Introduction
In the wide horizon of ophthalmologically rare
diseases among retinitis pigmentosa forms, Stargardt disease (OMIM
#248200) has gradually assumed an important role due to its
heterogeneity. Stargardt disease, known also as fundus
flavimaculatus in the late onset form, or heredomacular
degeneration, causes progressive bilateral decrease in vision
between childhood and teenage years, reaching a plateau phase
shortly after rapid reduction in visual acuity by the age of 50.
Most patients show a decrease of up to 6/60 or worse, reaching a
condition called 'legal blindness'. Stargardt patients develop
irregularly shaped yellowish-white flecks or spots in the macula,
causing decreased central vision. There is usually no problem
regarding peripheral vision, and therefore they rarely have issues
with bumping into objects when moving around (due to rod
apoptosis). In late stages of the disease, the involvement of cones
may also induce impairment of color vision. Other symptoms usually
include wavy vision, blind spots, blurriness, and difficulty
adapting to dim lighting (1).
Gene therapy could be a future solution (2). Stargardt disease is an inherited
condition mainly autosomal recessive, and the major causative gene
involved is ABCA4 (3),
also known as ABCR (4). It
is located on the short arm of chromosome 1 (1p22), and encodes for
a cytospecific member of the ATP-binding cassette (ABC) transporter
superfamily, retina photoreceptor specific. The protein consists of
two transmembrane domains (TMDs), also known as membrane-spanning
domains (MSDs) or integral membrane (IM) domains. It consists of
α-helices, embedded in the membrane bilayer, and is an allosteric
protein. The sequence and architecture of TMDs are variable,
reflecting the chemical diversity of substrates that can be
translocated. The nucleotide binding domain (NBD), on the other
hand, is located in the cytoplasm and has a highly conserved
sequence, and is the site for ATP binding (4). The structural architecture of ABC
transporters consists minimally of two TMDs and two NBDs (5).
The protein plays a fundamental role in the visual
cycle. To be precise, it is an inward-directed retinoid flipase,
which imports substrates from the lumen to the cytoplasmic side of
retinal disc membranes. The substrates are
all-trans-retinaldehyde (ATR) and
N-retinyl-phosphatidyl-ethanolamine (NR-PE), an intermediate
derived from the reaction of ATR with phosphatidyl-ethanolamine
(PE) located in disc membranes. ATR, once transported to the
cytoplasmic side, is reduced to vitamin A by trans-retinol
dehydrogenase (tRDH). Then, transferred to the retinal pigment
epithelium (RPE), it is converted to 11-cis-retinal. Abca4
protein is involved in photoresponse, removing ATR/NR-PE from the
extracellular photoreceptor surfaces during bleach recovery. More
than 700 mutations in the ABCA4 gene (OMIM #601691) have
been found to cause Stargardt macular degeneration, most of which
consist of single nucleotide variants (SNVs). An altered Abca4
protein cannot remove NR-PE from photoreceptor cells, thus it
combines with other ATR molecules. This, in turn, leads to
condensation, oxidation, hydrolysis and rearrangements. All of
these reactions produce the bis-retinoid
Di-retinoid-pyridinium-ethanolamine (A2E) (6), among which is lipofuscin, one of the
constituents of fatty yellow pigments that builds up in retinal
cells (7). This is toxic to the
retina, leading to photoreceptor apoptosis and Stargardt macular
degeneration progressive vision loss in patients. Different
phenotypes are associated with variable residual functions of the
protein, due to several variants of the ABCA4 gene. It is
therefore fundamental to analyze the gene in the most complete way,
in order to develop a correct differential diagnosis for each case
of Stargardt disease. This is one of the most difficult challenges
due to the very common overlapping symptoms of the pathology, and
contrasting data. An example is provided by the variant of our case
study, regarded as a non-pathogenic polymorphism (SNP), as a
high-penetrance disease-causing variant, or even as a possible
protecting factor. Similar to other pathologies, there is not just
one gene implicated in etiopathogenesis, and ABCA4 could
play a strong role in the development of retinitis pigmentosa.
In our hypothesis, the indirect effects of a mutated
ABCA4 could influence the activity of RP1, one of the
most frequent causative genes of syndromic or non-syndromic
retinitis pigmentosa (8).
Retinitis pigmentosa 1 (OMIM #180100), the most common form, shows
high involvement of the RP1 gene, located on 8q12. It is an
autosomal dominant form with relatively late onset of night
blindness, usually by the third decade of life, with slow
progression. Characteristic clinical findings include diffuse
retinal pigmentation, progressive decrease in recordable ERGs, and
concentric visual field loss. Funduscopic findings comprise retinal
atrophy, bone-spicule-like pigment deposits, and vascular
attenuation (9). Rp1 protein is
located in the region of the axoneme of rod and cone
photo-receptors. The photo-receptor axoneme begins at the basal
body in the distal inner segment and passes through the connecting
cilium. It is considered the primary pass for continuous polarized
transport of proteins and membrane needed in outer segments to
substitute older discs with new ones (10,11). The junction between the connecting
cilium and the outer segment is also where disc morphogenesis
occurs (12,13). It has been pointed out (14) that RP1 could play a role in
controlling the orientation and organization of outer segment
discs. It may function as a connection between newly formed discs
and the axoneme, and this interaction helps discs form in the
correct orientation and stack up into outer segments. Proteins
present in the disc rims, such as Abca4, Rom1 and peripherin are
potential candidates for such an interaction (14). In this study, we report the
genetic condition of a family where each carry several variants on
the ABCA4 gene and RP1.
Materials and methods
Clinical data
The target of our study is a Sicilian family with
three members. The proband, 54-year-old father, showed a
symptomatology common to syndromic retinitis pigmentosa and
Stargardt disease: high myopia and myopic chorioretinitis,
irregular astigmatism, incipient cataract and retinal dystrophy.
All of these disorders have left the patient severely visually
impaired, with a useful visual acuity of 1/20 in both eyes and
short perceptions of light and colors since pediatric age. Fundus
examination showed peripheral degeneration, an area of vitreous
traction and macular thickness reduced in the right eye. The left
eye showed degenerative myopia. Pattern evoked potential (PEP) and
flash evoked potential (FEP) confirmed typical signs of retinitis
pigmentosa, as shown in pattern electroretinogram (PERG) and flash
electroretinogram (FERG) (Figs.
1Figure 2Figure 3–4). The proband's wife, 65 years of age,
showed only a slight reduction in sensitivity on left eye
peripheral areas. Their 29-year-old daughter, instead, has revealed
no ophthalmologic symptoms (Fig.
5).
Following detailed genetic counseling, ABCA4
and RP1 gene analysis was requested. The research followed
the tenets of the Declaration of Helsinki and informed consent was
obtained from the subjects after explanation of the nature and
possible consequences of the study.
ABCA4 and RP1 genotyping
Genomic DNA was extracted from heparinized
peripheral blood using the salting out method and then stored in TE
buffer (l0 mM Tris-HCI, l mM EDTA, pH 8.0) until analysis. Coding
exons (50 for ABCA4 and 4 for RP1, respectively),
intron-exon boundaries and promoter regions of the 2 genes were
screened using primers designed according to the ABCA4 and
RP1 published nucleotide sequence of GenBank (accession no.
NG_009073.1 and NG_009840.1, respectively).
Polymerase chain reaction (PCR)
PCR amplifications were carried out in a 50
μl solution containing MgCl2 (2.5 mM), dNTPs (0.2
mM), 0.2 μl of each primer (10 μM), 0.8 μg of
genomic DNA and 1 unit of Euro Taq polymerase (EuroClone Spa Life
Sciences Division, Milan, Italy). DNA amplification was performed
on a thermal cycler Gene Amp PCR System 2700 (PE Applied
Biosystems, Foster City, CA, USA) as follows. After an initial
denaturation step at 94°C for 5 min, the samples were subjected to
35 cycles of amplification consisting of 40 sec of denaturation at
95°C, 35 sec of annealing and 45 sec of extension at 72°C. The
annealing temperature was optimized for each primer set. A final
extension at 72°C was carried out for l0 min. Following PCR, 5
μl of amplified product was examined by electrophoresis on a
2% agarose gel. PCR of RP1 (4 exons) and ABCA4 (50
exons) required 12 and 43 primer pairs. See Tables I and II for the sequences of primers.
 | Table IRP1 primer sequences. |
Table I
RP1 primer sequences.
| Exon | Forward primer | Reverse primer |
|---|
| 1 |
TGCAGAGCATGCTAGGAACT |
TATCAGCATATTGTGAAGGTTG |
| 2 |
TCTGGATGTCTGCAGCTATAT |
AGATGAGATTCCAGTCAGATTCT |
| 3 |
TGCTCAGTGATGATGTCTTTC |
TTTCTGTGGTGGAAGAAACTG |
| 4a |
GCTGCCTCTTCCTTTGGATAT |
GGCAAACCATTATTATGTGACAT |
| 4b |
ATCAAATGGAGGAGTCATCATTA |
TCTCAAATACCCAGATGCCACT |
| 4c |
CATCCTTGAGCAAAAACCCAA |
AGCATCAACTTGACAGAAGCTA |
| 4d |
CAAATGCCAGGTTCACTTGCA |
TGACATTTTGATGTGACACCAAT |
| 4e |
CTTGGATTCAACTGAAGAGTT |
AGCCTCTTACTGATTATTTCAT |
| 4f |
TTAATACAGTGGTAAATGGA |
TGAAATTCCACAGAATTATAA |
| 4g |
CATAGGATTTGTTAAAAGGGC |
AATAACAGTTAGTATTGGGCAAT |
| 4h |
TGTCTCTGATGATGCTATTAAA |
TACTGCTTTCAAGATCAGTTAAA |
| 4i |
ATCTCAACCAAGTAGTAAGAG |
TATATCATCATATAGTCATGCAG |
| Table IIABCA4 primer sequences. |
Table II
ABCA4 primer sequences.
| Exon | Forward primer | Reverse primer |
|---|
| 1 |
AACTAAGGGCTTATGTGTAAT |
CACTGCTTCAGTGCTAATC |
| 2 |
TCCTACTGCACACATGGGATC |
TTACATGCATCATAGACATGA |
| 3 |
ACACATGAGATGCTCCTGCT |
TCTGCTCCTAAGAGGTTAG |
| 4 |
TGTAAGGATACTCAATGTAGT |
TTCACCAAGGTGATGTTCAA |
| 5 |
AGTTGAGTTACAAGTGTTTCC |
TGAATGTGAACACAAGGAAG |
| 6 |
GATCTTAATTCCTGTCGCCA |
AAGGATTGTCCAGAACACCA |
| 7 |
AACATATAGGAGATCAGACTG |
TTGGGATGTGAACAGGTGCT |
| 8 |
TAAGGCTCATCCTAGTATTCT |
GTTCATGTCCAGAATTGCT |
| 9 |
TGCTACTAATGATGAGCTTGT |
CAGTGATGACTGTGGATGG |
| 10 |
CCATCCACAGTCATCACTG |
TGATCTAACTCCAATAGCG |
| 11 |
CGCTATTGGAGTTAGATCA |
AGACCACTTGACTTGCTAA |
| 12–13 |
ACCAGACTCTGGAGTTAAGC |
CATTAGCGTGTCATGGAGG |
| 14 |
AGAGTCCTCTGGTGGCTAG |
CTGCAGACTTGATGATGTG |
| 15 |
CACATCATCAAGTCTGCAG |
AAGCTAGATGTCACGCTCT |
| 16 |
ACTTGCAACTCCTCTGAGAG |
GCTGTTGCTAGTCAGATGT |
| 17 |
AGGAACTCAGCACATGGAGT |
TGAGGAGTCACTGTTGCAT |
| 18 |
GCTGACCTTACACTGAGAGA |
TCAAGTAGAGCCAGTAGGAT |
| 19 |
CAAGATTATTGGTCTTGCTGT |
ATCAGCCATTCATGATCACA |
| 20 |
AGATTGTGTGATCAGGCTTG |
TTCCACACACATGCAGATG |
| 21 |
AAGCAGTGCCTGGCATATAG |
CTCTCTGAATGAATGTCCAC |
| 22 |
TGGATGTATACACTGGTGCT |
TCTGAGCAGCAGAGGCAGA |
| 23–24 |
ACAGTGAGCATCTTGATTGC |
GTGGTTCCTGTACTCAGCT |
| 25 |
TACAGTATGTAGGAAGCTATG |
CTTCAGAATGTGTTCATCGA |
| 26 |
CACATAATTGATGACAAGCCA |
AGGAATGATGGCTTACTAAG |
| 27–28 |
GCAGACTTGATGGAGCATCA |
CTGGTCTCGAACTCAGGTG |
| 29 |
GATGATTAAGCTACCAGCCT |
ACAGAATGTTCTGGTGGCC |
| 30–31 |
GGCCACCAGAACATTCTGT |
CAACGCCTGCCATCTTGAA |
| 32 |
CAAGCTAGAGATGGTTATTC |
CTACTAGATCAAATAGGAAG |
| 33 |
TCAACTGTGTCATCTGTATG |
GCAGCCAGCTTGAACTATA |
| 34 |
ATCATTGAAGTGAGAACTAG |
CTTCTATGGTCTTCTGATAT |
| 35 |
CATATGACCTGACAACAGGA |
ACTTATGTCCTCCAAGAAGA |
| 36–37–38 |
AGAGAGCTACTAGTAGGCGT |
GAATCCTCTCAGGATGTTCA |
| 39 |
TAGTGGAGTGACAGCTTCAA |
CCTGCGGTGCAGTGATTAT |
| 40 |
GACTAGTGACAGCTTAACATA |
CTGGTTATCAGCTTCAGACC |
| 41 |
ACAGAGTATATACACAGCTAG |
ACAGCTGCTACATGTACGAT |
| 42–43 |
TACTTCATGACCTCCATTGC |
AGTGGATGCTCTTCACATAT |
| 44–45 |
CAGAAGGAAGCAGAAGCAAG |
TCTCATGTGGCTAGTGGAAG |
| 46 |
CAAGTGCTTAGTAGCCACAT |
CAACAGAGGAATCTCTTAAC |
| 47 |
CATGGAAGAATCTGACAGGA |
GAGATGCATCTTCAGGATAA |
| 48 |
TCATTCTGGAGGCGTGAGAT |
GTGGATTAAGGCAATGACAG |
| 49–50 |
CTGTCATTGCCTTAATCCAC |
TCTTATCAGCATGATGGCCT |
| 51 |
ATTCCTGAGCTCAAGTGATC |
AACACACCATAGCATCACAG |
| 52 |
GTAGGACACAAGCCATACCA |
GATGTGATGAGGATGTGGTG |
| 53 |
ACACATCTCGTATGTGTGTC |
AAGACTAGTCCATTCACTTC |
Sequencing
All PCR products were analyzed also by direct
nucleotide sequence analysis by the dideoxynucleotide method with
the BigDye Terminator 3.1 Cycle Sequencing kit on the 3500 Genetic
Analyzer (Applied Biosystems, Foster City, CA, USA).
Bioinformatic analysis
To clarify the hypothetical effects of the examined
variants, a deep bioinformatic analysis with CLC Genomics workbench
8.0.1 (www.clcbio.com) for primary structure
details, followed by PSIPRED secondary structure prediction
(http://bioinf.cs.ucl.ac.uk/psipred/)
was performed. Finally, RaptorX (http://raptorx.uchicago.edu) and Chimera software
(http://www.cgl.ucsf.edu/chimera/) were
used to highlight third structure aspects of ABCA4-mutated and
wild-type predicted proteins.
Results
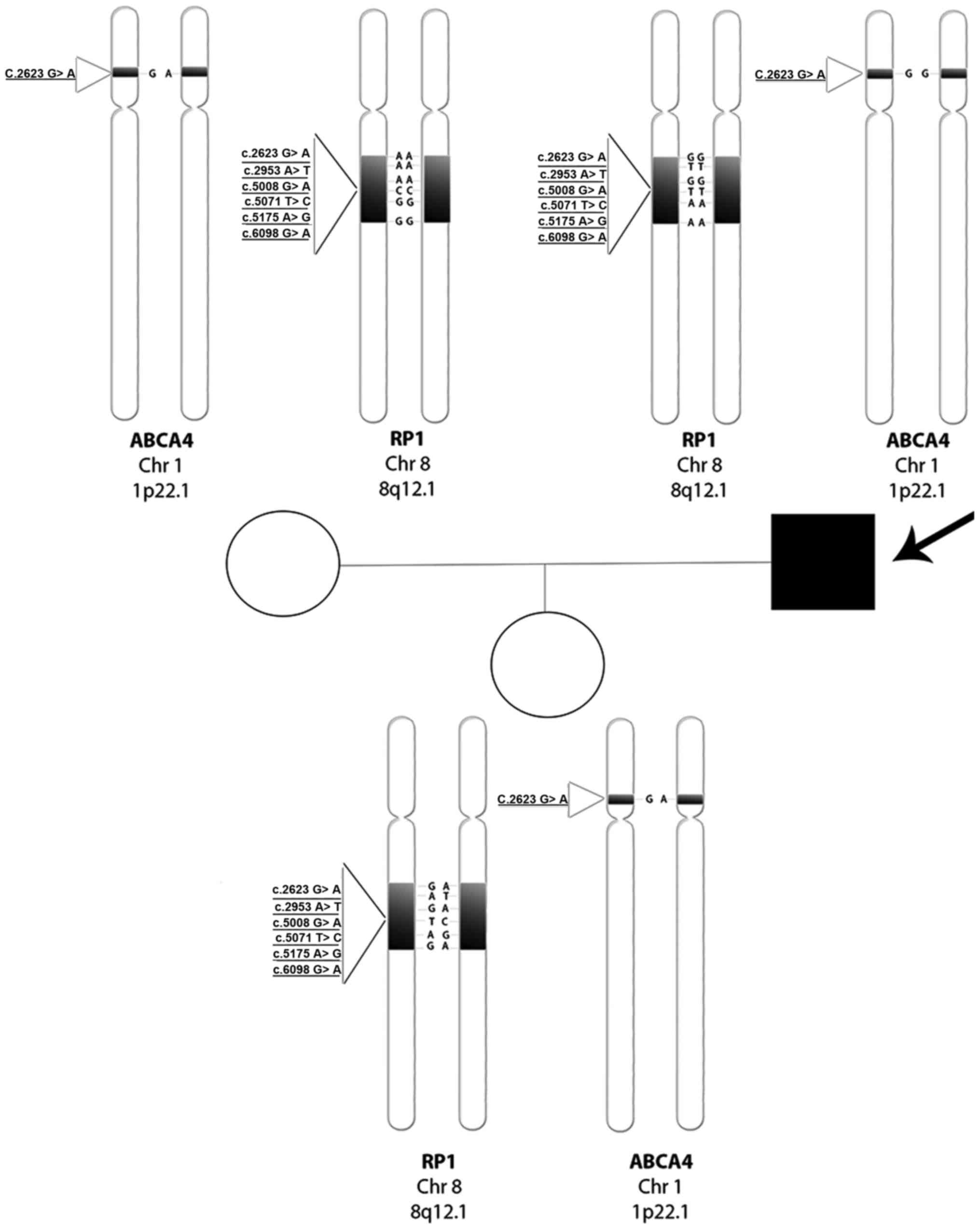
The entire genotype tree of the family is documented
in Fig. 6. Regarding the proband,
we report a wild-type condition for rs444772 (c.2623G>A) and for
three SNPs of RP1 'hot-spot' region in exon 4 (15): rs446227 (c.5008G>A), rs414352
(c.5071T>C) and rs441800 (c.5175A>G). In contrast, we found a
homozygous mutated condition regarding the other two RP1
SNPs, rs2293869 (c.2953A>T) and rs61739567 (c.6098G>A). The
proband's wife, instead, showed an opposite situation, a homozygous
mutated condition for the first four SNPs analyzed in her husband,
while the last two were wild-type. Their daughter, as expected from
the parents' genotypes, showed a heterozygous condition for all
examined SNPs. Regarding the ABCA4 gene, the proband showed
a wild-type condition for rs3112831 (c.1268A>G), while his wife
and daughter were both heterozygous.
We performed a search for Pfam domains on an
Abca4-mutated protein sequence, against a wild-type sequence using
CLC Genomics Workbench. The Pfam database, a large collection of
protein families, each represented by multiple sequence alignments
and hidden Markov models (HMMs), delivered the results shown in
Table III. The rs3112831
implies that one of two TMD domains starts from aa 515 instead of
513 of the wild-type, altering the recognition site of the protein
substrate (ATR or NR-PE).
| Table IIIPfam protein domain prediction for
wild-type and mutated Abca4. |
Table III
Pfam protein domain prediction for
wild-type and mutated Abca4.
| Sequence | Domain | Start | End | Accession | Score | E-value | Description | Predicted by |
|---|
ABCA4
wild-type |
ABC2_membrane_3 | 1597 | 1895 | PF12698.2 | 136.4 | 1.1E-39 | ABC-2 family
transporter protein | HMMER
3.1b1
(May 2013) |
| ABC_tran | 946 | 1090 | PF00005.22 | 105.3 | 2.9E-30 | ABC
transporter | HMMER
3.1b1
(May 2013) |
| ABC_tran | 1955 | 2099 | PF00005.22 | 74.5 | 9.1E-21 | ABC
transporter | HMMER
3.1b1
(May 2013) |
|
ABC2_membrane_3 | 513 | 856 | PF12698.2 | 61.1 | 8.6E-17 | ABC-2 family
transporter protein | HMMER
3.1b1
(May 2013) |
ABCA4
rs3112831
(c.1268A>G) |
ABC2_membrane_3 | 1597 | 1895 | PF12698.2 | 136.4 | 1.1E-39 | ABC-2 family
transporter protein | HMMER
3.1b1
(May 2013) |
| ABC_tran | 946 | 1090 | PF00005.22 | 105.3 | 2.9E-30 | ABC
transporter | HMMER
3.1b1
(May 2013) |
| ABC_tran | 1955 | 2099 | PF00005.22 | 74.5 | 9.1E-21 | ABC
transporter | HMMER
3.1b1
(May 2013) |
|
ABC2_membrane_3 | 515 | 856 | PF12698.2 | 61.2 | 8.3E-17 | ABC-2 family
transporter protein | HMMER
3.1b1
(May 2013) |
Examining the micromolecular meanings of these
alterations further, a deeper study with many bioinformatic
analyses and predictions of primary, secondary and tertiary
structures helped us visualize the potentially altered functions of
Abca4. Starting from a complete protein report from CLC Genomics
Workbench 8.0.1, we noted several important statistical differences
which reflect the amino acid change (Table IV).
| Table IVCLC Genomics Workbench Abca4
wild-type and mutated Protein Statistic Report. |
Table IV
CLC Genomics Workbench Abca4
wild-type and mutated Protein Statistic Report.
| Wild-type | rs3112831
(c.1268A>G) |
|---|
| Sequence
informations | | |
| Weight (kDa) | 255,941 | 255,901 |
| Isoelectric
point | 6.12 | 6.1 |
| Atomic
composition | | |
| Carbon (C) | 11.588 | 11.587 |
| Nitrogen (N) | 3.039 | 3.037 |
| Count of
hydrophobic and hydrophilic residues | | |
| Hydrophobic (A, F,
G, I, | 1.183 | 1.184 |
| L, M, P, V,
W) | | |
| Other | 503 | 502 |
| Amino acid
distribution table | | |
| Histidine (H) | 52 (0.023) | 51 (0.022) |
| Proline (P) | 129 (0.056) | 130 (0.057) |
| Counts of
di-peptides | | |
| Glu-His | 6 (0.03) | 5 (0.02) |
| Glu-Pro | 8 (0.02) | 9 (0.03) |
Furthermore, the substitution of the 423 histidine
with a proline brings about important changes in electrical
properties and solubility: the conjugate acid (protonated form) of
the imidazole side chain in histidine has a pKa of ~6.0; when
protonated, the imidazole ring bears two NH bonds and has a
positive charge, equally distributed between both nitrogens. The
distinctive cyclic structure of the proline side chain, instead,
gives proline exceptional conformational rigidity, which affects
the rate of peptide bond formation between proline and other amino
acids. When proline is bound as an amide in a peptide bond, its
nitrogen is not bound to any hydrogen, meaning it cannot act as a
hydrogen bond donor, but can be a hydrogen bond acceptor. Figs. 7 and 8 show these differences.
In order to highlight changes in the secondary
structure of Abca4, we chose PSIPRED (16), a popular structure prediction
method that incorporates two feed-forward neural networks to
perform an analysis of results obtained by the PSI-Blast homology
search algorithm (17). The
resulting scheme (Fig. 9)
underlines how the 423H>P causes the substitution of a coil
segment between position 504–505 with a helix, probably determining
a spatial misfolding which affects the protein function.
Since ATP binding triggers NBD dimerization, the
formation of the dimer may represent the 'power stroke'. Rotation
and tilting of transmembrane α-helices may both contribute to these
conformational changes, so it becomes a crucial forecast whether
the examining variant can modify the tertiary structure. RaptorX
(18–20) is a protein structure prediction
server excelling at predicting 3D structures for protein sequences
without close homologs in the Protein Data Bank (PDB). Given an
input sequence, RaptorX predicts its secondary and tertiary
structures as well as solvent accessibility and disordered regions.
We used this web-based application to carry out our aim, and
Chimera software (21) to get a
detailed 3D picture of the predicted mutated Abca4 from RaptorX pdb
exported files (Fig. 10). We
hypothesize that the basic N of imidazole side chain acts as a
nucleophile towards ATR or NR-PE atoms, constituting a crucial
component of the recognition site of Abca4. The substitution of
histidine with proline, due to atomic features of the latter, does
not permit a correct interaction with ligands: when proline is
bound as an amide in a peptide bond, its nitrogen is not bound to
any hydrogen, meaning it cannot act as a hydrogen bond donor. In
Fig. 11, we can see the entire
predicted 3D structure of Abca4 before dimerization and all
domains, emphasizing the 'transport channel' which involves the
423H>P substitution.
Discussion
We believe that RP1 homozygous variants found
in the proband could be responsible for his phenotype. His wife,
instead, although carrying a triple homozygous in the 'hot-spot'
region of RP1, normally associated with retinitis pigmentosa
pathology (15), was found to be
only mildly affected. Regarding the ABCA4 gene, she was
found to carry the c.1268A>G in heterozygosity. The non-affected
daughter inherited a condition of heterozygosity for all analyzed
variants of both genes, manifesting no typical symptoms of retinal
pathologies upon examination.
Online genetic database (EMBASE, ENSEMBL and PUBMED)
reports found variants as polymorphisms. The Human Gene Mutation
Database (HGMD) classified two of these (c.5008G>A for
RP1 and c.1268A>G for ABCA4) as disease-causing
mutations with a question mark (DM?), denoting a probable/possible
pathological mutation, reported to be pathogenic in the
corresponding report, but where the author has indicated that there
may be some degree of uncertainty.
The c.5008G>A, present in the wild-type condition
in the proband, implies the change of an alanine in position 1670
with a threonine implemented by this variation and represents a
regulatory region modification, due to its location in a promoter
flanking region. As with other RP1 analyzed SNPs, it would
appear to be implicated in retinitis pigmentosa phenotype of
Chinese (22–25) and Indian (26) populations, as well as indicated as
a member of the 'hot-spot' high causative region of RP1
(27).
The c.1268A>G, also found in the wild-type
condition in the proband, represents a missense variation, which
changes the histidine in position 423 with a proline, and is
located within a regulatory region, showing enhancer features,
involving one of TMD. It was regarded as a polymorphism found in
hetero-zygosity in 101/440 controls in a comprehensive survey of
sequence variation in the ABCA4 of a German population
(28). The same variant presented
as a high-penetrance disease-causing variant in a cohort of
patients with Stargardt disease in a study in 2004 (29), and as a reducing risk factor more
recently, also in a heterozygous model (30–32). Bioinformatic software predictions
(Sift, PolyPhen 2, PROVEAN), analyzing non-synonymous coding SNP
effects on protein function, give this variant the status of
tolerated or neutral. Furthermore, studies suggest c.1268A>G as
associated with late-onset Stargardt disease (33), with macular degeneration (34) and with retinitis pigmentosa
(35), depending on the severity
of the symptoms manifested. Despite all these studies, the
phenotype associated with this ABCA variant is not
clear.
According to our hypotheses, the c.1268A>G
missense variant may play a protective role against the damaging
RP1 'hot-spot' region variants in syndromic retinitis
pigmentosa. These findings suggest that, in our family case, the
variant examined led to an asymptomatic visual phenotype, without
any typical features of Stargardt disease or syndromic retinitis
pigmentosa. Our data are corroborated by the genetic (Fig. 6) and phenotypic (only a slight
reduction in sensitivity on peripheral areas) condition of the
proband's wife and daughter (the latter without any typical or
atypical symptomatology), suggesting the likely delaying effect of
the analyzed polymorphisms regarding pathology onset. We believe
that Rp1 and Abca4 could interact, directly or indirectly, in order
to extend the half-life of photoreceptors. In particular, we
speculate that the missense variant 1268A>G of ABCA4
induces a misfolding into an encoded protein, which decreases the
transport of ATR/NR-PE and, consequently, a lower quantity of PE
from disc membranes is consumed in spontaneous adduct formation
with ATR. This renewed stability of disc membrane lipids could
compensate for the lack due to RP1 homozygous variation in the
'hot-spot' region, which results in a misfolded protein unable to
guarantee the correct stacking of discs and, above all, proper
lipid transport from the inner to the outer segment, in order to
build new functional discs.
In conclusion, we analyzed the effects of
ABCA4 rs3112831 in a family with members showing a retinal
pathological genotypic condition for ABCA4 and RP1, but without any
evidence of phenotypic manifestations. Even thought the
c.1268A>G missense variant of the ABCA4 gene has often
been reported as causative of disease, and in other cases
protective of disease, in our family case, the variant appears to
reduce or delay the risk of onset of Stargardt disease.
References
|
1
|
Duno M, Schwartz M, Larsen PL and
Rosenberg T: Phenotypic and genetic spectrum of Danish patients
with ABCA4-related retinopathy. Ophthalmic Genet. 33:225–231. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Maugeri A, Klevering BJ, Rohrschneider K,
Blankenagel A, Brunner HG, Deutman AF, Hoyng CB and Cremers FP:
Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal
recessive cone-rod dystrophy. Am J Hum Genet. 67:960–966. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sun H and Nathans J: ABCR: Rod
photoreceptor-specific ABC transporter responsible for Stargardt
disease. Methods Enzymol. 315:879–897. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rees DC, Johnson E and Lewinson O: ABC
transporters: The power to change. Nat Rev Mol Cell Biol.
10:218–227. 2009. View
Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ambudkar SV, Dey S, Hrycyna CA,
Ramachandra M, Pastan I and Gottesman MM: Biochemical, cellular,
and pharmacological aspects of the multidrug transporter. Annu Rev
Pharmacol Toxicol. 39:361–398. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jang YP, Matsuda H, Itagaki Y, Nakanishi K
and Sparrow JR: Characterization of peroxy-A2E and furan-A2E
photooxidation products and detection in human and mouse retinal
pigment epithelial cell lipofuscin. J Biol Chem. 280:39732–39739.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Cideciyan AV, Aleman TS, Swider M,
Schwartz SB, Steinberg JD, Brucker AJ, Maguire AM, Bennett J, Stone
EM and Jacobson SG: Mutations in ABCA4 result in accumulation of
lipofuscin before slowing of the retinoid cycle: A reappraisal of
the human disease sequence. Hum Mol Genet. 13:525–534. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Pierrottet CO, Zuntini M, Digiuni M,
Bazzanella I, Ferri P, Paderni R, Rossetti LM, Cecchin S, Orzalesi
N and Bertelli M: Syndromic and non-syndromic forms of retinitis
pigmentosa: A comprehensive Italian clinical and molecular study
reveals new mutations. Genet Mol Res. 13:8815–8833. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chang S, Vaccarella L, Olatunji S, Cebulla
C and Christoforidis J: Diagnostic challenges in retinitis
pigmentosa: Genotypic multiplicity and phenotypic variability. Curr
Genomics. 12:267–275. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Young RW: The renewal of photoreceptor
cell outer segments. J Cell Biol. 33:61–72. 1967. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Anderson DH, Fisher SK and Steinberg RH:
Mammalian cones: Disc shedding, phagocytosis, and renewal. Invest
Ophthalmol Vis Sci. 17:117–133. 1978.PubMed/NCBI
|
|
12
|
Kinney MS and Fisher SK: The
photoreceptors and pigment epithelim of the adult Xenopus retina:
Morphology and outer segment renewal. Proc R Soc Lond B Biol Sci.
201:131–147. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Steinberg RH, Fisher SK and Anderson DH:
Disc morphogenesis in vertebrate photoreceptors. J Comp Neurol.
190:501–508. 1980. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Liu Q, Lyubarsky A, Skalet JH, Pugh EN Jr
and Pierce EA: RP1 is required for the correct stacking of outer
segment discs. Invest Ophthalmol Vis Sci. 44:4171–4183. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
El Shamieh S, Boulanger-Scemama E,
Lancelot ME, Antonio A, Démontant V, Condroyer C, Letexier M,
Saraiva JP, Mohand-Saïd S, Sahel JA, et al: Targeted next
generation sequencing identifies novel mutations in RP1 as a
relatively common cause of autosomal recessive rod-cone dystrophy.
Biomed Res Int. 2015:4856242015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Buchan DW, Minneci F, Nugent TC, Bryson K
and Jones DT: Scalable web services for the PSIPRED Protein
Analysis Workbench. Nucleic Acids Res. 41:W349–W357. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Altschul SF, Madden TL, Schäffer AA, Zhang
J, Zhang Z, Miller W and Lipman DJ: Gapped BLAST and PSI-BLAST: A
new generation of protein database search programs. Nucleic Acids
Res. 25:3389–3402. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Källberg M, Wang H, Wang S, Peng J, Wang
Z, Lu H and Xu J: Template-based protein structure modeling using
the RaptorX web server. Nat Protoc. 7:1511–1522. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma J, Wang S, Zhao F and Xu J: Protein
threading using context-specific alignment potential.
Bioinformatics. 29:i257–i265. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Peng J, Xu J and Raptor X: RaptorX:
Exploiting structure information for protein alignment by
statistical inference. Proteins. 79(Suppl 10): 161–171. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pettersen EF, Goddard TD, Huang CC, Couch
GS, Greenblatt DM, Meng EC and Ferrin TE: UCSF Chimera - a
visualization system for exploratory research and analysis. J
Comput Chem. 25:1605–1612. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Wang DY, Fan BJ, Chan WM, Tam OS, Chiang
WY, Lam SC and Pang CP: Digenic association of RHO and RP1 genes
with retinitis pigmentosa among Chinese population in Hong Kong.
Zhonghua Yi Xue Za Zhi. 85:1613–1617. 2005.In Chinese. PubMed/NCBI
|
|
23
|
Zhang X, Yeung KY, Pang CP and Fu W:
Mutation analysis of retinitis pigmentosa 1 gene in Chinese with
retinitis pigmentosa. Zhonghua Yi Xue Yi Chuan Xue Za Zhi.
19:194–197. 2002.In Chinese. PubMed/NCBI
|
|
24
|
Sheng X, Zhang X, Wu W, Zhuang W, Meng R
and Rong W: Variants of RP1 gene in Chinese patients with autosomal
dominant retinitis pigmentosa. Can J Ophthalmol. 43:208–212. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang X, Chen LJ, Law JP, Lai TY, Chiang
SW, Tam PO, Chu KY, Wang N, Zhang M and Pang CP: Differential
pattern of RP1 mutations in retinitis pigmentosa. Mol Vis.
16:1353–1360. 2010.PubMed/NCBI
|
|
26
|
Gandra M, Anandula V, Authiappan V,
Sundaramurthy S, Raman R, Bhattacharya S and Govindasamy K:
Retinitis pigmentosa: Mutation analysis of RHO, PRPF31, RP1, and
IMPDH1 genes in patients from India. Mol Vis. 14:1105–1113.
2008.PubMed/NCBI
|
|
27
|
Schwartz SB, Aleman TS, Cideciyan AV,
Swaroop A, Jacobson SG and Stone EM: De novo mutation in the RP1
gene (Arg677ter) associated with retinitis pigmentosa. Invest
Ophthalmol Vis Sci. 44:3593–3597. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Rivera A, White K, Stöhr H, Steiner K,
Hemmrich N, Grimm T, Jurklies B, Lorenz B, Scholl HP,
Apfelstedt-Sylla E, et al: A comprehensive survey of sequence
variation in the ABCA4 (ABCR) gene in Stargardt disease and
age-related macular degeneration. Am J Hum Genet. 67:800–813. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Oh KT, Weleber RG, Stone EM, Oh DM,
Rosenow J and Billingslea AM: Electroretinographic findings in
patients with Stargardt disease and fundus flavimaculatus. Retina.
24:920–928. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Aguirre-Lamban J, González-Aguilera JJ,
Riveiro-Alvarez R, Cantalapiedra D, Avila-Fernandez A,
Villaverde-Montero C, Corton M, Blanco-Kelly F, Garcia-Sandoval B
and Ayuso C: Further associations between mutations and
polymorphisms in the ABCA4 gene: Clinical implication of allelic
variants and their role as protector/risk factors. Invest
Ophthalmol Vis Sci. 52:6206–6212. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Brión M, Sanchez-Salorio M, Cortón M, de
la Fuente M, Pazos B, Othman M, Swaroop A, Abecasis G, Sobrino B
and Carracedo A; Spanish multi-centre group of AMD: Genetic
association study of age-related macular degeneration in the
Spanish population. Acta Ophthalmol. 89:e12–e22. 2011. View Article : Google Scholar
|
|
32
|
Webster AR, Héon E, Lotery AJ, Vandenburgh
K, Casavant TL, Oh KT, Beck G, Fishman GA, Lam BL, Levin A, et al:
An analysis of allelic variation in the ABCA4 gene. Invest
Ophthalmol Vis Sci. 42:1179–1189. 2001.PubMed/NCBI
|
|
33
|
Yatsenko AN, Shroyer NF, Lewis RA and
Lupski JR: Late-onset Stargardt disease is associated with missense
mutations that map outside known functional regions of ABCR
(ABCA4). Hum Genet. 108:346–355. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Baum L, Chan WM, Li WY, Lam DS, Wang PB
and Pang CP: ABCA4 sequence variants in Chinese patients with
age-related macular degeneration or Stargardt's disease.
Ophthalmologica. 217:111–114. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Valverde D, Riveiro-Alvarez R,
Aguirre-Lamban J, Baiget M, Carballo M, Antiñolo G, Millán JM,
Garcia Sandoval B and Ayuso C: Spectrum of the ABCA4 gene mutations
implicated in severe retinopathies in Spanish patients. Invest
Ophthalmol Vis Sci. 48:985–990. 2007. View Article : Google Scholar : PubMed/NCBI
|