease and phosphatase
inhibitor cocktails (Sigma). The protein content of the samples was
measured using BCA reagent (Pierce Biotechnology, Inc., Rockford,
IL, USA). Samples containing equal amounts of protein were
separated on 10% SDS-polyacrylamide gel and transferred onto
polyvinylidene difluoride membranes (Millipore Corp., Bedford, MA,
USA). After blocking with 1% BSA or 1% skim milk in T-TBS [50 mM
Tris-HCl (pH 7.2), 150 mM NaCl and 0.05% Tween-20], membranes were
incubated with the appropriate primary antibody, including
anti-Smad2/3 purified mouse monoclonal antibody (1:1,000; 610842;
BD Biosciences Transduction Laboratories, Franklin Lakes, NJ, USA)
or anti-phospho-Smad2/3 rabbit polyclonal antibody (1:1,000; Cell
Signaling Technology, Inc., Beverly, MA, USA). The blots were then
incubated with the appropriate alkaline phosphatase-conjugated
secondary antibody and signals were detected using an alkaline
phosphatase substrate kit (BCIP/NBT substrate kit; Vector
Laboratories Inc., Burlingame, CA, USA).
Immunofluorescence analysis of cultured
cells
For immunofluorescence analysis of cultured cells,
FLSs and NIH3T3 cells were subcultured on non-coated 8-well glass
culture slides (Thermo Fisher Scientific Inc.) at a density of
2×104 cells/well, and maintained in Ham's F-12
supplemented with 2 mM glutamine and 10% FBS. Cells were cultured
with or without Y-27632 (10 µM), CytB (5 µM),
CCG-100602 (10 µM) or FGF-1 (10 ng/ml) for the indicated
times. Then, the cells were fixed in 4% paraformaldehyde (Nacalai
Tesque, Inc., Kyoto, Japan) for 15 min and permeabilized with
Triton X-100 (Sigma). After background reduction with normal goat
serum, the cells were incubated with anti-MKL1/MRTF-A rabbit
polyclonal antibody (1:200; no. 14760; Cell Signaling Technology,
Inc.) or anti-SV40LT mouse monoclonal antibody (1:100; ab16879;
Abcam, Cambridge, UK) at room temperature for 1 h. After washing
with PBS to remove the excess primary antibody, the cells were
incubated with Alexa Fluor 568-conjugated goat anti-rabbit IgG or
Alexa Fluor 568-conjugated goat anti-mouse IgG as appropriate
(1:400; A-11011, and A-11031, respectively; Molecular Probes,
Leiden, The Netherlands). After washing with PBS to remove the
excess secondary antibody, the cells were stained with Alexa Fluor
488-conjugated phalloidin (1:1,000; Thermo Fisher Scientific Inc.),
which specifically detects F-actin. After washing with PBS to
remove excess phalloidin, nuclei were stained with the
DAPI-containing mounting medium, DAPI Fluoromount-G
(SouthernBiotech Inc., Birmingham, AL, USA). Then, the fluorescent
signal was detected using an Olympus IX70 fluorescence microscope
with the LCPIanFI 20 objective lens (Olympus Corp., Tokyo,
Japan).
Statistical analysis
The data are presented as the means ± SD (n=3, or
n=8). The data were statistically analyzed by Student's t-test, and
P<0.01 and P<0.05 are indicative of statistical significant
results. The results shown in all experiments are representative of
at least three independent experiments.
Results
Primary and immortalized synoviocytes
derived from TMJ synovial tissue exhibit myofibroblastic
characteristics
We first attempted to culture FLSs as a primary
culture as described in Materials and methods. However, as shown in
Fig. 1A, primary cultures of FLS
did not divide after 7 passages (28 divisions). To examine the
various characteristics of FLSs from TMJ synovial tissue, such as
growth, differentiation and metabolism, we immortalized FLSs by
overexpressing SV40LT. SL40LT overcomes cellular senescence in
fibroblastic cells (25). After
transfection of the SV40LT expression plasmid, we obtained one
clonal FLS population that was resistant to antibiotic selection.
The SV40LT-transfected FLSs exhibited an elongated fibroblastic
appearance (Fig. 1B), similar to
the appearance of FLSs in primary culture (data not shown). To
analyze the growth of the SV40LT-overexpressing FLSs, subculturing
and passaging were performed as described in Materials and methods.
SV40LT expression (red) was detected in the nuclei of the
SV40LT-transfectants using immunofluorescence analysis as described
in Materials and methods (Fig.
1C, upper left panel). Cumulative doubling times were plotted
against cumulative culture days (Fig.
1A). After SV40LT overexpression in FLSs, the cellular life
span was elongated compared to that of the primary FLS cultures,
and immortalized cells exhibited stable proliferative activity by
successful passaging at 3–4 day intervals for more than 120 days.
We named the established FLS cell line FLS1, and used this name
hereafter. Next, we compared characteristics of the FLS1 cells with
primary FLS cultures, mouse NIH3T3 embryonic fibroblasts as a
standard fibroblast control, and mouse monocyte/macrophage RAW264.7
cells as a negative fibroblast control. As shown in Fig. 1D, the mesenchymal cell marker
vimentin was strongly expressed in the primary FLSs, moderately
expressed in the FLS1 cells and NIH3T3 cells, and weakly expressed
in the RAW264.7 cells. MF marker α-SMA was vigorously expressed in
the primarily cultured FLSs and FLS1 cells, and was not detected in
the NIH3T3 and RAW264.7 cells (Fig.
1E). MF marker colIα1 was vigorously expressed in the primarily
cultured FLSs and FLS1 cells, very weakly expressed in the NIH3T3
cells, and was not detected in the RAW264.7 cells (Fig. 1F). Thus, the FLS1 cells more
potently expressed α-SMA and colIα1 mRNA than did the standard
NIH3T3 fibroblasts. On the other hand, FLS1 and NIH3T3 cells did
not express the leukocyte marker CD45 although primary FLSs
slightly expressed CD45, suggesting that the primary FLS cultures
contained a leukocyte fraction (Fig.
1G). Intriguingly, immunofluorescence analysis also revealed
that the FLS1 cells vigorously formed F-actin (green) (Fig. 1H, left panel), but NIH3T3 cells
did not (Fig. 1H, right panel),
suggesting that ROCK/actin/MRTF-mediated signaling for promoting MF
marker gene expression may be constitutively activated in the FLS1
cells.
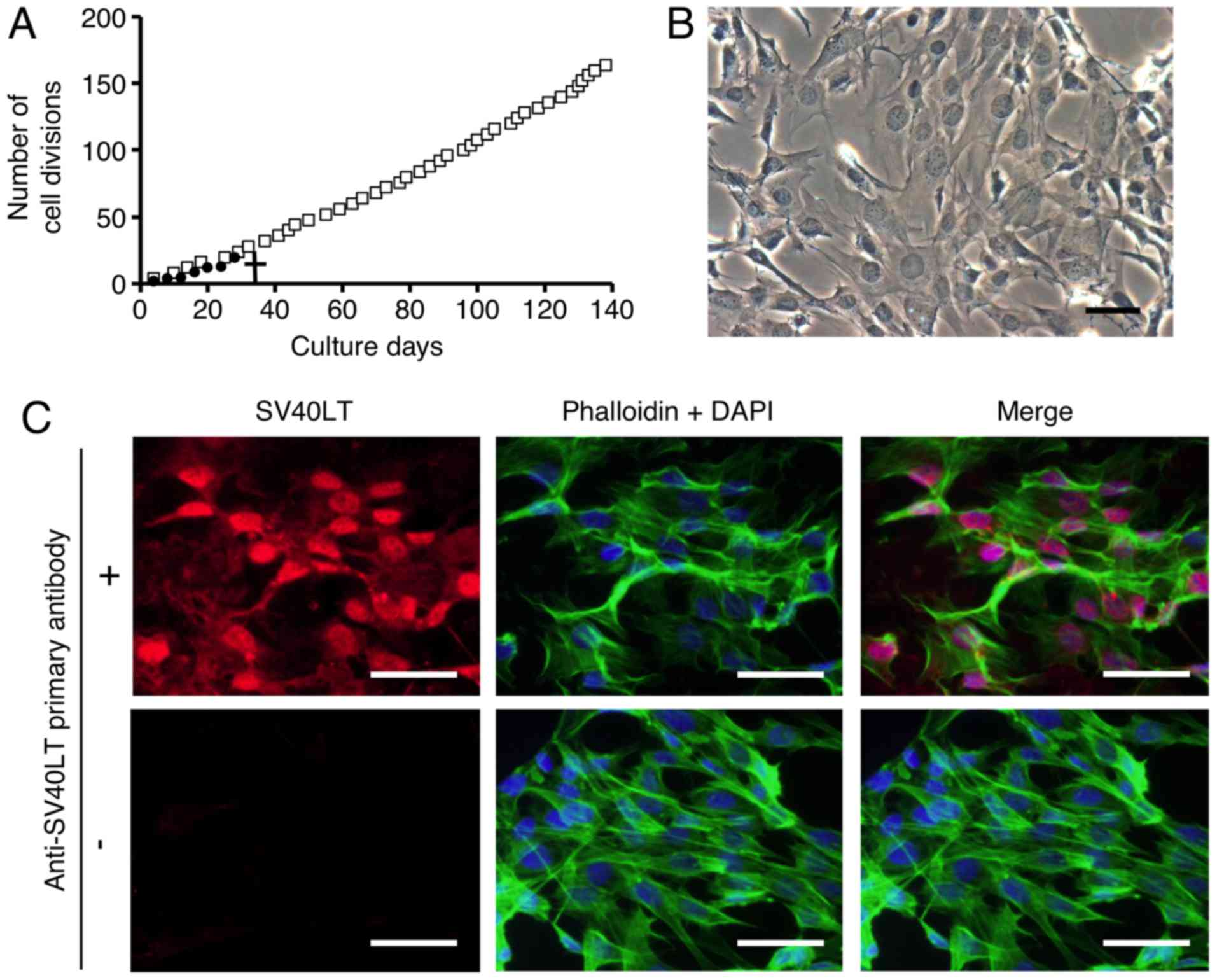 | Figure 1Primary and immortalized synoviocytes
derived from temporomandibular joint synovial tissue exhibit
myofibroblastic characteristics. After antibiotic selection, one
SV40LT-overexpressing fibroblast-like synoviocyte (FLS) cell line
was cloned and an FLS cell line was established. (A) Constant
proliferative activity was observed in the SV40LT-transfected
cells. Cumulative doubling times were plotted against cumulative
culture days (open squares, SV40LT-transfected cells; closed
circles, primary FLSs). A cross represents the proliferation limit
of the primary FLSs. (B) Morphological characteristics of
SV40LT-transfected FLSs on plastic culture plates was viewed by
phase-contrast microscopy (scale bar, 50 µm). (C) Expression
of SV40LT (red) in the SV40LT-transfected FLSs was evaluated at the
protein level using immunofluorescent analysis (upper left panel,
with the primary antibody against SV40LT, and the fluorescent
secondary antibody; lower left panel, without primary antibody
against SV40LT but with the fluorescent secondary antibody).
Filamentous actin (green) was stained with phalloidin, and nuclei
(blue) were labeled with DAPI (scale bar, 50 µm). (D-G) The
relative expression of mesenchymal, myofibroblast (MF), and
leukocyte markers were evaluated using RT-qPCR (D, mesenchymal cell
marker vimentin; E and F, MF markers α-smooth muscle actin (α-SMA)
and α1 chain of collagen type I (colIα1), respectively; and G,
leukocyte marker CD45). Data represent the means ± SD (n=3).
*P<0.01, and **P<0.05 are indicative of
statistical significance. (H) F-actin formation was examined using
immunofluorescence analysis. FLS1 cells (left panel), and NIH3T3
cells (right panel) were stained with phalloidin (green) and DAPI
(blue) (scale bar, 50 µm). |
These results indicated that immortalized FLSs
derived from mouse TMJ synovial tissue, FLS1 cells, showed
myofibroblastic characteristics, similar to primary FLS
cultures.
ROCK inhibition downregulates mRNA levels
of MF markers in FLS1 cells
We first evaluated the effect of the MF inducer
TGF-β1 on the status of MF marker expression in FLS1 cells.
Unexpectedly, TGF-β1 (10 ng/ml) did not affect the expression of MF
markers α-SMA or colIα1 in the FLS1 cells 24 h after TGF-β1
administration (data not shown). We evaluated the effect of
inhibiting the actin-polymerizing agent ROCK on MF marker
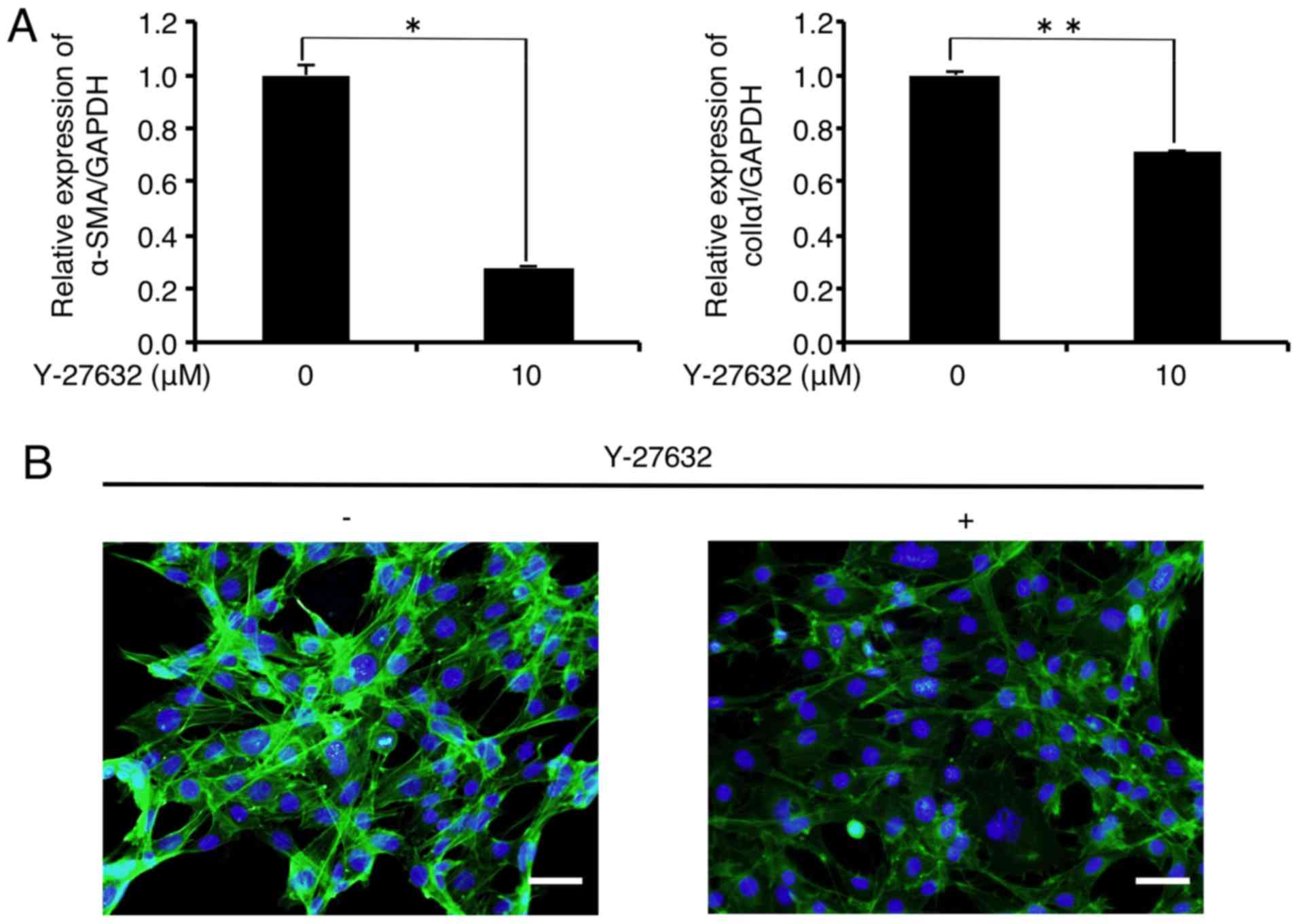
expression in the FLS1 cells using Y-27632. As shown in Fig. 2A, the mRNA levels of α-SMA and
colIα1 were significantly decreased to 28.2 and 70.9% of the
controls, respectively, 6 h after the administration of Y-27632 (10
µM). In addition, immunofluorescence analysis revealed that
Y-27632 (10 µM) inhibited F-actin formation (green) in the
FLS1 cells 6 h after administration (Fig. 2B). We also found that Y-27632 (10
µM) did not affect FLS1 cell viability 6 h after
administration (data not shown).
Actin-depolymerization downregulates mRNA
levels of MF differentiation markers in FLS1 cells
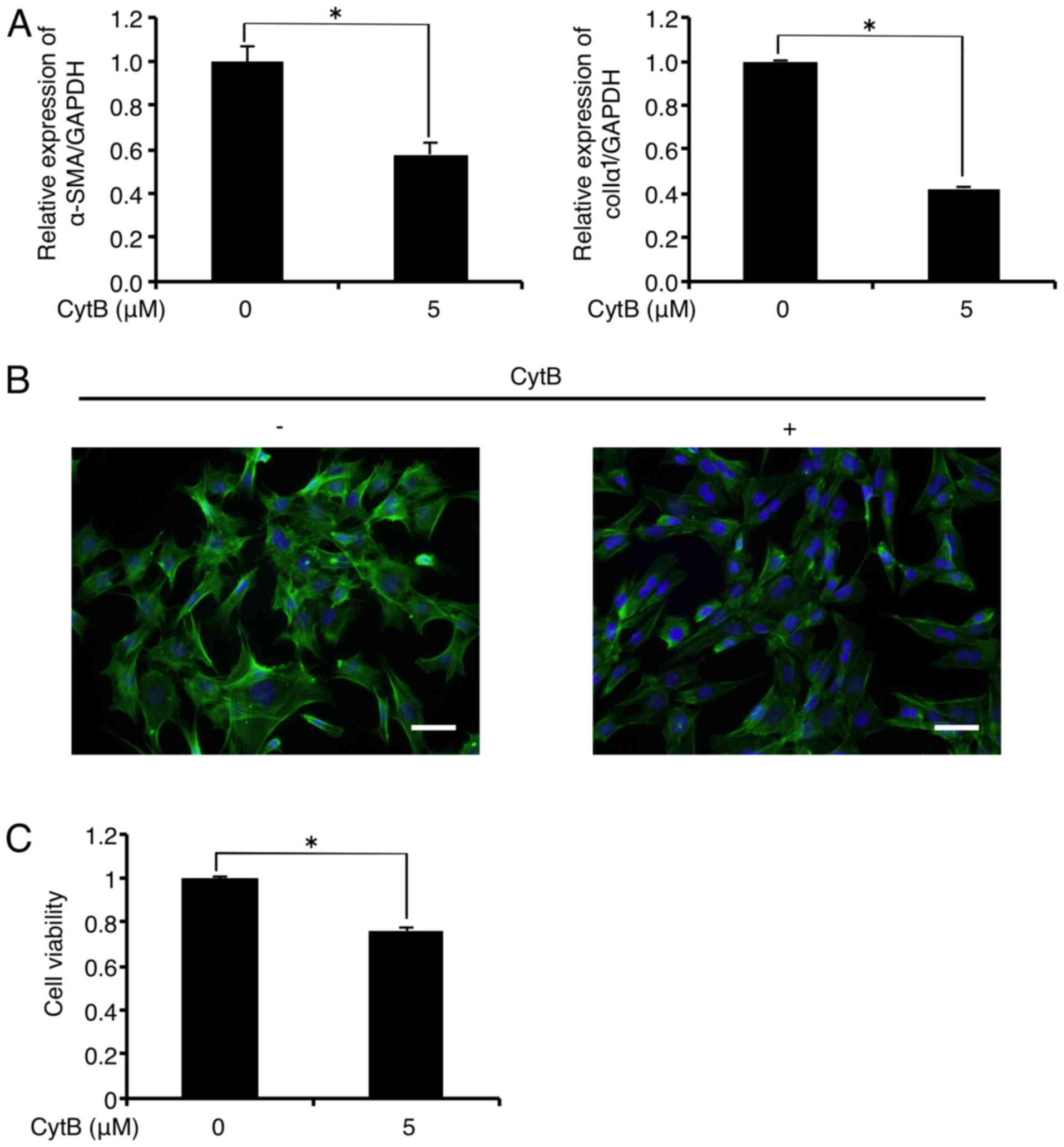
Treatment with the actin-depolymerizing agent CytB
(5 µM) significantly decreased mRNA levels of α-SMA and
colIα1 in the FLS1 cells to 57.8 and 42.3% of the controls,
respectively, 6 h after administration (Fig. 3A). In addition, immunofluorescence
analysis revealed that CytB (5 µM) treatment inhibited
F-actin formation (green) in the FLS1 cells 6 h after
administration (Fig. 3B). CytB (5
µM) also decreased FLS1 cell viability to 76.1% of the
control 6 h after administration (Fig. 3C).
Inhibition of MRTF/SRF-regulated
transcription downregulates mRNA levels of MF differentiation
markers in FLSs
CCG-100602 is a compound related to CCG-1423 that
blocks the serum-induced nuclear import of MRTF-A, and possesses a
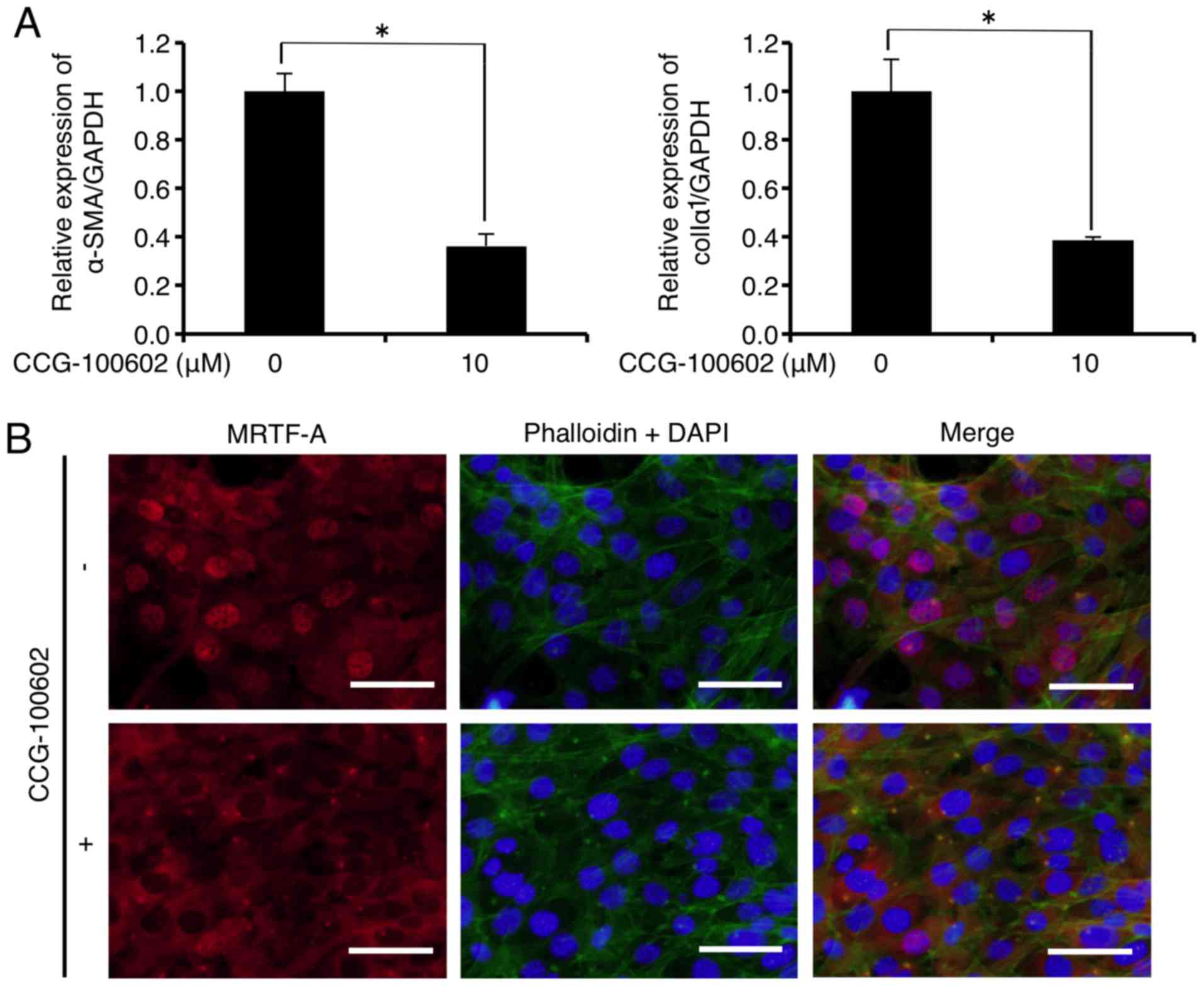
similar biological activity to CCG-1423 (26). As shown in Fig. 4A, mRNA levels of α-SMA and colIα1
were significantly decreased to 36.3 and 38.6% of the control,
respectively, 24 h after CCG-100602 (10 µM) administration.
In addition, immu-nofluorescence analysis showed that CCG-100602
(10 µM) did not inhibit F-actin formation (Fig. 4B upper and lower central panels,
green), but inhibited the MRTF-A nuclear localization (Fig. 4B upper and lower left panels, red)
in the FLS1 cells 24 h after administration. CCG-100602 treatment
did not affect FLS1 cell viability 24 h after administration (data
not shown).
MF differentiation attenuator FGF-1
downregulates mRNA expression levels of MF differentiation markers
in FLSs
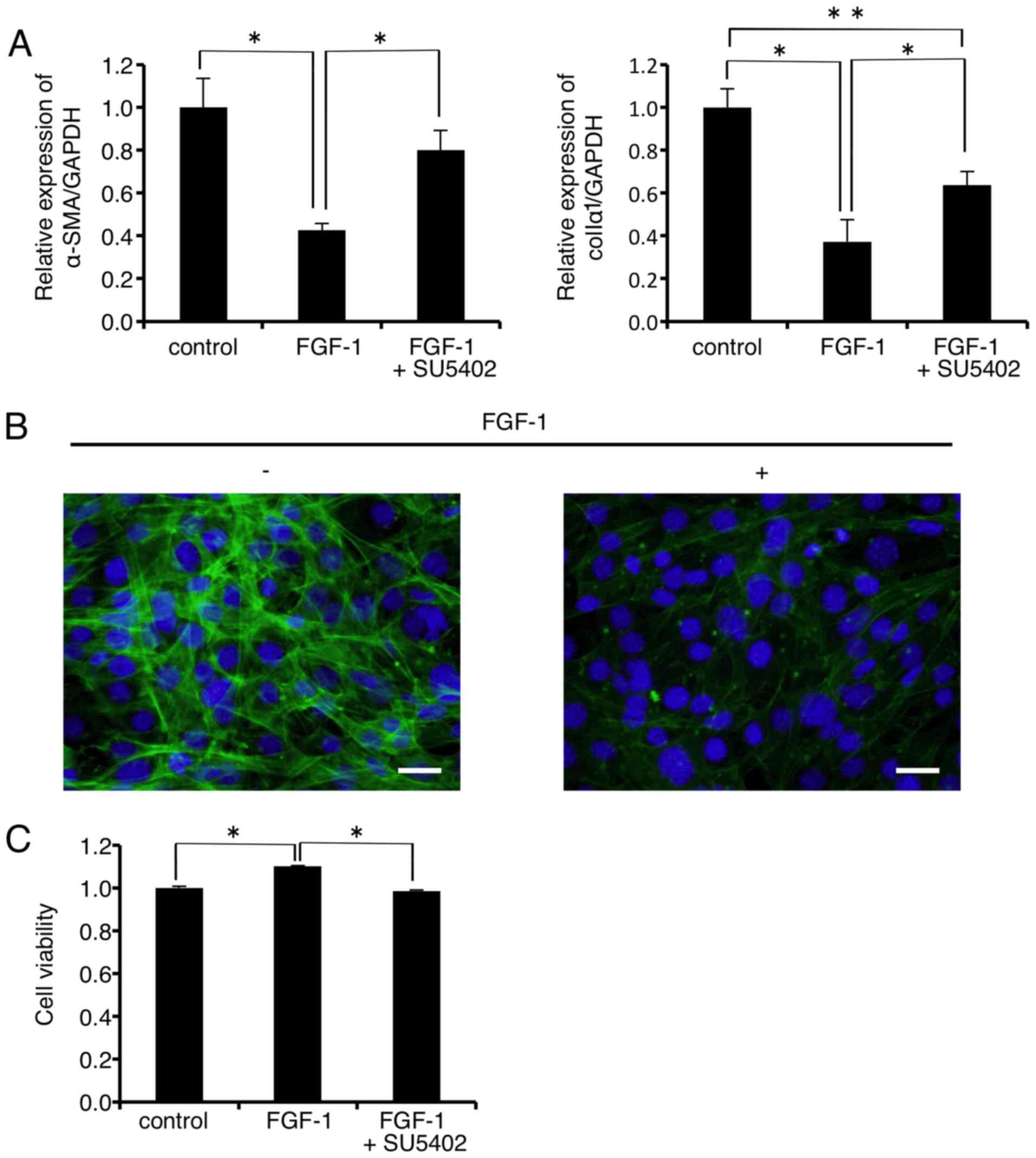
As shown in Fig.
5A, mRNA expression levels of α-SMA and colIα1 were
significantly decreased to 42.6 and 37.3% of the control,
respectively, 24 h after FGF-1 (10 ng/ml) administration. The
FGF-1-induced decrease in the mRNA level of α-SMA was almost
completely rescued by the FGFR1 inhibitor SU-5402 (Fig. 5A, left graph). On the other hand,
SU-5402 significantly but not completely rescued the FGF-1-induced
decrease in the mRNA level of colIα1 (Fig. 5A, right graph). In addition,
immunofluorescence analysis revealed that FGF-1 (10 ng/ml)
treatment inhibited F-actin formation in the FLS1 cells 24 h after
administration (Fig. 5B right
panel, green). FLS1 cell viability was significantly increased to
111.0% of the control 24 h after FGF-1 (10 ng/ml) administration
(Fig. 5C). In addition, the FGFR1
inhibitor SU-5402 abrogated the FGF-1-induced increase in FLS1 cell
viability.
Discussion
Synovial cell lines were established and are
valuable for investigating various synovial cell functions in
vitro. Some synovial cell lines (27–29) have been utilized for investigating
cellular and molecular mechanisms underlying the emergence of
rheumatoid arthritis (RA) and OA (30–32). However, synovial cell lines
derived from the TMJ are difficult to find; we found only one study
stating that fibroblast-like cells derived from baboon TMJs were
successfully immortalized by the ectopic expression of human
telomerase reverse transcriptase (hTERT) (33). It is well known that mammalian
primary cells possess finite proliferative activity in culture, and
accordingly, mouse primary FLS cultures derived from TMJs did not
divide after 7 passages (Fig.
1A). Here, we successfully obtained the immortalized mouse FLS
cell line, FLS1, by ectopic expression of SV40LT. Miyazawa et
al previously established the immortalized human rheumatoid FLS
cell line MH7A by ectopic expression of SV40LT (27). They demonstrated that their MH7A
cells retained the response to stimulation with the inflammatory
cytokine interleukin-1 (IL-1), suggesting that this cell line did
not lose the characteristics of rheumatoid FLS even after ectopic
SV40LT expression. We demonstrated that FLS1 cells expressed the
mesenchymal marker vimentin (Fig.
1D), and more vigorously expressed the MF markers α-SMA and
colIα1 than control NIH3T3 fibroblasts (Fig. 1E and F), suggesting that FLS1
cells retained their myofibroblastic characteristics.
As described above, TGF-β1 is well-known as a potent
inducer of MF differentiation in cells derived from mesenchymal
origin (9). We evaluated the
effect of TGF-β1 on the expression of MF markers α-SMA and colIα1
and found that TGF-β1 did not affect the expression of these
markers (data not shown). These results indicated that the FLS1
cells retained the characteristics of differentiated MFs that
vigorously express α-SMA and colIα1 even when these cells were not
stimulated with TGF-β1. We previously reported that TGF-β1 promoted
α-SMA expression in PDL-derived fibroblastic cells in a
Smad2-dependent manner (34).
Therefore, we also evaluated the effect of TGF-β1 on Smad2
phosphorylation in FLS1 cells using western blot analysis. We found
that TGF-β1 treatment (10 ng/ml) did not induce Smad2/3
phosphorylation in FLS1 cells, suggesting that FLS1 cells were not
responsive to TGF-β1 stimulation (data not shown).
TGF-β1 activates the ROCK/actin/MRTF-A signal
transduction pathway, which results in the expression of α-SMA in
cardiac fibroblasts (11).
Although FLS1 cells did not exhibit increased α-SMA mRNA expression
after TGF-β1 stimulation as described above,
ROCK/actin/MRTF-mediated signaling seemed to be activated in FLS1
cells even when the cells were not stimulated with TGF-β1 (Fig. 1H, left panel). Therefore, we
investigated whether ROCK-mediated signal positively regulated the
MF differentiation status of FLS1 cells. Treatment with the ROCK
inhibitor Y-27632 inhibited F-actin formation (Fig. 2B), and significantly decreased
mRNA expression of MF markers α-SMA and colIα1 (Fig. 2A). These results indicated that
ROCK inhibition attenuated the incorporation of G-actin into
growing F-actin stress fibers in FLS1 cells, possibly resulting in
the sequestration of MRTF-A on G-actin in the cytoplasm. As a
consequence, the sequestered MRTF-A cannot enter the nucleus,
attenuating the transcription of MF marker gene expression.
Importantly, Y-27632 treatment did not affect FLS1 cell viability
(data not shown), indicating that the Y-27632-induced
downregulation of MF marker expression in FLS1 cells was not
related to cytotoxicity. These results strongly suggest that
Y-27632 may be a good candidate as an anti-fibrosis drug around the
TMJ. Honjo et al previously reported that Y-27632 was
effective in preventing fibro-proliferation and scar formation in a
rabbit model of glaucoma surgery (35), suggesting a medical application of
this ROCK inhibitor for preventing abnormal fibrogenesis.
We also evaluated the effects of the
actin-depolymerizing agent CytB on MF differentiation status in
FLS1 cells. We demonstrated that CytB treatment attenuated the
incorporation of G-actin into growing F-actin stress fibers in FLS1
cells as expected (Fig. 3B),
resulting in downregulated α-SMA and col1αI mRNA levels (Fig. 3A). These results indicated that
CytB-induced actin depolymerization may increase the
G-actin/F-actin ratio in the cytoplasm, resulting in decreased
MRTF-A nuclear entry. As a result, α-SMA and colIα1 mRNA levels
were suppressed in the FLS1 cells. On the other hand, CytB
significantly decreased FLS1 cell viability to 76.1% of the control
(Fig. 3C), suggesting that
CytB-mediated cytotoxicity might secondarily and non-specifically
decrease α-SMA and colIα1 expression. Patel et al previously
demonstrated that the actin depolymerizing agents CytB, latrunculin
and Y-27632 decreased α-SMA promoter activity and MF
differentiation in human and rat mesangial cells (36), which is consistent with our
results.
We also demonstrated that the MRTF-A inhibitor
CCG-100602 attenuated MRTF-A nuclear translocation (Fig. 4B, red), but did not affect F-actin
formation (Fig. 4B, green) in the
FLS1 cells. As a result, CCG-100602 treatment resulted in decreased
mRNA levels of α-SMA and colIα1 (Fig.
4A). Johnson et al reported that CCG-100602 inhibited
MRTF-A nuclear translocation and suppressed TGF-β-induced
expression of MF markers in human colonic fibroblasts (37). Importantly, CCG-100602 treatment
did not affect FLS1 cell viability (data not shown), indicating
that CCG-100602-induced downregulation of MF marker expression in
FLS1 cells was not related to cytotoxicity. These results strongly
suggest that CCG-100602 can be utilized as a drug to prevent
fibrosis around the TMJ.
FGF-1 suppressed F-actin formation (Fig. 5B), and decreased mRNA levels of
α-SMA and colIα1 (Fig. 5A),
suggesting that FGF-1 may reverse MF differentiation of FLS1 cells
by disrupting ROCK/actin/MRTF-A-mediated signaling. Importantly,
FGF-1 did not decrease FLS1 cell viability (Fig. 5C), indicating that the
FGF-1-induced downregulation of MF marker expression in FLS1 cells
was not related to cytotoxicity. Interestingly, FGF-1 treatment
actually increased FLS1 cell viability (Fig. 5C), suggesting that FGF-1 may not
be an anti-fibrosis drug around the TMJ in itself: FGF-1 reverses
MF differentiation of FLS1 cells, but increases the population of
pre-MFs that retain the ability to differentiate into mature MFs.
In addition, as shown in Fig. 5A and
C, the FGFR1 inhibitor SU-5402 suppressed not only the
FGF-1-induced inhibition of mRNA expression of MF markers α-SMA and
colIα1 but also the FGF-1-induced promotion of cell viability of
FLS1 cells. These results indicated that FGF-1 not only suppressed
mRNA expression of MF markers in FLS1 cells, but also promoted cell
viability of the cells through FGFR1-mediated signal transduction
pathway. We also found that FLS1 cells expressed mRNA of FGFR1-4
(data not shown). As shown in Fig.
5A, the FGF-1-induced decrease in the mRNA level of α-SMA was
almost completely rescued by the FGFR1 inhibitor SU-5402 (Fig. 5A, left graph). In contrast,
SU-5402 significantly but not completely rescued the FGF-1-induced
decrease in the mRNA level of colIα1 (Fig. 5A, right graph), implicating that
FGF-1 might activate cellular signalling for the dedifferentiation
of the cells partially through FGFR2-4. On the other hand, FGF-2
treatment was found to suppress the TGF-β-induced expression of
α-SMA in human airway SMCs (38),
and in mouse pluripotent 10T1/2 cells (39) in an ERK-dependent manner. However,
it remains to be clarified which intracellular signals downstream
of FGF-1 negatively regulate ROCK/actin/MRTF-A axis-mediated signal
transduction in FLS1 cells. In addition, elucidating the molecular
mechanisms underlying the FGF-1-induced suppression of MF marker
expression in FLS1 cells may enable researchers to find other drug
targets in addition to the ROCK/actin/MRTF-A axis in the
future.
Taken together, these results strongly suggest that
the ROCK/actin/MRTF gene regulatory axis promotes the fibrogenic
activity of synoviocytes around the TMJ. Our findings partly
clarified the molecular mechanisms underlying the emergence of
TMJ-OA, and further investigation is warranted to identify drug
targets for treating this condition at the molecular level.
Abbreviations:
|
TMJ
|
temporomandibular joint
|
|
OA
|
osteoarthritis
|
|
MFs
|
myofibroblasts
|
|
ECM
|
extracellular matrix
|
|
α-SMA
|
α-smooth muscle actin
|
|
TGF-β
|
transforming growth factor-β
|
|
FGF
|
fibroblast growth factor
|
|
FGFRs
|
FGF receptors
|
|
RTK
|
receptor tyrosine kinase
|
|
MAPKs
|
mitogen-activated protein kinases
|
|
PI3K
|
phosphoinositide 3-kinase
|
|
SMC
|
smooth muscle cell
|
|
PDL
|
periodontal ligament
|
|
ERK
|
extracellular signal-regulated
kinase
|
|
EGF
|
epidermal growth factor
|
|
FLSs
|
fibroblast-like synoviocytes
|
|
ROCK
|
Rho-associated coiled-coil forming
kinase
|
|
MRTF
|
myocardin-related transcription
factor
|
|
SRF
|
serum response factor
|
|
G-actin
|
globular actin
|
|
F-actin
|
filamentous actin
|
|
CytB
|
cytochalasin B
|
|
Ham's F-12
|
Nutrient Mixture F-12 Ham
|
|
PBS
|
phosphate-buffered saline
|
|
FBS
|
fetal bovine serum
|
|
SV40LT
|
simian virus 40 large T antigen
|
|
α-MEM
|
minimum essential medium Eagle's
α-modification
|
|
colIα1
|
α1 chain of collagen type I
|
|
RA
|
rheumatoid arthritis
|
|
hTERT
|
human telomerase reverse
transcriptase
|
|
IL-1
|
interleukin-1
|
Acknowledgments
The present study was supported in part by JSPS
KAKENHI grant nos. 26670852 and 16H05534 to A.I.; 25463053 and
16K11654 to N.C.; 26462823 to S.K.; 15K20606 to H.K.; 22592076 to
M.K.; and a Grant-in-Aid for Strategic Medical Science Research
Centre from the Ministry of Education, Culture, Sports, Science and
Technology of Japan, 2010–2014.
References
|
1
|
Tanaka E, Detamore MS and Mercuri LG:
Degenerative disorders of the temporomandibular joint: Etiology,
diagnosis, and treatment. J Dent Res. 87:296–307. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Matsumoto R, Ioi H, Goto TK, Hara A,
Nakata S, Nakasima A and Counts AL: Relationship between the
unilateral TMJ osteoarthritis/osteoarthrosis, mandibular asymmetry
and the EMG activity of the masticatory muscles: A retrospective
study. J Oral Rehabil. 37:85–92. 2010. View Article : Google Scholar
|
|
3
|
Krisjane Z, Urtane I, Krumina G, Neimane L
and Ragovska I: The prevalence of TMJ osteoarthritis in
asymptomatic patients with dentofacial deformities: A cone-beam CT
study. Int J Oral Maxillofac Surg. 41:690–695. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Hill CL, Hunter DJ, Niu J, Clancy M,
Guermazi A, Genant H, Gale D, Grainger A, Conaghan P and Felson DT:
Synovitis detected on magnetic resonance imaging and its relation
to pain and cartilage loss in knee osteoarthritis. Ann Rheum Dis.
66:1599–1603. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wang XD, Zhang JN, Gan YH and Zhou YH:
Current understanding of pathogenesis and treatment of TMJ
osteoarthritis. J Dent Res. 94:666–673. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Krieg T, Abraham D and Lafyatis R:
Fibrosis in connective tissue disease: The role of the
myofibroblast and fibroblast-epithelial cell interactions.
Arthritis Res Ther. 9(Suppl 2): S42007. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hill JA and Olson EN: Cardiac plasticity.
N Engl J Med. 358:1370–1380. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kehat I and Molkentin JD: Molecular
pathways underlying cardiac remodeling during pathophysiological
stimulation. Circulation. 122:2727–2735. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Tomasek JJ, Gabbiani G, Hinz B, Chaponnier
C and Brown RA: Myofibroblasts and mechano-regulation of connective
tissue remodelling. Nat Rev Mol Cell Biol. 3:349–363. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Desmoulière A, Geinoz A, Gabbiani F and
Gabbiani G: Transforming growth factor-beta 1 induces alpha-smooth
muscle actin expression in granulation tissue myofibroblasts and in
quiescent and growing cultured fibroblasts. J Cell Biol.
122:103–111. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Small EM: The actin-MRTF-SRF gene
regulatory axis and myofibroblast differentiation. J Cardiovasc
Transl Res. 5:794–804. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Hinz B, Phan SH, Thannickal VJ, Galli A,
Bochaton-Piallat ML and Gabbiani G: The myofibroblast: One
function, multiple origins. Am J Pathol. 170:1807–1816. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Fahlgren A, Andersson B and Messner K:
TGF-beta1 as a prognostic factor in the process of early
osteoarthrosis in the rabbit knee. Osteoarthritis Cartilage.
9:195–202. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
van den Berg WB: Growth factors in
experimental osteoarthritis: Transforming growth factor beta
pathogenic? J Rheumatol Suppl. 43:143–145. 1995.PubMed/NCBI
|
|
15
|
Sandbo N and Dulin N: Actin cytoskeleton
in myofibroblast differentiation: Ultrastructure defining form and
driving function. Transl Res. 158:181–196. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Beenken A and Mohammadi M: The FGF family:
Biology, pathophysiology and therapy. Nat Rev Drug Discov.
8:235–253. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Liu F and Zhuang S: Role of receptor
tyrosine kinase signaling in renal fibrosis. Int J Mol Sci.
17:9722016. View Article : Google Scholar :
|
|
18
|
Ornitz DM, Xu J, Colvin JS, McEwen DG,
MacArthur CA, Coulier F, Gao G and Goldfarb M: Receptor specificity
of the fibroblast growth factor family. J Biol Chem.
271:15292–15297. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Raju R, Palapetta SM, Sandhya VK, Sahu A,
Alipoor A, Balakrishnan L, Advani J, George B, Kini KR, Geetha NP,
et al: A network map of FGF-1/FGFR signaling system. J Signal
Transduct. 2014:9629622014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shimbori C, Bellaye PS, Xia J, Gauldie J,
Ask K, Ramos C, Becerril C, Pardo A, Selman M and Kolb M:
Fibroblast growth factor-1 attenuates TGF-β1-induced lung fibrosis.
J Pathol. 240:197–210. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Maltseva O, Folger P, Zekaria D, Petridou
S and Masur SK: Fibroblast growth factor reversal of the corneal
myofibroblast phenotype. Invest Ophthalmol Vis Sci. 42:2490–2495.
2001.PubMed/NCBI
|
|
22
|
Takahashi M, Okubo N, Chosa N, Takahashi
N, Ibi M, Kamo M, Mizuki H, Ishisaki A and Kyakumoto S: Fibroblast
growth factor-1-induced ERK1/2 signaling reciprocally regulates
proliferation and smooth muscle cell differentiation of
ligament-derived endothelial progenitor cell-like cells. Int J Mol
Med. 29:357–364. 2012.
|
|
23
|
Kimura H, Okubo N, Chosa N, Kyakumoto S,
Kamo M, Miura H and Ishisaki A: EGF positively regulates the
proliferation and migration, and negatively regulates the
myofibroblast differentiation of periodontal ligament-derived
endothelial progenitor cells through MEK/ERK- and JNK-dependent
signals. Cell Physiol Biochem. 32:899–914. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Davis J and Molkentin JD: Myofibroblasts:
Trust your heart and let fate decide. J Mol Cell Cardiol. 70:9–18.
2014. View Article : Google Scholar
|
|
25
|
Ozer HL, Banga SS, Dasgupta T, Houghton J,
Hubbard K, Jha KK, Kim SH, Lenahan M, Pang Z, Pardinas JR, et al:
SV40-mediated immortalization of human fibroblasts. Exp Gerontol.
31:303–310. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hayashi K, Watanabe B, Nakagawa Y, Minami
S and Morita T: RPEL proteins are the molecular targets for
CCG-1423, an inhibitor of Rho signaling. PLoS One. 9:e890162014.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Miyazawa K, Mori A and Okudaira H:
Establishment and characterization of a novel human rheumatoid
fibroblast-like synoviocyte line, MH7A, immortalized with SV40 T
antigen. J Biochem. 124:1153–1162. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yamazaki T, Yokoyama T, Akatsu H, Tukiyama
T and Tokiwa T: Phenotypic characterization of a human synovial
sarcoma cell line, SW982, and its response to dexamethasone. In
Vitro Cell Dev Biol Anim. 39:337–339. 2003. View Article : Google Scholar
|
|
29
|
Georgescu HI, Mendelow D and Evans CH:
HIG-82: An established cell line from rabbit periarticular soft
tissue, which retains the 'activatable' phenotype. In Vitro Cell
Dev Biol. 24:1015–1022. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Nakayama H, Yaguchi T, Yoshiya S and
Nishizaki T: Resveratrol induces apoptosis MH7A human rheumatoid
arthritis synovial cells in a sirtuin 1-dependent manner. Rheumatol
Int. 32:151–157. 2012. View Article : Google Scholar :
|
|
31
|
Sommerfelt RM, Feuerherm AJ, Skuland T and
Johansen B: Cytosolic phospholipase A2 modulates TLR2 signaling in
synoviocytes. PLoS One. 10:e01190882015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hsu HC, Chang WM, Wu JY, Huang CC, Lu FJ,
Chuang YW, Chang PJ, Chen KH, Hong CZ, Yeh RH, et al: Folate
deficiency triggered apoptosis of synoviocytes: Role of
overproduction of reactive oxygen species generated via NADPH
oxidase/mitochondrial complex II and calcium perturbation. PLoS
One. 11:e01464402016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
McDaniel JS, Akula Suresh Babu R, Navarro
MM and LeBaron RG: Transcriptional regulation of proteoglycan 4 by
17β-estradiol in immortalized baboon temporomandibular joint disc
cells. Eur J Oral Sci. 122:100–108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yoshida M, Okubo N, Chosa N, Hasegawa T,
Ibi M, Kamo M, Kyakumoto S and Ishisaki A: TGF-β-operated growth
inhibition and translineage commitment into smooth muscle cells of
periodontal ligament-derived endothelial progenitor cells through
Smad- and P38 MAPK-dependent signals. Int J Biol Sci. 8:1062–1074.
2012. View Article : Google Scholar :
|
|
35
|
Honjo M, Tanihara H, Kameda T, Kawaji T,
Yoshimura N and Araie M: Potential role of Rho-associated protein
kinase inhibitor Y-27632 in glaucoma filtration surgery. Invest
Ophthalmol Vis Sci. 48:5549–5557. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Patel K, Harding P, Haney LB and Glass WF
II: Regulation of the mesangial cell myofibroblast phenotype by
actin polymerization. J Cell Physiol. 195:435–445. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Johnson LA, Rodansky ES, Haak AJ, Larsen
SD, Neubig RR and Higgins PD: Novel Rho/MRTF/SRF inhibitors block
matrix-stiffness and TGF-β-induced fibrogenesis in human colonic
myofibroblasts. Inflamm Bowel Dis. 20:154–165. 2014. View Article : Google Scholar
|
|
38
|
Schuliga M, Javeed A, Harris T, Xia Y, Qin
C, Wang Z, Zhang X, Lee PV, Camoretti-Mercado B and Stewart AG:
Transforming growth factor-β-induced differentiation of airway
smooth muscle cells is inhibited by fibroblast growth factor-2. Am
J Respir Cell Mol Biol. 48:346–353. 2013. View Article : Google Scholar :
|
|
39
|
Kawai-Kowase K, Sato H, Oyama Y, Kanai H,
Sato M, Doi H and Kurabayashi M: Basic fibroblast growth factor
antagonizes transforming growth factor-beta1-induced smooth muscle
gene expression through extracellular signal-regulated kinase 1/2
signaling pathway activation. Arterioscler Thromb Vasc Biol.
24:1384–1390. 2004. View Article : Google Scholar : PubMed/NCBI
|